
Abstract
CD4+ T cells play an essential role in orchestrating adequate immunity, but their overactivity has been associated with the development of immune-mediated inflammatory diseases, including liver inflammatory diseases. These cells can be subclassified according to their maturation stage, cytokine profile, and pro or anti-inflammatory functions, i.e., functional heterogeneity. In this review, we summarize what has been discovered so far regarding the role of the different CD4+ T cell polarization states in the progression of two prominent and still different liver inflammatory diseases: non-alcoholic steatohepatitis (NASH) and autoimmune hepatitis (AIH). Finally, the potential of CD4+ T cells as a therapeutic target in both NASH and AIH is discussed.
Similar content being viewed by others

Introduction
In healthy conditions, the liver can tolerate the influx of food- and bacterial-derived antigens and pathogen-associated molecular patters (PAMPs). This is possible due to several immunoregulatory mechanisms including a tight control of T cell activation by, for example, regulatory T cells [14, 68, 90]. However, a variety of environmental and genetic factors such as viral infection, alcohol, obesity, and HLA risk alleles can favor inflammatory liver diseases of which non-alcoholic steatohepatitis (NASH) and autoimmune hepatitis (AIH) are among the most common ones creating a severe public health challenge [2, 86, 109, 113, 138].
Obesity and metabolic syndrome promote accumulation of lipids in the liver and thereby cause NAFLD (non-alcoholic fatty liver disease). The accumulation of lipids is accompanied by cellular stress and leads, in some patients, to tissue damage and inflammation (NASH, non-alcoholic steatohepatitis) [49]. NASH development has been associated with high intake of nutrients, but also with an altered microbiota [11, 131]. A potentially detrimental effect of the intestinal microbiota on the progression from NAFLD to NASH has so far only been shown in mouse models. While germ-free mice on a high-fat diet (HFD) are protected from NASH, transplantation of stool from dysbiotic mice accelerates disease [4, 42, 121]. Translocation of bacterial antigens due to increased gut leakiness has also been suggested to link the intestine and the liver and thereby to further enhance inflammation and disease progression [30, 99].
AIH is characterized by destruction of the hepatic parenchyma by an autoreactive immune response. Clinical manifestations of early AIH are rather heterogeneous across patients, but characteristic to all of them is a progressive and detrimental disease with high titers of auto-antibodies and liver infiltrating plasma cells [5, 71, 82]. Tissue damage in AIH is directly mediated by immune cells and is usually accompanied by stronger infiltration of lymphocytes compared to NASH patients [125].
Both NASH and AIH can be followed by cirrhosis and hepatocellular carcinoma (HCC) [1, 122]. HCC caused roughly 782 000 deaths worldwide in 2018 [13].
The mechanism driving both NASH and AIH is not clear, but there is evidence of an important role of T cells.
T cells are the central orchestrators of inflammatory responses. Indeed, in an experimental mouse model of NASH, the blockade of CD4+ T cell infiltration into liver and small intestine protects the mice from the development of NASH [100]. In this model, an increased number of peripheral T cells express the integrins α4β5 when comparing MCD diet fed mice to those on a normal diet. At the same time, the expression of the α4β5 ligand MAdCAM-1 is elevated in the gut and liver tissue. The expression of MAdCAM-1 is dependent on the microbiota since antibiotic treatment reduces its expression. Infiltration of CD4+ T cells in both tissues can be blocked by α4β5 antibodies and protect from liver inflammation [100].
Evidence of the role of CD4+ T cells in AIH were provided using different mouse models. Conditional expression of autoantigen in the liver was shown to cause spontaneous development of AIH by autoreactive CD4+ T cells. Similarly, a defect in central tolerance due to a deletion of medullary thymic epithelial cells or by thymectomy of neonatal PD1−/− mice caused AIH in a T cell-dependent mechanism. Finally, transfer of CD4+ T cells of mice suffering from AIH could induce liver inflammation in recipient mice.
Considering that T cells, in particular CD4+ T cells, play a key role in both NASH and AIH, here we will dissect the contribution of the different subsets of CD4+ T cells in the pathogenesis of these immune-mediated inflammatory liver diseases.
Naïve CD4+ T cells
Priming of naïve CD4+ T cells usually occurs in secondary lymphoid organs such as spleen and lymph nodes. Naïve CD4+ T cells are found in circulation, and by expressing a particular combination of receptors (e.g., CCR7 and CD62L), they are able to home to the lymphnodes (e.g., CCR7) but not to enter tissue [18, 23, 47, 66, 102]. However, in contrast to other tissues, the architecture of the liver allows interaction of blood circulating T cells with antigen-presenting cells of the liver in the sinusoids [8, 22, 129]. Consequently, the liver might not only represent an additional site of T cell priming, but its unique environment might also predetermine the fate of CD4+ T effector cells during inflammatory liver disease. The anatomical site of priming of naïve CD4+ T cells is proposed as an important factor determining subsequent CD4+ T cell polarization and their capacity to infiltrate tissues. Recently, we have demonstrated the presence of resident naïve like T cells in human livers; however whether these cells are primed in the tissue and whether this determines their potential pathogenic fate remains still unclear, especially in the context of liver inflammation. Despite the lack of data on the above mentioned concept, there is data on the possibility that naïve T cells can be directly primed in the liver.
Professional antigen-presenting cells in the liver are liver sinusoidal endothelial cells (LSEC) and Kupffer cells, both lining the liver sinusoids which makes them easily accessible for blood circulating naïve T cells. In in vitro culture, LSEC are able to efficiently present antigens and activate naïve CD4+ T cells. In this system, CD4+ T cells start producing the cytokines IL-10, IL-4, and IFN-γ [60]. However, CD45− CD31bright cells, which might either represent LSEC or vascular endothelial cells, were not able to activate naïve CD4+ T cells [58]. Therefore, further studies are needed to fully elucidate the role of LSEC in priming naïve CD4+ T cells in vivo.
Kupffer cells are macrophages specialized to the liver environment. They reside in the sinusoids and express high levels of MHCII and co-stimulatory molecules and are able to activate naïve CD4+ T cells even though to a lesser extent than splenic dendritic cells [75, 135].
Transgenic mouse models expressing specific antigens in the liver have been used to study T cell activation in vivo. Using a mouse model of antigen (i.e., ovalbumin) specific activation, the activation of antigen-specific CD8+ T cells was observed, while the activation of CD4+ T cells failed [25, 130]. However, another study suggests that the abovementioned effect is at least partially dependent on the type of antigen, since in a similar transgenic mouse model in which antigen derived from mycobacterium instead of ovalbumin is expressed, naïve CD4+ T cells could be activated in the liver by Kupffer cells [118].
Additionally, ectopic expression of neural antigen in the liver leads to development of naive CD4+ T cells into Foxp3+ TREG cells, which in turn protect from experimental autoimmune encephalomyelitis (EAE)/immunopathology in the central nervous system (CNS) [19, 78]. This suggests that naïve CD4+ T cells can infiltrate the liver and, at least under physiological conditions, will acquire a regulatory phenotype promoting tolerance to antigen present in the liver.
Less accessible than LSECs and Kupffer cells are hepatic stellate cells (HSC) which are located in the perisinusoidal space. Despite being less accessible, in vitro cultures and adoptive transfer experiments in which mice are lacking MHCI showed that HSC cells can present antigen to T cells [128].
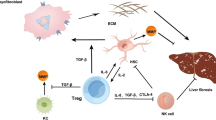
During inflammatory conditions, the portfolio of antigen-presenting cells in the liver might be expanded to hepatocytes. Hepatocytes express no or low levels of MHCII during physiological conditions. However, MHCII expression on hepatocytes can be detected in alcoholic and non-alcoholic hepatitis [77]. In a transgenic mouse model expressing MHCII on hepatocytes, CD4+ T cells can indeed be activated [45] (Fig. 1). Whether priming of naïve T cells by hepatocytes is taking place in NASH or AIH still needs to be confirmed.

Liver CD4+ T cells and their role in NASH and AIH. Naïve CD4+ T cells can be primed directly in the liver by different types of antigen-presenting cells (APC) and then mature into effector T cells with different polarization states, namely TH1, TH2, and TH17. The cytokine profile of the effector cells has been associated with the development of NASH and AIH. Effector T cells can form effector memory (TEM) and central memory T (TCM) cells, and their cytokine profile has also been associated with the development of NASH. In AIH, the frequency of memory TREG cells was found to be not significantly different between AIH patients and healthy subjects. Finally, it has been proposed that Foxp3+ TREG cells can undergo apoptosis in the inflamed liver and thus, probably, unleash the pathogenic activity of the effector T cells
Additionally, the impact of naïve T cells activated by hepatocytes during chronic inflammation on the disease progression needs further investigation. A study performed in a transgenic mouse model expressing antigen on hepatocytes observed indeed an impaired T cell response during LCMV infection. In this model, T cells showed a decreased INF-γ production and transgenic mice were impaired in virus control [127].
In summary, data from in vitro cultures and transgenic mouse models suggest that naïve CD4+ T cell can be primed in the liver. However, the consequences of this ectopic activation for human inflammatory liver diseases need to be further investigated.
Effector T cells
It has been suggested that CD4+ effector T cells play important roles in both protecting the liver from infections and also causing hepatocellular damage and autoimmunity [97]. Early stages of liver inflammation are dominated by CD4+ effector T cells and followed by a cytotoxic CD8+ T cell response [91, 112, 116]. Effector CD4+ T cells can acquire different cell states (i.e., TH1, TH2, and TH17 cells), here referred to as subsets, which are characterized by different cytokine profiles. Moreover, it has been shown that CD4+ T cell subsets can display a mixed phenotype characterized by the concomitant features of different polarization states, e.g., cytokines, and even potentially loose their originally polarization state acquiring a different one. For example, TH17 cells can acquire both a TH1 phenotype under chronic inflammation [39, 46, 57] and an anti-inflammatory phenotype during the resolution of the inflammation [34, 132]. The above described cellular phenomenon is here defined as T cell plasticity.
The different role of CD4+ T cell subsets in NASH and AIH patients has begun to be elucidated (Fig. 1). However, the role of plasticity in the context of AIH and NASH has scarcely been investigated.
Below we provide a summary of the role of the different CD4+ T cells subsets in NASH and AIH.
TH1 cells
The infiltration of the liver by TH1 cells, which are characterized by the production of IFN-γ, was shown to correlate also strongly with disease progression and liver injury of AIH patients [108].
TH1 cells were found to be enriched in the liver of NASH patients [7, 50]. Furthermore, investigating the NASH hepatic gene signature, IFN-γ response pathway genes showed the highest enrichment [38]. In the peripheral blood of NAFLD patients, Rau et al. also showed an increase in TH1 cells compared to healthy controls [101]. The potential pathogenic role of IFN-γ in the liver is probably attributed to its multiple detrimental functions, including induction of hepatocyte apoptosis and cell cycle arrest [111], induction of expression of chemokines such as CCR2 and their receptors on liver cells [51], and activation of Kupffer cells [142].
CD4+ T cells can adapt in response to a changing environment and therefore exhibit different polarization states [34, 132]. However, studies exploring the role of T cells in NASH are so far limited to selected key cytokines, such as IFN-γ, and do not investigate the potential plasticity of T cells.
In the liver of AIH patients, an increase of IFN-γ–producing cells was observed [74, 139, 140]. In addition, in a concanavalin A (ConA) mouse model of immune-mediated liver injury, a reduction of serum IFN-γ levels lead to decreased liver injury [141].
Furthermore, the presence of TH17 cells and T cells co-producing IFN-γ and TNF-α was reported in AIH [12, 103]: Findings indicated that TNF-α-producing CD4+ T cells were significantly expanded, both in blood and liver of AIH patients. However, the majority of the TNF-α-producing CD4+ T cells in AIH also co-produced IFN-γ, suggesting that these cells might represent a pathogenic activation state of TH1 cells [12].
CD4+ T cells co-producing IFN-γ and IL-17A (TH1/TH17 cells) were found to be decreased in the early stages of AIH pathogenesis in the blood, consistent with a working hypothesis of an enhanced recruitment of cells into the liver. Interestingly, AIH patients under standard immunosuppression (corticosteroids, azathioprine) failed to correct these TH1/TH17 imbalances in the blood and a persistent infiltration in the liver was observed, demonstrating that a deeper immunological restoration of tolerance does not occur despite satisfactory resolution of hepatitis [103].
These findings indicate that AIH is not only associated with classical TH cell subsets, but rather with a larger spectrum of mixed TH cell subsets with different polarization states, which should be further investigated (Fig. 1).
TH2 cells
TH2 cells ensure protective immunity against helminthic infections and play a key role in the pathogenesis of allergic diseases [124]. In the liver, TH2 cells were shown to have a strong pro-fibrogenic effect, and inhibition of IL-13 signaling blocks fibrosis development [95]. Few studies have thoroughly investigated this subset in the context of NAFLD and AIH and their role in these diseases remains unknown.
Rau et al. described an increase in circulating TH2 cells of NAFLD patients compared to healthy normal-weight controls, who were not matched for age [101]. Interestingly, 12 months after bariatric surgery, the TH2/ Foxp3+ TREG ratio was decreased. However, other authors did not find any differences in TH2 numbers, neither in peripheral blood nor in the liver when they compared NASH patients and NAFLD patients or controls [31, 50]. To our knowledge, the involvement of the TH2 subset has not been thoroughly investigated in an animal model of NAFLD.
In AIH, the role of TH2 cells still remains elusive. Early studies showed that the TH2 cytokines IL-5 and IL-13 were present in the late cirrhotic stage of AIH patients [24, 26, 91, 92]. In type I autoimmune hepatitis in children, an increased mRNA expression of Il4 was observed in liver samples [21]. Furthermore, it is known that the cytokines IL-4 and IL-6 can regulate B-cell activation and promote the production of antinuclear antibodies (ANA) and anti-smooth muscle antibodies (SMA) [110]. However, no significant differences in Il4 mRNA expression levels between patients and healthy subjects were observed in peripheral blood mononuclear cells (PBMCs) [6].
In mouse models, IL-4 producing TH2 cells play an essential role in inducing ConA-immune-mediated liver injury via activation of STAT6. STAT6 upregulates the expression of the chemoattractant eotaxin in hepatocytes and sinusoidal endothelial cells and induces IL-5 expression, resulting in eosinophil and neutrophil recruitment into the liver and leading to hepatitis [52] (Fig. 1).
TH17 cells
TH17 cells are characterized by the production of IL-17A, IL-17F, and IL-22 and are believed to play an important role in the development of a variety of autoimmune diseases [64].
TH17 cells were shown to be present in larger numbers in the liver of NASH patients in comparison to healthy controls [36, 101, 123]. Moreover, Rau et al. report a decrease of TH17 cells in peripheral blood, as well as in the TH17/ Foxp3+ TREG ratio, when NASH patients were re-evaluated 12 months after bariatric surgery [101].
Furthermore, TH17 cells were also shown to be present in larger numbers in the liver and peripheral blood of NAFLD mouse models [81, 106, 115]. The IL-17A and IL-17F axis was shown to be important in the development and progression of NASH. IL-17RA−/−, IL-17A−/−, and IL-17F−/− mice exhibited decreased steatohepatitis and hepatocellular damage [35, 41, 106]. In line with these findings, the use of an anti-IL-17 monoclonal antibody significantly improved liver function, attenuated hepatic lipid accumulation, suppressed Kupffer cell activation, and decreased pro-inflammatory cytokine levels in a model of HFD induced NAFLD [133, 134]. Simultaneous blocking of CD25 or IL-17A and IL-17F shifted the TH17/Foxp3+ TREG imbalance from MCD diet-induced TH17 dominance to Foxp3+ TREG dominance and also decreased hepatic steatosis and inflammation [73].
Moreover, multiple experimental murine and in vitro models showed that IL-17A administration can lead to an increase in hepatic steatosis [41, 44]. IL-17A was reported to have a pro-fibrotic effect through activation of hepatic stellate cells [114] and an in vitro study showed that IL-17A enhances the expression of pro-fibrotic genes (e.g., ACTA2 and COL1A1) through an upregulation of TGF-β receptor [28].
AIH has been associated with IL-17A expression [12, 37, 63, 70, 139, 140], although the role of TH17 cells in the pathogenesis of AIH remains controversial.
The frequency of circulating TH17 cells and the expression of the key transcription factor for these cells, RORɣt, were elevated in PBMCs of AIH patients [6, 136, 139, 140]. In the liver of AIH patients, the frequency of IL-17A producing cells and the expression of TH17-related cytokines (IL-23, IL-21, IL-1β, and IL-6) was also significantly elevated [139, 140]. Interestingly, the duration and severity of hepatitis may be dependent on TH17 cells in AIH [117].
In ConA-induced liver injury, IL-17A-deficient mice develop the same level of liver injury as wild-type mice [137]. In contrast, the results from two independent research studies indicated that IL-17A-deficient mice had a significant reduction in liver injury compared with wild-type mice [63, 88]. The reason for the discrepancy between the findings is not clear, but the authors speculate that they could be attributed to the different environment of the animal facilities that may affect IL-17A−/− mice.
While IL-17A and IL-17F seem to play an important role in inducing liver inflammation via stimulating multiple types of liver non-parenchymal cells to produce pro-inflammatory cytokines and chemokines, IL-22 appears to be an important factor in promoting hepatocyte survival and proliferation. It was demonstrated that treatment with IL-22 prevents, while treatment with IL-22 neutralizing antibodies enhances ConA-induced liver injury [98]. The hepatoprotective role of IL-22 in T-cell hepatitis was also confirmed by other studies using IL-22-deficient mice [59, 137] (Fig. 1).
Memory T cells
Following the expansion phase of effector T cells, three main populations of memory cells can be recognized: central memory T cells (TCM), effector memory T cells (TEM), and tissue-resident memory T cells (TRM). At present, these memory T cell subsets are primarily characterized by their phenotype, migratory properties, and tissue homing patterns, which in many instances imply unique functional attributes [3, 87]. Memory T cells are not only involved in promoting physiological immunity, but also in promoting autoimmune responses. Due to rapid pro-inflammatory qualities of TRM cells, they can lead a misguided action and result in immunopathology [62]. The role of CD4+ memory T cells in NASH and AIH has scarcely been investigated and the phenotypic markers used to define memory T cells are not consistent between studies.
In NASH, patients showed increased numbers of IFN-γ+ memory (CD45RO+) CD4+ and CD8+ T cells compared with controls, while numbers of CD4+ and CD8+ CD45RA+ subsets were decreased [50]. One of the molecular mechanisms driving T cell infiltration into the liver is increased chemotaxis, as peripheral CD4+ T cells from obese mice and NASH patients migrate more readily toward the chemokine CXCL12 compared to T cells from healthy mice or healthy donors [10]. In line with this finding, a longitudinal analysis of peripheral blood of humanized mice showed that central memory (CCR7+CD45RA–) and effector memory (CCR7–CD45RA–) CD4+ T cells and their associated cytokines IL-17A and IFN-γ expanded with time and infiltrated the liver (Her et al.,2020).
Regulatory T cells
Regulatory T cells and the capacity of some effector cells to convert into regulatory T cells provide, among others, key mechanisms to establish peripheral tolerance. CD4+ T cells with regulatory function can be divided into at least two subsets: Foxp3+ TREG cells and Foxp3− IL10+ type 1 regulatory T (TR1) cells. Both cell subsets have been described in the context of liver tolerance.
In an hepatitis B virus (HBV) carrier mouse model, Kupffer cells induce TR1 cells rather than Foxp3+ TREG cells [133, 134]. Transfer of CD4+ T cells from HBV carrier mice confers systemic tolerance to HBV antigen in recipient mice. This tolerogenic effect is dependent on IL-10 expression. However, the majority of reports on regulatory T cell subsets in the liver focuses on Foxp3+ TREG cells. Specifically in the context of NASH and AIH, the role of TR1 cells has not yet been investigated.
A first indication for the importance of Foxp3+ TREG cells in liver tolerance is provided by the observation that injection of anti-CD25 antibodies leads to rejection of liver transplants in mice [67]. Foxp3+ TREG cells can be induced both by hepatocytes and HSC in in vitro cultures. The induction is favored by the presence of TGF-β and Notch signaling by hepatocytes and IL-2 and retinoid acid receptor in the context of HSC co-culture [17, 27, 54]. In vivo, expression of antigen delivered by AAV vectors in hepatocytes leads to tolerance towards the antigen which is presumably mediated by Foxp3+ TREG and Kupffer cells [14]. The tolerogenic function of the liver can be utilized for therapeutic purposes in mouse models. Indeed, application of nanoparticles delivering neuronal peptide to LSEC protects from immunopathology in EAE through the conversion of T cells to Foxp3+ TREG in a TGF-β-dependent mechanism [19]. Hence, Foxp3+ TREG cells are generated in the tolerogenic liver environment and can support tolerance in the CNS.
During the inflammatory condition of NASH, Foxp3+ TREG cells play a key role in disease control. Depletion of Foxp3+ TREG cells in a mouse model of NASH aggravates disease [105]. The inflammatory environment during NASH impairs Foxp3+ TREG cell survival. Oxidative stress, TNF-α, and type I interferon produced by Kupffer cells and dendritic cells during NASH promote apoptosis of Foxp3+ TREG cells [80, 105]. Oxidative stress seems to preferentially induce apoptosis in Foxp3+ TREG cells and consequently shifts the ratio of effector T cells to Foxp3+ TREG cells towards effector T cells [80]. More precisely, both the ratio of TH17 and TH2 effector cells to TREG cells have been associated with severity of inflammation, i.e., the progression of NAFLD to NASH. Bariatric surgery could recover the imbalance of Foxp3+ TREG and effector T cells [101]. Overall, Foxp3+ TREG cells seem to have a protective role during disease progression in mouse models of NASH. However, the protective function of Foxp3+ TREG cells might be limited by their increased apoptosis rate during inflammation.
The role of Foxp3+ TREG in AIH disease progression is still discussed.
Clearly, adoptive transfer of Foxp3+ TREG cells can overcome inflammation in experimental mouse models of AIH including a model of AIRE-mutation, xenoimmunization with human autoantigen, and ConA-induced liver injury [40, 48, 65]. However, it remains unclear whether Foxp3+ TREG cells in AIH patients are impaired in number. In part, conflicting results can be explained by different approaches to define Foxp3+ TREG cells. In a study investigating Foxp3+ TREG defined by CD25hi expression, the authors observed diminished Foxp3+ TREG cells in AIH patients in comparison to healthy controls [76]. Importantly, CD25 is not only expressed by Foxp3+ TREG but also by activated effector T cells. Hence, other studies have distinguished Foxp3+ TREG and T effector cells in human more precisely by combining CD25, CD127, and Foxp3. Using this more stringent definition of Foxp3+ TREG cells, no difference in the frequency of Foxp3+ TREG in AIH patients compared to healthy controls was observed [94]. Also, the frequency of memory Foxp3+ TREG cells defined as CD25+CD127‐FOXP3+ CD45RA− were not significantly different between AIH patients and healthy subjects [103].
Moreover, the frequency of Foxp3+ TREG cells in the blood positively correlates with severity of inflammation within the group of AIH patients [94]. In line with this observation, intrahepatic Foxp3+ TREG cells in untreated AIH patients are rather enriched, and the number of these cells decreases during immunosuppression [116].
Another study focused on the Foxp3+ TREG to effector T cell ratio rather than the plain number of these cells. The authors found a dysbalance of Foxp3+ TREG and effector T cells in AIH patients [69]. One possible explanation comes from data suggesting that Foxp3+ TREG cells are more prone to undergo apoptosis in active AIH patients [55]. In addition, Foxp3+ TREG cells from a proinflammatory enviroment exhibit lower levels of the anti-apoptotic molecule c-Flip and high expression of CD95, indicating elevated susceptibility to Fas-mediated apoptosis [20, 96]. Additionally, Foxp3+ TREG cells require the cytokine IL-2 for survival, and the concentration of IL-2 was shown be lower in diseased liver as compared to healthy liver [20]. Indeed, in vitro studies stimulating PBMCs or liver infiltrating lymphocytes from patients with autoimmune liver diseases with low doses of IL-2 showed improved survival and function of Foxp3+ TREG [53]. In line with this, in an experimental mouse model of AIH, treatment with complexed IL-2/anti-IL-2 could increase the number of Foxp3+ TREG and diminish disease severity [16] (Fig. 1).
In conclusion, on the one hand, an increased susceptibility of Foxp3+ TREG to undergo apoptosis during a pathological liver inflammation might explain the restrict capacity of these cells to expand in equal proportion to effector T cells. On the other hand, the preliminary and promising preclinical and clinical studies testing either Foxp3+ TREG cell therapy or complexed IL-2 suggest that Foxp3+ TREG are definitely an interesting therapeutic target for AIH and probably also for NASH patients.
Therapeutic approaches
While homeostatic inflammation is an aspect of an healthy liver, a lack of resolution or chronic liver injury leads to progressive liver fibrosis and permanent liver damage. Eventually a chronic liver inflammation leads to HCC and to the death of the patients.
For the treatment of NASH, there is not a single drug approved by the Food and Drug Administration (FDA) or European Medicines Agency (EMA). Standard AIH treatment consists of immunosuppressive therapy; however more than 70% of patients relapse when treatment withdrawal is attempted, suggesting a persistence of pathogenic cells, such as autoreactive CD4+ T cells [43, 117].
Approaching liver disease as a range of overlapping pathways leading to the dysregulation of homeostatic inflammatory processes provides novel avenues for the development of future therapies targeting inflammation and resolution within the liver.
There are only a few therapeutic approaches in NASH targeting T cells. CCR2 was shown to play an important role in T cell differentiation [79]. In hepatic inflammation and fibrosis the dual CCR2/CCR5 chemokine receptor antagonist (Cenicriviroc) has been efficient [29] and is therefore been investigated in current phase III clinical trials in patients with NASH and fibrosis (ClinicalTrials.gov Identifier: NCT03028740). However, a decrease in fibrosis but no NASH resolution was observed in a phase IIb trial (ClinicalTrials.gov Identifier: NCT02217475).
Since TNF-α producing cells, including T cells, were shown to be involved in the pathogenesis of AIH, a study by Weiler-Norman and colleagues reported the first series of AIH patients who were treated with infliximab, an antibody targeting TNF-α. The study included 11 difficult-to-treat AIH patients to whom the standard treatment did not lead to remission. Here, infliximab treatment led to a reduction of inflammation [56, 126].
Of note, a retrospective statistical analysis of different clinical studies all including patients treated with anti-TNF-α, and a single center report of 8 cases, showed that anti-TNF-α treatment associates with liver damage [33, 104]. Therefore, further clinical studies testing the effect of anti-TNF-α treatment are urgently needed in AIH patients.
Anecdotal clinical observations with off-label use of ustekinumab, a pharmacological antagonist of the IL-23/IL-17 axis [32], do not indicate a significant effect on AIH activity.
The association between regulatory T cell deficiency and inadequate immune tolerance in AIH sparked rationale to treat autoimmune diseases by the administering autologous Foxp3+ TREG cells. Foxp3+ TREG cell directed therapy, though ex vivo expansion or IL-2 administration, is increasingly tested in the context of posttransplant tolerance [15, 107, 119, 120] and type 1 diabetes mellitus [9, 84]. Here, the safety and feasibility of Foxp3+ TREG cell therapy in humans was shown, and the evidence suggested potential improvements in clinical, biochemical, and immunological status with Foxp3+ TREG cell therapy. In AIH patients, it has been shown that autologous Foxp3+ TREG cell therapy is feasible and safe, and interestingly a strong preferential homing of Foxp3+ TREG cells to the liver and spleen was observed for up to 72 h. However, this study was neither designed nor had the statistical power to demonstrate an effect on AIH activity [93]. Also, administration of IL-2 to AIH patients was shown to increase the pool of circulating Foxp3+ TREG cells, and it was proven to be safe in the two treated patients [72]. In short, despite these preliminary encouraging data, larger clinical trials targeting Foxp3+ TREG cells in AIH patients are urgently needed.
Finally, CXCL9 and CXCL10 were shown to regulate the differentiation of naïve T cells to TH1 cells and lead to the migration of immune cells to inflammatory sites [61, 85]. Plasma levels of CXCL9 and CXCL10 increase with advancing disease stage in AIH [89], although this can be reduced with administration of ursodeoxycholic acid (UDCA) in some patients [83]. A multicenter phase-II clinical trial of a humanized anti-CXCL10 antibody in the treatment of primary biliary cholangitis is currently underway. If this shows promise, AIH would also be a potential indication for future therapeutic study using this agent.
In conclusion, there are only few therapeutic approaches targeting T cells in NASH. However, due to the strong T cell activation within this disease, T cells could be a promising target for future therapies. Furthermore, in AIH patients, cell-based therapies, such as regulatory T cell therapy, could finally replace long-term immunosuppression treatments which are still characterized by serious side effects.
References
Anstee QM, Reeves HL, Kotsiliti E, Govaere O, & Heikenwalder M (2019) From NASH to HCC: current concepts and future challenges. In Nature Reviews Gastroenterology and Hepatology (Vol. 16, Issue 7, pp. 411–428). Nature Publishing Group. https://doi.org/10.1038/s41575-019-0145-7
Asrani SK, Devarbhavi H, Eaton J, & Kamath PS (2019) Burden of liver diseases in the world. In Journal of Hepatology (Vol. 70, Issue 1, pp. 151–171). Elsevier B.V. https://doi.org/10.1016/j.jhep.2018.09.014
Baaten BJG, Cooper AM, Swain SL, & Bradley LM (2013) Location, location, location: the impact of migratory heterogeneity on T cell function. In Frontiers in Immunology (Vol. 4, Issue OCT). Front Immunol. https://doi.org/10.3389/fimmu.2013.00311
Bäckhed F, Ding H, Wang T, Hooper LV, Gou YK, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101(44):15718–15723. https://doi.org/10.1073/pnas.0407076101
Balitzer D, Shafizadeh N, Peters MG, Ferrell LD, Alshak N, Kakar S (2017) Autoimmune hepatitis: review of histologic features included in the simplified criteria proposed by the international autoimmune hepatitis group and proposal for new histologic criteria. Mod Pathol 30(5):773–783. https://doi.org/10.1038/modpathol.2016.267
Behfarjam F, Nasseri-Moghaddam S, Jadali Z (2019) Enhanced Th17 responses in patients with autoimmune hepatitis. Middle East J Digest Dis 11(2):98–103. https://doi.org/10.15171/mejdd.2018.134
Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, Gugenheim J, Barr J, Mato JM, Le Marchand-Brustel Y, Tran A, & Gual P (2010) Hepatic expression patterns of inflammatory and immune response genes associated with obesity and nash in morbidly obese patients. PLoS ONE 5(10). https://doi.org/10.1371/journal.pone.0013577
Bertolino P, McCaughan GW, & Bowen DG (2002) Role of primary intrahepatic T-cell activation in the “liver tolerance effect.” In Immunology and Cell Biology (Vol. 80, Issue 1, pp. 84–92). Immunol Cell Biol. https://doi.org/10.1046/j.0818-9641.2001.01048.x
Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, & Tang Q (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7(315). https://doi.org/10.1126/scitranslmed.aad4134
Boujedidi H, Robert O, Bignon A, Cassard-Doulcier AM, Renoud ML, Gary-Gouy H, Hemon P, Tharinger H, Prévot S, Bachelerie F, Naveau S, Emilie D, Balabanian K, Perlemuter G (2015) CXCR4 dysfunction in non-alcoholic steatohepatitis in mice and patients. Clin Sci 128(4):257–267. https://doi.org/10.1042/CS20130833
Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, Hunault G, Oberti F, Calès P, Diehl AM (2016) The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63(3):764–775. https://doi.org/10.1002/hep.28356
Bovensiepen CS, Schakat M, Sebode M, Zenouzi R, Hartl J, Peiseler M, Li J, Henze L, Woestemeier A, Schramm C, Lohse AW, Herkel J, Weiler-Normann C (2019) TNF-producing Th1 cells are selectively expanded in liver infiltrates of patients with autoimmune hepatitis. J Immunol 203(12):3148–3156. https://doi.org/10.4049/jimmunol.1900124
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. https://doi.org/10.3322/caac.21492
Breous E, Somanathan S, Vandenberghe LH, Wilson JM (2009) Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 50(2):612–621. https://doi.org/10.1002/hep.23043
Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, DeFor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE (2011) Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: Safety profile and detection kinetics. Blood 117(3):1061–1070. https://doi.org/10.1182/blood-2010-07-293795
Buitrago-Molina LE, Pietrek J, Noyan F, Schlue J, Manns MP, Wedemeyer H, Hardtke-Wolenski M, Jaeckel E (2021) Treg-specific IL-2 therapy can reestablish intrahepatic immune regulation in autoimmune hepatitis. J Autoimmun 117:102591. https://doi.org/10.1016/j.jaut.2020.102591
Burghardt S, Claass B, Erhardt A, Karimi K, Tiegs G (2014) Hepatocytes induce Foxp3 + regulatory T cells by notch signaling. J Leukoc Biol 96(4):571–577. https://doi.org/10.1189/jlb.2ab0613-342rr
Butcher EC, Williams M, Youngman K, Rott L, Briskin M (1999) Lymphocyte trafficking and regional immunity. Adv Immunol 72(72):209–253. https://doi.org/10.1016/s0065-2776(08)60022-x
Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, Wraith DC, Korn T, Schramm C, Lohse AW, Heeren J, Herkel J (2014) TGF-β-dependent induction of CD4+CD25+Foxp3 + Tregs by liver sinusoidal endothelial cells. J Hepatol 61(3):594–599. https://doi.org/10.1016/j.jhep.2014.04.027
Chen YY, Jeffery HC, Hunter S, Bhogal R, Birtwistle J, Braitch MK, Roberts S, Ming M, Hannah J, Thomas C, Adali G, Hübscher SG, Syn WK, Afford S, Lalor PF, Adams DH, Oo YH (2016) Human intrahepatic regulatory T cells are functional, require IL-2 from effector cells for survival, and are susceptible to Fas ligand-mediated apoptosis. Hepatology 64(1):138–150. https://doi.org/10.1002/hep.28517
Cherñavsky AC, Paladino N, Rubio AE, De Biasio MB, Periolo N, Cuarterolo M, Goñi J, Galoppo C, Cañero-Velasco MC, Muñoz AE, Fainboim H, Fainboim L (2004) Simultaneous expression of Th1 cytokines and IL-4 confers severe characteristics to type I autoimmune hepatitis in children. Hum Immunol 65(7):683–691. https://doi.org/10.1016/j.humimm.2004.03.004
Crispe IN (2011) Liver antigen-presenting cells. In Journal of Hepatology (Vol. 54, Issue 2, pp. 357–365). J Hepatol. https://doi.org/10.1016/j.jhep.2010.10.005
Cyster JG (2005) Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. In Annual Review of Immunology (Vol. 23, pp. 127–159). Annu Rev Immunol. https://doi.org/10.1146/annurev.immunol.23.021704.115628
Czaja AJ, Carpenter HA, Santrach PJ, Moore SB (1993) Significance of HLA DR4 in type 1 autoimmune hepatitis. Gastroenterology 105(5):1502–1507. https://doi.org/10.1016/0016-5085(93)90157-8
Derkow K, Loddenkemper C, Mintern J, Kruse N, Klugewitz K, Berg T, Wiedenmann B, Ploegh HL, Schott E (2007) Differential priming of CD8 and CD4 T-cells in animal models of autoimmune hepatitis and cholangitis. Hepatology 46(4):1155–1165. https://doi.org/10.1002/hep.21796
Doherty DG, Donaldson PT, Underhill JA, Farrant JM, Duthie A, Mieli-Vergani G, McFarlane IG, Johnson PJ, Eddleston ALWF, Mowat AP, Williams R (1994) Allelic sequence variation in the HLA class II genes and proteins in patients with autoimmune hepatitis. Hepatology 19(3):609–615. https://doi.org/10.1002/hep.1840190311
Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, Grakoui A (2013) Hepatic stellate cells preferentially induce Foxp3 + regulatory t cells by production of retinoic acid. J Immunol 190(5):2009–2016. https://doi.org/10.4049/jimmunol.1201937
Fabre T, Kared H, Friedman SL, Shoukry NH (2014) IL-17A Enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol 193(8):3925–3933. https://doi.org/10.4049/jimmunol.1400861
Fantuzzi L, Tagliamonte M, Gauzzi MC, & Lopalco L (2019) Dual CCR5/CCR2 targeting: opportunities for the cure of complex disorders. In Cellular and Molecular Life Sciences (Vol. 76, Issue 24, pp. 4869–4886). Springer. https://doi.org/10.1007/s00018-019-03255-6
Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarzian A (2008) Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int 28(7):1026–1033. https://doi.org/10.1111/j.1478-3231.2008.01723.x
Ferreyra Solari NE, Inzaugarat ME, Baz P, De Matteo E, Lezama C, Galoppo M, Galoppo C, Cherñavsky AC (2012) The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J Clin Immunol 32(3):611–621. https://doi.org/10.1007/s10875-011-9635-2
Fragoulis GE, Siebert S, Mcinnes IB (2016) Therapeutic targeting of IL-17 and IL-23 cytokines in immune-mediated diseases. Annu Rev Med 67:337–353. https://doi.org/10.1146/annurev-med-051914-021944
French JB, Bonacini M, Ghabril M, Foureau D, & Bonkovsky HL (2016) Hepatotoxicity associated with the use of anti-TNF-α agents. In Drug Safety (Vol. 39, Issue 3, pp. 199–208). Springer International Publishing. https://doi.org/10.1007/s40264-015-0366-9
Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, De Zoete MR, Licona-Limón P, Paiva RS, Ching T, Weaver C, Zi X, Pan X, Fan R, Garmire LX, Cotton MJ, Drier Y, Bernstein B, Geginat J, … Flavell RA (2015) TH17 cells transdifferentiate into regulatory T cells uring resolution of inflammation. Nature 523(7559):221–225. https://doi.org/10.1038/nature14452
Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Cappelletti M, Huppert SS, Iwakura Y, Dong C, Shanmukhappa SK, & Divanovic S (2016) Regulation of inflammation by IL-17A and IL-17F modulates non-alcoholic fatty liver disease pathogenesis. PLoS ONE 11(2). https://doi.org/10.1371/journal.pone.0149783
Gomes AL, Teijeiro A, Burén S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, Perna C, Djouder N (2016) Metabolic Inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30(1):161–175. https://doi.org/10.1016/j.ccell.2016.05.020
Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC, Mieli-Vergani G, Vergani D, Longhi MS (2014) Dysfunctional CD39POS regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology 59(3):1007–1015. https://doi.org/10.1002/hep.26583
Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, Fleury S, Deprince A, Nikitin A, Woitrain E, Ducrocq-Geoffroy L, Pic S, Derudas B, Dehondt H, Gheeraert C, Van Gaal L, Driessen A, Lefebvre P, Staels B, Francque S, Dombrowicz D (2019) Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab 1(6):604–614. https://doi.org/10.1038/s42255-019-0076-1
Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT (2015) Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA 112(22):7061–7066. https://doi.org/10.1073/pnas.1415675112
Hardtke-Wolenski M, Taubert R, Noyan F, Sievers M, Dywicki J, Schlue J, Falk CS, Ardesjö Lundgren B, Scott HS, Pich A, Anderson MS, Manns MP, Jaeckel E (2015) Autoimmune hepatitis in a murine autoimmune polyendocrine syndrome type 1 model is directed against multiple autoantigens. Hepatology 61(4):1295–1305. https://doi.org/10.1002/hep.27639
Harley ITW, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, Sheridan R, Xanthakos SA, Steinbrecher KA, Sartor RB, Kohli R, Karp CL, Divanovic S (2014) IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 59(5):1830–1839. https://doi.org/10.1002/hep.26746
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482(7384):179–185. https://doi.org/10.1038/nature10809
Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS (2013) Autoimmune hepatitis. The Lancet 382(9902):1433–1444. https://doi.org/10.1016/S0140-6736(12)62163-1
Herck MAV, Weyler J, Kwanten WJ, Dirinck EL, Winter BYD, Francque SM, & Vonghia L (2019) The differential roles of T-cells in non-alcoholic fatty liver disease and obsity. In Frontiers in Immunology (Vol. 10, Issue FEB). Frontiers Media S.A. https://doi.org/10.3389/fimmu.2019.00082
Herkel J, Jagemann B, Wiegard C, Garcia Lazaro JF, Lueth S, Kanzler S, Blessing M, Schmitt E, Lohse AW (2003) MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocytes. Hepatology 37(5):1079–1085. https://doi.org/10.1053/jhep.2003.50191
Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B (2011) Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 12(3):255–263. https://doi.org/10.1038/ni.1993
Horst AK, Neumann K, Diehl L, & Tiegs G (2016) Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. In Cellular and Molecular Immunology (Vol. 13, Issue 3, pp. 277–292). Chinese Soc Immunology. https://doi.org/10.1038/cmi.2015.112
Huang H, Deng Z (2019) Adoptive transfer of regulatory T cells stimulated by allogeneic hepatic stellate cells mitigates liver injury in mice with concanavalin a-induced autoimmune hepatitis. Biochem Biophys Res Commun 512(1):14–21. https://doi.org/10.1016/j.bbrc.2019.02.147
Ibrahim SH, Hirsova P, Gores GJ (2018) Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67(5):963–972. https://doi.org/10.1136/gutjnl-2017-315691
Inzaugarat ME, Ferreyra Solari NE, Billordo LA, Abecasis R, Gadano AC, Cherñavsky AC (2011) Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol 31(6):1120–1130. https://doi.org/10.1007/s10875-011-9571-1
Jaruga B, Hong F, Kim WH, Gao B (2004) IFN-γ/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol - Gastrointest Liver Physiol 287(5):50–55. https://doi.org/10.1152/ajpgi.00184.2004
Jaruga B, Hong F, Sun R, Radaeva S, Gao B (2003) Crucial role of IL-4/STAT6 in T cell-mediated hepatitis: up-regulating eotaxins and IL-5 and recruiting leukocytes. J Immunol 171(6):3233–3244. https://doi.org/10.4049/jimmunol.171.6.3233
Jeffery HC, Jeffery LE, Lutz P, Corrigan M, Webb GJ, Hirschfield GM, Adams DH, Oo YH (2017) Low-dose interleukin-2 promotes STAT-5 phosphorylation, Treg survival and CTLA-4-dependent function in autoimmune liver diseases. Clin Exp Immunol 188(3):394–411. https://doi.org/10.1111/cei.12940
Jiang G, Yang HR, Wang L, Wildey GM, Fung J, Qian S, Lu L (2008) Hepatic stellate cells preferentially expand allogeneic CD4+CD25+FoxP3+ regulatory T cells in an IL-2-dependent manner. Transplantation 86(11):1492–1502. https://doi.org/10.1097/TP.0b013e31818bfd13
John K, Hardtke-Wolenski M, Jaeckel E, Manns MP, Schulze-Osthoff K, & Bantel H (2017) Increased apoptosis of regulatory T cells in patients with active autoimmune hepatitis. In Cell Death and Disease (Vol. 8, Issue 12, p. 3219). Nature Publishing Group. https://doi.org/10.1038/s41419-017-0010-y
Karampetsou MP, Liossis SNC, & Sfikakis PP (2010) TNF-α antagonists beyond approved indications: stories of success and prospects for the future. In QJM (Vol. 103, Issue 12, pp. 917–928). QJM. https://doi.org/10.1093/qjmed/hcq152
Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TLM, Xu B, Dhungana Y, Rankin S, Chen W, Rosencrance C, Yang K, Fan Y, Cheng Y, Easton J, Neale G, Vogel P, Chi H (2019) Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 565(7737):101–105. https://doi.org/10.1038/s41586-018-0806-7
Katz SC, Pillarisetty VG, Bleier JI, Shah AB, DeMatteo RP (2004) Liver sinusoidal endothelial cells are insufficient to activate T cells. J Immunol 173(1):230–235. https://doi.org/10.4049/jimmunol.173.1.230
Kleinschmidt D, Giannou AD, McGee HM, Kempski J, Steglich B, Huber FJ, Ernst TM, Shiri AM, Wegscheid C, Tasika E, Hübener P, Huber P, Bedke T, Steffens N, Agalioti T, Fuchs T, Noll J, Lotter H, Tiegs G, … Huber S (2017) A protective function of Il-22BP in ischemia reperfusion and acetaminophen-induced liver injury. J Immunol 199(12):4078–4090. https://doi.org/10.4049/jimmunol.1700587
Knolle PA, Schmitt E, Jin S, Germann T, Duchmann R, Hegenbarth S, Gerken G, Lohse AW (1999) Induction of cytokine production in naive CD4+ T cells by antigen- presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward T(h1) cells. Gastroenterology 116(6):1428–1440. https://doi.org/10.1016/S0016-5085(99)70508-1
Korniejewska A, Mcknight AJ, Johnson Z, Watson ML, Ward SG (2011) Expression and agonist responsiveness of CXCR3 variants in human T lymphocytes. Immunology 132(4):503–515. https://doi.org/10.1111/j.1365-2567.2010.03384.x
Krebs CF, Reimers D, Zhao Y, Paust HJ, Bartsch P, Nuñez S, Rosemblatt MV, Hellmig M, Kilian C, Borchers A, Enk LUB, Zinke M, Becker M, Schmid J, Klinge S, Wong MN, Puelles VG, Schmidt C, Bertram T, … Mittrücker HW (2020) Pathogen-induced tissue-resident memory TH17 (TRM17) cells amplify autoimmune kidney disease. Sci Immunol 5(50). https://doi.org/10.1126/SCIIMMUNOL.ABA4163
Lafdil F, Wang H, Park O, Zhang W, Moritoki Y, Yin S, Fu XY, Gershwin ME, Lian ZX, & Gao B (2009) Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology 137(6). https://doi.org/10.1053/j.gastro.2009.08.004
Lafdil F, Miller AM, Ki SH, & Gao B (2010) Th17 cells and their associated cytokines in liver diseases. In Cellular and Molecular Immunology (Vol. 7, Issue 4, pp. 250–254). Cell Mol Immunol. https://doi.org/10.1038/cmi.2010.5
Lapierre P, Béland K, Yang R, Alvarez F (2013) Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology 57(1):217–227. https://doi.org/10.1002/hep.26023
Ley K, Laudanna C, Cybulsky MI, & Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. In Nature Reviews Immunology (Vol. 7, Issue 9, pp. 678–689). Nat Rev Immunol. https://doi.org/10.1038/nri2156
Li W, Kuhr CS, Zheng XX, Carper K, Thomson AW, Reyes JD, Perkins JD (2008) New insights into mechanisms of spontaneous liver transplant tolerance: the role of Foxp3-expressing CD25+CD4+ regulatory T cells. Am J Transplant 8(8):1639–1651. https://doi.org/10.1111/j.1600-6143.2008.02300.x
Li M, Zhao W, Wang Y, Jin L, Jin G, Sun X, Wang W, Wang K, Xu X, Hao J, Jin R, Fu W, Sun Y, Chang Y, Huang X, Zhou X, Wu H, Zhang K, Ge Q (2020) A wave of Foxp3+ regulatory T cell accumulation in the neonatal liver plays unique roles in maintaining self-tolerance. Cell Mol Immunol 17(5):507–518. https://doi.org/10.1038/s41423-019-0246-9
Liang M, Liwen Z, Yun Z, Yanbo D, & Jianping C (2018) The imbalance between Foxp3+Tregs and Th1/Th17/Th22 cells in patients with newly diagnosed autoimmune hepatitis. J Immunol Res 2018. https://doi.org/10.1155/2018/3753081
Liberal R, Grant C, Mieli-Vergani G, Vergani D, Longhi M (2012) PWE-281 Different effector T cell responses may account for different patterns of liver injury in childhood autoimmune liver disease. Gut 61(Suppl 2):A412.1-A412. https://doi.org/10.1136/gutjnl-2012-302514d.281
Liberal, Rodrigo, Krawitt, E. L., Vierling, J. M., Manns, M. P., Mieli-Vergani, G., & Vergani, D. (2016). Cutting edge issues in autoimmune hepatitis. In Journal of Autoimmunity (Vol. 75, pp. 6–19). Academic Press. https://doi.org/10.1016/j.jaut.2016.07.005
Lim TY, Martinez-Llordella M, Kodela E, Gray E, Heneghan MA, Sanchez-Fueyo A (2018) Low-dose interleukin-2 for refractory autoimmune hepatitis. Hepatology 68(4):1649–1652. https://doi.org/10.1002/hep.30059
Liu Y, She W, Wang F, Li J, Wang J, Jiang W (2014) 3, 3′-diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol 23(2):489–498. https://doi.org/10.1016/j.intimp.2014.09.024
Lohr HF, Schlaak JF, Lohse AW, Bocher WO, Arenz M, Gerken G, Buschenfelde KM (1996) Autoreactive CD4+ LKM-specific and anticlonotypic T-cell responses in LKM-1 antibody-positive autoimmune hepatitis. Hepatology 24(6):1416–1421. https://doi.org/10.1002/hep.510240619
Lohse AW, Knolle PA, Bilo K, Uhrig A, Waldmann C, Ibe M, Schmitt E, Gerken G, Buschenfelde KHMZ (1996) Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology 110(4):1175–1181. https://doi.org/10.1053/gast.1996.v110.pm8613007
Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D (2004) Impairment of CD4+CD25+ regulatory T-cells in autoimmune liver disease. J Hepatol 41(1):31–37. https://doi.org/10.1016/j.jhep.2004.03.008
Lu JG, Iyasu A, French B, Tillman B, French SW (2020) Overexpression of MHCII by hepatocytes in alcoholic hepatitis (AH) compared to non-alcoholic steatohepatitis (NASH) and normal controls. Alcohol 84:27–32. https://doi.org/10.1016/j.alcohol.2019.08.008
Lüth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C, Brück W, Wraith DC, Herkel J, Lohse AW (2008) Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Investig 118(10):3403–3410. https://doi.org/10.1172/JCI32132
Luther SA, & Cyster JG (2001) Chemokines as regulators of T cell differentiation. In Nature Immunology (Vol. 2, Issue 2, pp. 102–107). Nat Immunol. https://doi.org/10.1038/84205
Ma X, Hua J, Mohamood AR, Hamad ARA, Ravi R, Li Z (2007) A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 46(5):1519–1529. https://doi.org/10.1002/hep.21823
Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, Han M, Thornton AM, Zhang H, Egger M, Luo J, Felsher DW, McVicar DW, Weber A, Heikenwalder M, Greten TF (2016) NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531(7593):253–257. https://doi.org/10.1038/nature16969
Manns MP, Lohse AW, & Vergani D (2015) Autoimmune hepatitis - update 2015. In Journal of Hepatology (Vol. 62, Issue S1, pp. S100–S111). Elsevier. https://doi.org/10.1016/j.jhep.2015.03.005
Manousou P, Kolios G, Drygiannakis I, Koulentaki M, Pyrovolaki K, Voumvouraki A, Notas G, Bourikas L, Papadaki HA, Kouroumalis E (2013) CXCR3 axis in patients with primary biliary cirrhosis: a possible novel mechanism of the effect of ursodeoxycholic acid. Clin Exp Immunol 172(1):9–15. https://doi.org/10.1111/cei.12032
Marek-Trzonkowska N, Wujtewicz MA, Myśliwiec M, Witkowski P, Dobyszuk A, Møynarski W, Grabowska M, Balcerska A, Techmańska I, Myśliwska J, Juścińska J, Trzonkowski P (2012) Administration of CD4 +CD25 highCD127 - regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care 35(9):1817–1820. https://doi.org/10.2337/dc12-0038
Matloubian M, & Cyster JG (2012) Th1 cell induction in lymph nodes according to a red-blue chemokine map. In Immunity (Vol. 37, Issue 6, pp. 954–956). Immunity. https://doi.org/10.1016/j.immuni.2012.11.007
Mitra S, De A, & Chowdhury A (2020) Epidemiology of non-alcoholic and alcoholic fatty liver diseases. In Translational Gastroenterology and Hepatology (Vol. 5). AME Publishing Company. https://doi.org/10.21037/TGH.2019.09.08
Mueller SN, Gebhardt T, Carbone FR, & Heath WR (2013) Memory T cell subsets, migration patterns, and tissue residence. In Annual Review of Immunology (Vol. 31, pp. 137–161). Annu Rev Immunol. https://doi.org/10.1146/annurev-immunol-032712-095954
Nagata T, Mckinley L, Peschon JJ, Alcorn JF, Aujla SJ, Kolls JK (2008) Requirement of IL-17RA in Con A induced hepatitis and negative regulation of IL-17 production in mouse T cells. J Immunol 181(11):7473–7479. https://doi.org/10.4049/jimmunol.181.11.7473
Nishioji K, Okanoue T, Itoh Y, Narumi S, Sakamoto M, Nakamura H, Morita A, Kashima K (2001) Increase of chemokine interferon-inducible protein-10 (IP-10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin Exp Immunol 123(2):271–279. https://doi.org/10.1046/j.1365-2249.2001.01391.x
Oo YH & Sakaguchi S (2013) Regulatory T-cell directed therapies in liver diseases. In Journal of Hepatology (Vol. 59). https://doi.org/10.1016/j.jhep.2013.05.034
Oo YH, Hubscher SG, & Adams DH (2010) Autoimmune hepatitis: New paradigms in the pathogenesis, diagnosis, and management. In Hepatology International (Vol. 4, Issue 2, pp. 475–493). Hepatol Int. https://doi.org/10.1007/s12072-010-9183-5
Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, Shetty S, Harki J, Shaw JC, Eksteen B, Hubscher SG, Walker LSK, Adams DH (2010) Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol 184(6):2886–2898. https://doi.org/10.4049/jimmunol.0901216
Oo YH, Ackrill S, Cole R, Jenkins L, Anderson P, Jeffery HC, Jones N, Jeffery LE, Lutz P, Wawman RE, Athwal AK, Thompson J, Gray J, Guo K, Barton D, Hirschfield GM, Wong T, Guest P, Adams DH (2019) Liver homing of clinical grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. JHEP Reports 1(4):286–296. https://doi.org/10.1016/j.jhepr.2019.08.001
Peiseler M, Sebode M, Franke B, Wortmann F, Schwinge D, Quaas A, Baron U, Olek S, Wiegard C, Lohse AW, Weiler-Normann C, Schramm C, Herkel J (2012) FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol 57(1):125–132. https://doi.org/10.1016/j.jhep.2012.02.029
Pellicoro A, Ramachandran P, Iredale JP, & Fallowfield JA (2014) Liver fibrosis and repair: immune regulation of wound healing in a solid organ. In Nature Reviews Immunology (Vol. 14, Issue 3, pp. 181–194). Nat Rev Immunol. https://doi.org/10.1038/nri3623
Plaza-Sirvent C, Schuster M, Neumann Y, Heise U, Pils MC, Schulze-Osthoff K, Schmitz I (2017) c-FLIP Expression in Foxp3-expressing cells Is essential for survival of regulatory T cells and prevention of autoimmunity. Cell Rep 18(1):12–22. https://doi.org/10.1016/j.celrep.2016.12.022
Racanelli V, & Rehermann B (2006) The liver as an immunological organ. In Hepatology (Vol. 43, Issue 2 SUPPL. 1). Hepatology. https://doi.org/10.1002/hep.21060
Radaeva S, Sun R, Pan HN, Hong F, Gao B (2004) Interleukin 22 (IL-22) Plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 39(5):1332–1342. https://doi.org/10.1002/hep.20184
Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, Smith T, Neish AS, Li H, Tan S, Wu P, Liu X, Yu Y, Farris AB, Nusrat A, Parkos CA, Anania FA (2016) Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 151(4):733-746.e12. https://doi.org/10.1053/j.gastro.2016.06.022
Rai RP, Liu Y, Iyer SS, Liu S, Gupta B, Desai C, Kumar P, Smith T, Singhi AD, Nusrat A, Parkos CA, Monga SP, Czaja MJ, Anania FA, Raeman R (2020) Blocking integrin α4β7-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J Hepatol 73(5):1013–1022. https://doi.org/10.1016/j.jhep.2020.05.047
Rau M, Schilling A-K, Meertens J, Hering I, Weiss J, Jurowich C, Kudlich T, Hermanns HM, Bantel H, Beyersdorf N, Geier A (2016) Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol 196(1):97–105. https://doi.org/10.4049/jimmunol.1501175
Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK (2001) Visualizing the generation of memory CD4 T cells in the whole body. Nature 410(6824):101–105. https://doi.org/10.1038/35065111
Renand A, Habes S, Mosnier J, Aublé H, Judor J, Vince N, Hulin P, Nedellec S, Métairie S, Archambeaud I, Brouard S, Gournay J, Conchon S (2018) Immune alterations in patients with type 1 autoimmune hepatitis persist upon standard immunosuppressive treatment. Hepatology Communications 2(8):972–985. https://doi.org/10.1002/hep4.1202
Rodrigues S, Lopes S, Magro F, Cardoso H, Horta e Vale AM, Marques M, Mariz E, Bernardes M, Lopes J, Carneiro F, Macedo G (2015) Autoimmune hepatitis and anti-tumor necrosis factor alpha therapy: a single center report of 8 cases. World J Gastroenterol 21(24):7584–7588. https://doi.org/10.3748/wjg.v21.i24.7584
Roh YS, Kim JW, Park S, Shon C, Kim S, Eo SK, Kwon JK, Lim CW, Kim B (2018) Toll-like receptor-7 signaling promotes nonalcoholic steatohepatitis by inhibiting regulatory T cells in mice. Am J Pathol 188(11):2574–2588. https://doi.org/10.1016/j.ajpath.2018.07.011
Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, Mombello C, Follenzi A, Novelli F, Carini R (2016) The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci 130(3):193–203. https://doi.org/10.1042/CS20150405
Sánchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, Thirkell S, Lowe K, Fry L, Heward J, Kerr A, Ali J, Fisher C, Lewis G, Hope A, Kodela E, Lyne M, Farzaneh F, Kordasti S, … Lombardi G (2020) Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant 20(4):1125–1136. https://doi.org/10.1111/ajt.15700
Schlaak JF, Lohr H, Gallati H, Meyer-Zum-Buschenfelde KH, Fleischer B (1993) Analysis of the in vitro cytokine production by liver-infiltrating T cells of patients with autoimmune hepatitis. Clin Exp Immunol 94(1):168–173. https://doi.org/10.1111/j.1365-2249.1993.tb05996.x
Schramm C, Lohse AW (2014) Autoimmune hepatitis on the rise. J Hepatol 60:478–479. https://doi.org/10.1016/j.jhep.2013.10.020
Shao X, Qian Y, Xu C, Hong B, Xu W, Shen L, Jin C, Wu Z, Tong X, & Yao H (2013) The protective effect of intrasplenic transplantation of Ad-IL-18BP/IL-4 gene-modified fetal hepatocytes on cona-induced hepatitis in mice. PLoS ONE 8(3). https://doi.org/10.1371/journal.pone.0058836
Sun R, Park O, Horiguchi N, Kulkarni S, Jeong WI, Sun HY, Radaeva S, Gao B (2006) STAT1 contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice. Hepatology 44(4):955–966. https://doi.org/10.1002/hep.21344
Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, Albano E (2014) Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 59(3):886–897. https://doi.org/10.1002/hep.26749
Takahashi A, Ohira H, Abe K, Zeniya M, Abe M, Arinaga-Hino T, Torimura T, Yoshizawa K, Takaki A, Kang JH, Suzuki Y, Nakamoto N, Inui A, Tanaka A, Takikawa H (2020) Increasing incidence of acute autoimmune hepatitis: a nationwide survey in Japan. Sci Rep 10(1):14250. https://doi.org/10.1038/s41598-020-71296-0
Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, Wang X, Ryffel B, Sun B (2013) IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol 191(4):1835–1844. https://doi.org/10.4049/jimmunol.1203013
Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, Han X, Peng Y, Chen X, Shen L, Qiu D, Li Z, Ma X (2011) Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol 166(2):281–290. https://doi.org/10.1111/j.1365-2249.2011.04471.x
Taubert R, Hardtke-Wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J, Olek S, Falk CS, Manns MP, Jaeckel E (2014) Intrahepatic regulatory T cells in autoimmune hepatitis are associated with treatment response and depleted with current therapies. J Hepatol 61(5):1106–1114. https://doi.org/10.1016/j.jhep.2014.05.034
Taubert R, Hupa-Breier KL, Jaeckel E, & Manns MP (2018) Novel therapeutic targets in autoimmune hepatitis. In Journal of Autoimmunity (Vol. 95, pp. 34–46). Academic Press. https://doi.org/10.1016/j.jaut.2018.10.022
Tay SS, Wong YC, Roediger B, Sierro F, Lu B, McDonald DM, McGuffog CM, Meyer NJ, Alexander IE, Parish IA, Heath WR, Weninger W, Bishop GA, Gamble JR, McCaughan GW, Bertolino P, Bowen DG (2014) Intrahepatic activation of naive CD4 + T cells by liver-resident phagocytic cells. J Immunol 193(5):2087–2095. https://doi.org/10.4049/jimmunol.1400037
Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, Watanabe M, Aoyagi T, Suzuki T, Shimamura T, Kamiyama T, Sato N, Sugita J, Hatanaka K, Bashuda H, Habu S, Demetris AJ, Okumura K (2016) A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology 64(2):632–643. https://doi.org/10.1002/hep.28459
Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, Myśliwska J, Hellmann A (2009) First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol 133(1):22–26. https://doi.org/10.1016/j.clim.2009.06.001
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444(7122):1027–1031. https://doi.org/10.1038/nature05414
Valean S, Acalovschi M, Dumitrascu DL, Ciobanu L, Nagy G, & Chira R (2019) Hepatocellular carcinoma in patients with autoimmune hepatitis – a systematic review of the literature published between 1989–2016. Med Pharm Rep 92(2). https://doi.org/10.15386/mpr-1228
Vonghia L, Magrone T, Verrijken A, Michielsen P, Van Gaal L, Jirillo E, & Francque S (2015) Peripheral and hepatic vein cytokine levels in correlation with non-alcoholic fatty liver disease (NAFLD)-related metabolic, histological, and haemodynamic features. PLoS ONE 10(11). https://doi.org/10.1371/journal.pone.0143380
Walker JA, & McKenzie ANJ (2018) TH2 cell development and function. In Nature Reviews Immunology (Vol. 18, Issue 2, pp. 121–133). Nature Publishing Group. https://doi.org/10.1038/nri.2017.118
Webb GJ, Hirschfield GM, Krawitt EL, Gershwin ME (2018) Cellular and molecular mechanisms of autoimmune hepatitis. Annu Rev Pathol 13:247–292. https://doi.org/10.1146/annurev-pathol-020117-043534
Weiler-Normann C, Schramm C, Quaas A, Wiegard C, Glaubke C, Pannicke N, Möller S, Lohse AW (2013) Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J Hepatol 58(3):529–534. https://doi.org/10.1016/j.jhep.2012.11.010
Wiegard C, Wolint P, Frenzel C, Cheruti U, Schmitt E, Oxenius A, Lohse AW, Herkel J (2007) Defective T helper response of hepatocyte-stimulated CD4 T cells impairs antiviral CD8 response and viral clearance. Gastroenterology 133(6):2010–2018. https://doi.org/10.1053/j.gastro.2007.09.007
Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, Modlin RL, Liblau RS, Gressner AM, Kaufmann SHE (2007) Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 26(1):117–129. https://doi.org/10.1016/j.immuni.2006.11.011
Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, Kubes P (1997) A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Investig 99(11):2782–2790. https://doi.org/10.1172/JCI119468
Wuensch SA, Spahn J, Crispe IN (2010) Direct, help-independent priming of CD8+ T cells by adeno-associated virus-transduced hepatocytes. Hepatology 52(3):1068–1077. https://doi.org/10.1002/hep.23745
Xie G, Wang X, Liu P, Wei R, Chen W, Rajani C, Hernandez BY, Alegado R, Dong B, Li D, Jia W (2016) Distinctly altered gut microbiota in the progression of liver disease. Oncotarget 7(15):19355–19366. https://doi.org/10.18632/oncotarget.8466
Xu H, Agalioti T, Zhao J, Steglich B, Wahib R, Vesely MCA, Bielecki P, Bailis W, Jackson R, Perez D, Izbicki J, Licona-Limón P, Kaartinen V, Geginat J, Esplugues E, Tolosa E, Huber S, Flavell RA, & Gagliani N (2020) The induction and function of the anti-inflammatory fate of TH17 cells. Nat Commun 11(1). https://doi.org/10.1038/s41467-020-17097-5
Xu R, Tao A, Zhang S, Zhang M (2013) Neutralization of interleukin-17 attenuates high fat diet-induced non-alcoholic fatty liver disease in mice. Acta Biochim Biophys Sin 45(9):726–733. https://doi.org/10.1093/abbs/gmt065
Xu L, Yin W, Sun R, Wei H, Tian Z (2013) Liver type i regulatory T cells suppress germinal center formation in HBV-tolerant mice. Proc Natl Acad Sci USA 110(42):16993–16998. https://doi.org/10.1073/pnas.1306437110
You Q, Cheng L, Kedl RM, Ju C (2008) Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 48(3):978–990. https://doi.org/10.1002/hep.22395
Yu H, Huang J, Liu Y, Ai G, Yan W, Wang X, Ning Q (2010) IL-17 contributes to autoimmune hepatitis. Journal of Huazhong University of Science and Technology Medical Sciences = Hua Zhong Ke Ji Da Xue Xue Bao Yi Xue Ying De Wen Ban = Huazhong Keji Daxue Xuebao Yixue Yingdewen Ban 30(4):443–446. https://doi.org/10.1007/s11596-010-0446-0
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA (2007) Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27(4):647–659. https://doi.org/10.1016/j.immuni.2007.07.023
Zhai M, Liu Z, Long J, Zhou Q, Yang L, Zhou Q, Liu S, Dai Y (2021) The incidence trends of liver cirrhosis caused by nonalcoholic steatohepatitis via the GBD study 2017. Sci Rep 11(1):5195. https://doi.org/10.1038/s41598-021-84577-z
Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, Qiu D, Wei J, Liu Y, Shen L, Chen X, Peng Y, Li Z, & Ma X (2011) Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS ONE 6(4). https://doi.org/10.1371/journal.pone.0018909
Zhao Y, Zhang Y, Liu Y-M, Liu Y, Feng X, Liao H-Y, Vergani D, Ma Y, Yan H-P (2011) Identification of T cell epitopes on soluble liver antigen in Chinese patients with auto-immune hepatitis. Liver Int 31(5):721–729. https://doi.org/10.1111/j.1478-3231.2011.02487.x
Zheng C, Yin S, Yang Y, Yu Y, Xie X (2018) CD24 aggravates acute liver injury in autoimmune hepatitis by promoting IFN-γ 3 production by CD4+ T cells. Cell Mol Immunol 15(3):260–271. https://doi.org/10.1038/cmi.2016.57
Zocco MA, Carloni E, Pescatori M, Saulnier N, Lupascu A, Nista EC, Novi M, Candelli M, Cimica V, Mihm S, Gasbarrini G, Ramadori G, Gasbarrini A (2006) Characterization of gene expression profile in rat Kupffer cells stimulated with IFN-α or IFN-γ. Dig Liver Dis 38(8):563–577. https://doi.org/10.1016/j.dld.2006.04.015
Funding
Open Access funding enabled and organized by Projekt DEAL. This review and the authors have been supported by grants from the Deutsche Forschungsgemeinschaft (DFG; SFB841 and KFO306).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the Special issue on: Mediators of liver inflammation and carcinogenesis - Guest Editors: Johannes Herkel and Dirk Schmidt-Arras
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muscate, F., Woestemeier, A. & Gagliani, N. Functional heterogeneity of CD4+ T cells in liver inflammation. Semin Immunopathol 43, 549–561 (2021). https://doi.org/10.1007/s00281-021-00881-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-021-00881-w