
Abstract
Six silica-supported phosphotungstic acid catalysts (PTA/SiO2) of different composition (20–70 wt% PTA content) have been synthesized and characterized by elemental analysis, BET, BJH, NH3-TPD methods, FT-IR spectroscopy of adsorbed pyridine and 1H MAS NMR techniques. The new composite catalysts were first applied in the Friedel–Crafts alkylation of toluene with 1-octene as a benchmark process under batch conditions in order to screen their activity and recyclability. The combined analytical techniques together with the catalytic studies enabled the identification of the main factors affecting the activity of the catalysts. Based on these preliminary experiments, the best performing catalyst system (50 wt% PTA/SiO2) was investigated in continuous flow mode using an in-house-made flow reactor. The thorough screening of the reaction conditions (temperature, toluene/1-octene molar ratio and flow rate) provided firm evidence that the 50 wt% PTA/SiO2 composite is highly active, selective and stable catalyst under mild reaction conditions even at elevated flow rate. Additionally, the catalyst used in the flow mode could successfully be regenerated and reused in the alkylation process.
Similar content being viewed by others
1 Introduction
The acid catalyzed alkylation of aromatic hydrocarbons represent one of the most fundamental and strategic synthetic methodologies leading to fine or bulk chemicals on an industrial scale [1,2,3,4]. The alkylation of benzene with ethene, propene or higher alkenes are amongst the most widely used processes to produce ethyl-, isopropyl- or linear alkyl-benzenes (LABs), respectively, as valuable synthetic organic chemicals [5]. In addition to the synthetic application of these bulk intermediates, the subsequent hydrogenation of the alkylated aromatics leads to alkyl cycloalkanes, an alternative liquid fuel component with high hydrogen content [6]. The high hydrogen-to-carbon ratio in liquid fuels is essential concerning the reduction of CO2 emission [7]. The application of traditional Lewis- (e.g. ZnCl2, AlCl3) or Brönsted–Lowry-acids (HF or H2SO4) to alkylation reactions, however, frequently accompanies with time and energy consuming separation processes, lower selectivity, reduced process safety and the generation of hazardous, highly corrosive acidic wastes necessitating additional treatment and maintenance protocols [8].To overcome these difficulties a number of solid or supported acid catalysts have been developed and applied in these types of organic transformations [5, 9, 10].
Amongst solid acid catalysts, heteropolyacids (HPAs) are a unique class of compounds due to their high Brönsted-acidity, relatively high thermal stability, low corrosivity and high structural modularity as well as the ease of their preparation [11, 12]. They are insoluble in non-polar reaction media and generally exhibit much stronger acidity than traditional mineral acids (H2SO4, HClO4 or HI), and therefore HPAs are usually considered as superacids. In particular, phosphotungstic acid (H3PW12O40, PTA) exhibits the highest Brönsted-acidity (Hammett acidity H0 = − 13.16) [13] and stability amongst the Keggin-series of heteropolyacids. Despite the strong acidic nature of HPAs, their inherent low surface area (~ 5–10 m2/g) often demands support on a porous carrier to provide the desirable catalytic turnover. Similarly to the heteropolyacid itself, the nature of the support can also easily be fine-tuned to achieve the highest activity and selectivity in the catalytic process. Accordingly, a number of different acidic or neutral materials were used as carriers for HPAs, including activated carbon [14,15,16,17], zirconia [18,19,20], or other mesoporous supports (e.g. SBA-15, MCM-41) [21,22,23,24,25,26,27]. Nevertheless, silica is one of the most widespread carriers for HPAs that is attributed to its commercial availability, neutral or slightly acidic nature and tunable structure. Additionally, it has been reported that silica supported HPAs are thermodynamically more stable compared to their unsupported form [28]. Based on the above facts, the catalytic application of supported HPAs opened up new horizons in acid catalyzed chemical transformations. Indeed, in the last several decades supported HPAs proved to be highly active in a plethora of acid catalyzed reactions, such as alkylation [29,30,31,32], acylation [33,34,35], isomerization [26, 36, 37], esterification/ester hydrolysis [38,39,40,41], conversion of carbohydrates into 5-hydroxymethylfurfural [42] or in oxidation reactions [43,44,45,46]. Since the first successful utilization of silica supported heteropolyacids in the liquid phase Friedel–Crafts alkylation of aromatic compounds by alkenes [47], great efforts have been devoted to fine tune the structural characteristics of the catalyst and the support for these types of reactions [48,49,50,51]. Despite the high activities and selectivities thus achieved in the alkylation of aromatics with alkenes, the recycling of these catalyst proved to be less promising due to their gradual deactivation in batch systems. In order to exclude the possibility of catalyst contamination between successive catalytic runs and to increase the operational simplicity of the process, continuous flow technology could be mentioned as a rational solution to overcome these difficulties. Interestingly, however, the use of supported heteropolyacids for the liquid phase alkylation of aromatics with alkenes has scarcely been studied under continuous flow conditions [52,53,54]. Additionally, the available examples are limited to the kinetic investigation of the process instead of the optimization and the thorough screening of the reaction conditions in the flow mode [52, 53]. To the best of our knowledge, there are no experimental data available in the literature focusing on the practical application of these catalytic systems in continuous flow liquid phase alkylation reactions.
As the continuous flow technique could also ensure reduced reaction times, better control of reaction parameters, high reproducibility, rapid optimization, screening and analysis as well as improved process safety, we decided to unite environmentally benign heteropolyacid catalysis with flow technology for the intensification of the Friedel–Crafts alkylation of aromatics with alkenes. The present report focuses on the structural and catalytic investigation of silica supported Keggin-type phosphotungstic acid, a catalyst that could easily be prepared using commercially available starting materials. Specifically, (i) the structural characterization of the catalysts of different composition, (ii) their application in batch systems and (iii) their potential in continuous flow mode under diverse reaction conditions have been investigated. The main goal of the present study was to determine the limitations of the catalytic system under flow conditions and thereby to decrease the energy consumption and environmental impact of the catalytic process while maximizing efficiency.
2 Results and Discussion
2.1 Synthesis and Characterization of the Catalysts
The catalysts were synthesized by the simple wet impregnation of silica with an aqueous solution of phosphotungstic acid at room temperature. The composites were prepared over a range of 20–70 wt% nominal PTA loading. After the evaporation of the solvent, the catalysts were dried at 150 °C for 10 h in high vacuum and were used without further treatment. The immobilized heteropolyacid catalysts were first evaluated by elemental analysis using ICP-AES technique. As the data of Table 1 reveal, the tungsten content of the samples successively increases with the nominal PTA loading indicating the successful support on the carrier.
Next, the textural properties of the composites were studied by low temperature nitrogen adsorption. The isotherms were of type II with H2 hysteresis loop, characteristic for mesoporous silica. The average pore diameter (Daverage) was found to be between 6.8 and 5.8 nm (Table 1). The data clearly show the gradual decrease of both BET surface area (SBET) and pore volume (V1.7–300) with increasing PTA loadings. Additionally, the decrease in the pore volume is more significant at higher loadings. At the same time, however, no change in the average pore diameter can be observed by increasing the amount of the PTA. Although the theoretical surface coverage (Φ, calculated from the average diameter of the Keggin-unit (approx. 12 Å), at monolayer saturation Φ = 1) [55] suggests that the composites do not reach the monolayer saturation even at 70 wt% nominal loading, the drastic reduction of the BET area indicates that significant amount of bulk PTA appears on the surface at higher heteropolyacid concentrations. This phenomenon can be explained by the blockage of some pores by small aggregates of PTA units that decrease the available surface area, thus facilitating the formation of bulk crystallites leading to an even more intense reduction of the BET surface at higher PTA loadings.
The IR spectra of the PTA/SiO2 composites were particularly informative about the structure of the heteropolyacid on the silica surface. Despite the partial overlaps of the corresponding signals, the characteristic IR bands of the PTA Keggin structure observed at 1080 (υas(P–O)), 982 (υas(W=O)), 888 (υs(W–O–W)) and 804 cm−1 (υas(W–O–W) appear in the spectra together with the signals of the silica support (υas(Si–O–Si) 1200–1000 cm−1, υas(Si–OH) ~ 960 cm−1, υs(Si–O–Si) 802 cm−1, δ(Si–O–Si) 460 cm−1) (Fig. 1) [56]. The most intense change can be observed in the case of the internal W–O–W signal at 888 cm−1 that clearly grows with the PTA content, indicating the evolution of the surface heteropolyacid phases. Additionally, the infrared spectra of the supported heteropolyacids provided firm evidence for the successful immobilization of the Keggin-units without their decomposition.

IR spectra of PTA, PTA/SiO2 composites with different heteropolyacid loadings and pure silica
It is generally accepted that PTA reacts with hydroxylated silica surface by the protonation of one or more silanol groups to form ion pairs (Fig. 2) [57, 58].

Interaction of PTA with hydroxylated silica surface
Based on the pioneering work of Kozhevnikov [59] and Lefebvre [60], MAS NMR spectroscopy proved to be a valuable methodology to investigate the nature of different supported heteropolyacid species on silica surface. As the host-PTA interaction involves the acidic protons of the latter, the comparison of 1H MAS NMR spectra of composites with different PTA loadings might provide convincing proof for the presence of the crystalline heteropolyacid. Indeed, the effect of increasing PTA loading on the 1H MAS spectra is quite apparent as is illustrated in Fig. 3. The 1H MAS spectrum of the silica support exhibits a broad resonance centered at 1.8 ppm with a shoulder at ~ 3.0 ppm, that can be assigned to the isolated (non-hydrogen bonded) and hydrogen bonded silanols, respectively [61,62,63]. Clearly, the addition of PTA to silica changes the shapes and the relative areas of these signals, that can be attributed to the protonation of the surface silanol OH’s and the appearance of new protonic environments [64]. More importantly, however, a new signal appears at 9.3 ppm at 50 wt% PTA concentration and becomes more intense at higher loadings. This new signal can be assigned to the protons of the dehydrated crystalline PTA lacking interaction with the silica surface.

1H MAS NMR spectra of PTA/SiO2 composites with different PTA loading (reference signal (silicone rubber): 0.05 ppm)
These observations are in accord with the results provided by nitrogen gas porosimetry. The relative amount of the heteropolyacid interacting with the silica surface decreases significantly as its loading increases. The formation of PTA crystallites starts at 40–50 wt% concentrations. This conclusion was also supported by scanning electron microscopy (SEM). As it is displayed in Fig. 4, surface particles with anomalous morphology appears at 50% PTA/SiO2 compositions that start to aggregate at 60% PTA loading. These observations suggest the relatively high dispersion of the PTA on the silica surface at low PTA concentrations (up to 40%) and the appearance of the bulk heteropolyacid at higher loadings. The relative amount of the well-dispersed surface heteropolyacid and the PTA crystallites together with the surface area of the composites have crucial importance in determining catalytic activity.

SEM images of SiO2 support and PTA/SiO2 composites with different PTA loading
The acidity of the catalysts was characterized by NH3-TPD (temperature-programmed desorption) measurements and by FT-IR spectroscopy of adsorbed pyridine. NH3-TPD measurements give information about the amount and strength of acid sites, whereas by adsorbed pyridine (Py) we can differentiate between the type of acidity, i.e. Lewis (L) or Brønsted (B). The amount of desorbed ammonia, corresponding to the acid site concentration in the sample, is listed in Table 2. By ammonia TPD experiments it can be observed that PTA/silica catalysts have medium and strong acid sites. The acidity of PTA/silica catalysts can be associated with various species with well-known acidic nature, such as phosphate (PO43−) and tungstate (WO42−) based oxoanions.
The TPD curves (Fig. 5) show similar, overlapping peaks between 200 and 600 °C. However, with increasing amount of PTA, the high temperature peak between 550 and 600 °C becomes more intensive. This can be explained by the formation of WO3, which can be due to decomposition of bulk PTA over 400 °C. The amount of desorbed ammonia is increasing with the increasing amount of PTA loaded, but it reaches a maximum over 50% of PTA content. This phenomenon is in connection with the dispersion of PTA on the silica support, the accessibility of acid centers. In spite of the low coverage of silica with PTA, with already 40% PTA content the appearance of crystalline H3PW12O40 phase can be detected by XRPD method (Figure S7), which becomes more pronounced for PTA/SiO2-70 sample. This phenomenon is in accordance with the results provided by 1H MAS NMR and SEM techniques. The low dispersion of acid centers in the bulk phase limits their accessibility, and the relatively narrow pores of silica support also have adverse effect on the availability of active species by pore clogging. However, taking into account the amount of adsorbed ammonia only in the 200–450 °C temperature range, by omitting the high temperature peak over 500 °C, the 50% PTA sample shows the highest acidity (Fig. 5). Decomposition of the Keggin-ion starts over 460 °C, therefore the temperature region above it cannot be taken into consideration [65].

NH3-TPD curves of the silica supported PTA catalysts
The acidic properties of selected catalysts were characterized also by FT-IR spectroscopy of adsorbed pyridine (Py) in order to compare their Brønsted and Lewis acidity. Py adsorption bands are associated with the 8a, 8b, 19a, and 19b ring vibrations of Py. Pyridine can be coordinated to Lewis acid sites (L-Py) or protonated on Brønsted acid sites (B-Py). The bands at about 1450 cm−1 and 1610–1620 cm−1 are assigned to L-Py (νs19a and νs8a). The protonated Py coordinated to the conjugated base of the solid Brønsted acid (B-Py), gives bands around 1530–1560 cm−1, around 1610 cm−1 and at around 1635 cm−1 (νs19b, νs8b and νs8a). The bands can be shifted to higher/lower wavenumbers by increasing/decreasing acidic strength. Figure 6 shows the spectra of some selected PTA/SiO2 catalysts, and the ratio of Brønsted/Lewis sites, calculated by the area under the corresponding FT-IR bands, is listed in Table 2. On all samples, bands characteristic for both Lewis (L-Py) and Brønsted acid sites (B-Py) can be observed. In a bulk crystalline phase Py can mainly access only the surface species. In the two catalysts with lower amount of PTA, the intensity of Brønsted and Lewis acid bands are quite similar. However, in PTA/SiO2-50 sample one can observe increased Brønsted acidity and decreased amount of Lewis acid centers accessible to Py, resulting in the highest B/L ratio (Table 2). By 60% PTA catalyst the acidity is slightly increased, however the B/L ratio is lower compared to 50% PTA sample. Investigating the Py desorption temperature (not shown) all the catalysts retained Py on B and L sites up to 300 °C, but was totally desorbed at 400 °C. The latter results suggest the presence of moderately strong acid centers.

FT-IR spectra of adsorbed pyridine on some selected PTA/SiO2 catalysts. Py (6 mbar) was adsorbed on 250 °C dehydrated samples at 100 °C, and spectra with Py desorbed at 200 °C in high vacuum are compared
In summary, the evaluation of acidity evidenced that PTA/SiO2-50 catalyst shows the highest amount of acidity among the studied samples, and moreover has relatively the highest amount of Brønsted acid centers, which can predict its outstanding catalytic performance.
2.2 Catalytic Investigations
In the first set of catalytic experiments the activity of the catalysts of different composition were compared in the alkylation of toluene with 1-octene as a benchmark reaction (Fig. 7) in batch mode.

Supported heteropolyacid-catalyzed Friedel–Crafts alkylation of toluene with 1-octene
The tests were conducted under the same reaction conditions, at 70 °C and at a toluene/1-octene molar ratio (T/O) of 10/1 using the same amount of catalyst. The activity strongly depends on the composition of the catalyst and a conversion maximum can be observed at 50 wt% PTA loading (Diagram 1). This reactivity pattern can be explained by the fact that non-polar substrates mainly react at the surface of the catalysts with the protons of isolated PTA units and cannot get into the highly polar bulk crystallites. As it was expected, at low loadings the conversion of the alkylation steadily increases with the amount of the PTA. However, as the bulk crystallite becomes dominant at higher catalyst loadings, the activity tends to decrease. It is also important to note that the surface area also decreases with the PTA loading, and therefore it can also be mentioned as an additional factor responsible for the reduced activity at higher loadings.

Effect of the catalyst composition on the activity. Reaction conditions: toluene 15 mmol (1.6 mL), 1-octene 1.5 mmol (0.235 mL), PTA/SiO2 catalyst 23 mg. Temperature: 60 °C, reaction time: 5 min, stirring rate: 900 rpm
The main purpose of using immobilized catalysts is their easy separation from the product and their recycling to the next catalytic run. Therefore, we investigated the recyclability of the best performing catalyst (50 wt% PTA/SiO2) in the alkylation of toluene with 1-octene at 70 °C and at a T/O ratio of 10. The catalyst was recycled four times, the reaction time of each run was 15 min. In order to model the poisoning effect of water as a feed impurity two sets of experiments (A and B) were realized using (A) predried toluene (15 ppm of water content according to Karl-Fisher titration) and (B) toluene without preliminary drying (160 ppm of water content according to Karl-Fisher titration), respectively. The tests demonstrated the rapid deactivation of the catalyst upon recycling (Diagram 2). Additionally, the comparison of the experiments A and B clearly indicates the detrimental effect of water on the activity. The presence of water reduces the catalytic activity as water competes with the alkene for acidic sites.

Effect of recycling and water content on the catalytic activity of 50 wt% PTA/SiO2. Reaction conditions: toluene 15 mmol (1.6 mL), 1-octene 1.5 mmol (0.235 mL), 50 wt% PTA/SiO2 catalyst 23 mg. Temperature: 70 °C, reaction time: 15 min, stirring rate: 900 rpm. (A) Performed with predried toluene containing 15 ppm of water; (B) performed with toluene containing 160 ppm of water
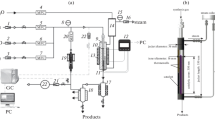
The best performing catalyst, 50 wt% PTA/SiO2 was then tested under flow conditions in the same alkylation process as model reaction. As it is shown in Fig. 8, a pre-drying column containing 4 Å molecular sieve has been inserted between the feed and the catalyst in order to prevent catalyst deactivation by the introduction of water.

Schematic view of the in-house-constructed continuous flow system
First, the feed ratio (T/O) has been studied in the continuous catalytic process: in addition to the 10/1 toluene/1-octene molar ratio, 5/1, 2/1 and 1/1 mixtures were applied in the alkylation reaction (Diagram 3). The flow reactions were performed at 70 °C and 0.9 mL/min flow rate. At T/O molar ratios lower than 10/1, the octene conversion significantly decreased during the 4 h long period of continuous operation. Additionally, a slow reduction in the activity could be observed in the case of T/O ratio lower than 10/1 during this time that seemed to be somewhat more intense at lower T/O values. Nevertheless, the conversion of octene was higher than 45% after the 4 h long time-on-stream even at T/O = 1. The selectivity of the formation of monooctyl-toluene decreased with decreasing T/O ratio due to the more pronounced dialkylation in these cases. It is, however, important to note that the amount of the dialkylated product was 6.2% at T/O = 1 and only 2.6% at T/O = 10 after 4 h continuous operation.

Effect of the feed composition on the conversion of octene and on the ratio of dioctyl-toluene in the alkylated product after 240 min. Reaction conditions: catalyst: 626 mg of 50 wt% PTA/SiO2; temperature: 70 °C; flow rate: 0.9 mL/min. The percentage of the dialkylated product has been determined from the samples taken at 240 min. A fresh catalyst bed was used for each run
Although the conversion of octene dynamically changed during the 4 h long continuous operation, the results are suitable to uncover the high potential of the catalyst in Friedel–Crafts alkylation as well as to explore its limitations. Indeed, the amount of the product monooctyl-toluene could be increased by a factor of 2.64 during the 4 h long time-on-stream when changing the molar ratio from 10/1 to 1/1 (Diagram 4). Furthermore, the latter continuous run produced the least by-products relative to monooctyl-toluene compared to the experiments performed with higher T/O ratios. This change can nicely be expressed by the environmental factor (E-factor) defined as the mass of by-products (unreacted toluene, octene and dioctyl-toluene) per mass of the product monooctyl-toluene. Decreasing the toluene/1-octene molar ratio in the feed from 10/1 to 1/1 results in the reduction of the E-factor from a value of 4.4 to 0.9. Although the unreacted components in the product can easily be regenerated by distillation and thus reused, the possibility of the significant reduction in the E-factor further increases the environmentally benign nature of this catalytic process.

The effect of the feed-composition on the production of monooctyl-toluene and on the environmental factor. Reaction conditions: catalyst: 626 mg of 50 wt% PTA/SiO2; temperature: 70 °C; flow rate: 0.9 mL/min. The continuous catalytic experiments were performed with fresh catalyst at each feed compositions investigated. The E-factor has been calculated as the mass of by-products (unreacted toluene, octene and dioctyl-toluene) per mass of the product monooctyl-toluene after each continuous run
The next catalytic tests were conducted at T/O ratio of 10, due to the fact that the selectivity of the alkylation proved to be the highest at this feed composition. First, the effect of time-on-stream on the catalytic activity was investigated using the 50 wt% PTA/SiO2 composite. The catalyst proved to be highly stable at 0.9 mL/min flow rate and at 70 °C (Diagram 5) as it provided complete octene conversion for 16 h long continuous operation. A marginally slow deactivation (~ 1% decrease in octene conversion) could be observed after 20 h. Nevertheless, under these reaction conditions more than 800 mmol of monooctyl-toluene could be produced using 634 mg of 50 wt% PTA/SiO2 during the 20 h long continuous run.

The effect of time-on-stream on the catalytic activity. Reaction conditions: catalyst: 634 mg of 50 wt% PTA/SiO2; toluene/1-octene molar ratio: 10; temperature: 70 °C, flow rate: 0.9 mL/min
The energy consumption of chemical processes can be significantly decreased by the application of catalytic systems that are able to operate under mild reaction conditions, even at low temperature. As the continuous experiment depicted in Diagram 5 demonstrated the high stability of the catalyst, we decided to investigate the effect of the temperature in the same run, as a continuation of this experiment. In addition to the 70 °C reaction temperature, the catalytic system was screened at 60, 50 and 40 °C. In each case, 3 h long periods were evaluated to compare the octene conversion. Diagram 6 displays the gradual change in the conversion due to varying temperature. Most importantly, the catalyst was able to efficiently produce monooctyl-toluene even at temperatures as low as 50 or 40 °C, with 77 and 45% conversion, respectively. The amount of the dioctyl-toluene in the alkylated product decreased from 1.2 to 0.6% by changing the temperature from 70 to 40 °C, respectively. After finishing the experiment at 40 °C, the reaction temperature was reset to 70 °C to prove the stability of the system. The conversion of octene returned to a value of 98% that provided further evidence for the stability of the flow system as well as for the slow deactivation of the catalyst, i.e. 1% decrease in octene conversion during a period of 15 h.

The effect of the temperature on the production of monooctyl-toluene. Reaction conditions: catalyst: 634 mg of 50 wt% PTA/SiO2; toluene/1-octene molar ratio: 10, flow rate: 0.9 mL/min
After returning to the initial conditions (70 °C, 0.9 mL/min) we decided to investigate the effect of the flow rate in this same continuous run, starting at 98% octene conversion. The variation of the flow rate was screened in 1.5 h long periods at 0.9, 1.2, 1.5 and 1.8 mL/min velocities, respectively (Diagram 7). By doubling the flow rate (from 0.9 to 1.8 mL/min) the conversion decreased to 94% under these reaction conditions. Clearly, in this high conversion regime the decreased residence time barely affects the conversion and therefore the production rate of monooctyl-toluene can significantly be enhanced. Again, by resetting the system to the initial conditions, the octene conversion returned to a value of 97%.

The effect of the flow rate on the production of monooctyl-toluene. Reaction conditions: catalyst: 634 mg of 50 wt% PTA/SiO2; toluene/1-octene molar ratio: 10, temperature: 70 °C
The reaction mixture produced in the 43 h long continuous experiment was concentrated by distillation. The distillation residue was then analyzed by ICP. This method indicated that only a marginal leaching of the PTA occurred: 0.1 mg of the heteropolyacid could be detected in the effluent after the 43 h long experiment.
After the 43 h long continuous run the catalyst was washed with toluene, removed from the column and regenerated under mild conditions by heating for 10 h at 150 °C in high vacuum. The catalyst was then quantitatively refilled into the cartridge and was subjected to an additional 24 h long continuous experiment. The regeneration of the catalyst was successful as full conversion could be observed for 7 h long time-on-stream (Diagram 8). After this period of time, however, the activity started to fall more rapidly than in the case of the freshly prepared catalyst. Based on these experiments, it is surmised that the catalyst is reversibly deactivated by water that can be removed by the treatment of the composite at elevated temperature under reduced pressure. This assumption is also corroborated by the fact that in the 1H NMR spectrum of the recycled catalyst (Figure S8) no signal for the dehydrated bulk PTA can be observed, suggesting the appearance of the catalytically less active hydrated form (~ 2 ppm) [66]. Although, no direct evidence could be provided at the present stage, it is reasonable to assume that additional effects are also responsible for catalyst deactivation. For instance, the formation of high molecular weight polymeric species even at low concentration may lead to pore blockage thus reducing the number of available acidic sites.

Continuous flow alkylation with regenerated catalyst. Reaction conditions: catalyst: 634 mg of 50 wt% PTA/SiO2; toluene/1-octene molar ratio: 10, temperature: 70 °C, flow rate: 0.9 mL/min
It is important to emphasize that the 50% PTA/SiO2 composite proved to be a highly active, selective and stable catalyst, compared to zeolites, as one of the most frequently used acidic catalysts for liquid phase Friedel–Crafts alkylation reactions under flow conditions [67]. The silica supported heteropolyacid were able to work under mild reaction conditions (≤ 70 °C) even at relatively high flow rates and low T/O ratios (from 10/1 to 1/1), thus decreasing environmental impact and increasing efficiency. Additionally, conversions up to 97% could be obtained during the 43 h long continuous flow process. The high activity compared to zeolites can mainly be attributed to the generally higher acidity of HPA’s. The selectivity towards the monoalkylated product is commensurate to that of zeolites in the alkylation of toluene [68]. These features make PTA silica composites a promising, greener alternative for zeolite type alkylation catalysts. Furthermore, the present study also demonstrates that the application of silica supported PTA under flow conditions offers a reliable methodology for liquid phase Friedel–Crafts alkylation reactions compared to the use of immobilized heteropolyacids in batch systems.
3 Conclusions
The present study demonstrates the beneficial use of silica supported phosphotungstic acid catalysts in a continuous flow system under mild reaction conditions for the Friedel–Crafts alkylation of toluene with 1-octene as a model reaction. Six silica-supported catalysts were synthesized by using commercially available starting materials by simple wet impregnation method. The combined analytical methodologies (ICP, porosimetry, FT-IR, NH3-TPD, SEM and 1H MAS NMR) together with catalytic experiments in batch system enabled the identification of the main factors influencing catalytic activity of the catalysts of different composition. Namely, (i) the relative amount of the well-dispersed surface heteropolyacid and the bulk PTA crystallites and (ii) the surface area of the composites have crucial importance in determining activity. The best performing composite, 50 wt% PTA/SiO2 proved to be highly active, selective and stable in continuous flow system under mild reactions conditions. It should be noted that using 634 mg 50 wt% PTA/SiO2 catalyst more than 216 g (0.99 mol) of monooctyl-toluene could be synthesized at 70 °C temperature and 0.9 mL/min flow rate under a 24 h long continuous operation. In addition, the deactivation of the catalyst was marginal during this experiment, as the conversion of octene was > 98% after 24 h. Moreover, the catalyst was tested at low temperature (as low as 40 °C), at high flow rates (up to 1.8 mL/min) and with different toluene/1-octene ratios to have a deeper insight into the limits of its working conditions. The system remained active at 40 °C, i.e. higher than 45% octene conversion could be observed, and the increasing flow rate did not affect the octene conversion significantly. Furthermore, it has been demonstrated that the catalyst can be used with a toluene/1-octene ratio of 1 and could provide high monooctyl-toluene selectivity under these conditions with the significant reduction of the environmental factor in the alkylation process.
The basic catalytic results presented in the current report suggest that the application of supported heteropolyacid catalysts under flow conditions offer an environmentally benign, green alternative for liquid phase Friedel–Crafts alkylation reactions. The low energy consumption, the mild reaction conditions, the operational simplicity associated with the flow mode, the non-corrosive nature of the catalyst as well as its easy preparation make this synthetic strategy an economically and ecologically valuable methodology for the alkylation of aromatic hydrocarbons.
4 Experimental
4.1 General Experimental Details
All synthetic manipulations were carried out under argon using Schlenk techniques. Solvents were purified, dried and deoxygenated by standard methods. All reagent transfers (aromatic and aliphatic hydrocarbons) were carried out via cannula under argon pressure or by the use of gas tight syringes. The silica (Silicagel-60, particle size: 0.045–0.063 mm) used as support was purchased from Molar Chemicals. PTA (tungstophosphoric acid, H3PW12O40·xH2O, cryst. extra pure) was obtained from Merck. Molecular sieve (4 Å) was purchased form Aldrich. ICP elemental analyses were carried out using a GBC Integra XM spectrometer. The specific surface area and pore size distribution were determined by nitrogen physisorption measured at − 196 °C with Thermo Scientific Surfer-type instrument on samples previously outgassed in vacuum at 120 °C for 1 h followed by 250 °C for 2 h. The surface areas of the samples (SBET) were determined by the BET (Brunauer–Emmett–Teller) method from the corresponding nitrogen adsorption isotherm. The pore size was calculated from the desorption branch using the BJH (Barret–Joyner–Halenda) theory. The total pore volume was calculated from the adsorbed volume at 0.95 relative pressure. NMR measurements were carried out on a Bruker Avance 400 spectrometer (NMR Laboratory, University of Pannonia) operating at 400.13 MHz, 161.98 MHz for 1H and 31P, respectively. 1H solid state experiments were carried out using a 4 mm MAS probe and the rotor spin rate synchronized hahnecho.av T2 filter pulse sequence. In a typical experiment 8000 Hz spin rate (CNST31) along with 24 rotor cycles (L1) effectively removed the broad water signals. The solidecho.av sequence with 1 ms echo delay gave the same qualitative result at 20 kHz spinning rate measured with a 2.5 mm MAS probe. Signal to noise ratio ~ 500 spectra were recorded with only 16 transients.
XRPD patterns were recorded by a Philips PW 1810/3710 diffractometer applying monochromatized CuKα radiation (40 kV, 35 mA), between 3 and 75°2θ, with 0.02° step size for 1 s. For the identification of crystalline H3PW12O40 phase ICDD card No. 75-2125 was used.
The acidity of the catalysts was characterized by ammonia thermo-desorption (NH3-TPD) measurements. About 100 mg of catalyst was placed into a quartz flow-through microreactor (φ = 4 mm) and activated in He-flow at 250 °C for 1 h. Then it was evacuated at the same temperature for 30 min and cooled to room temperature under evacuation. Ammonia adsorption was carried out by contacting the sample with 13.3 kPa NH3 at room temperature for 15 min. It was followed by a 15 min evacuation at room temperature, then the system was flushed by He in a two-step temperature program. The reactor was heated by 10 °C min−1 rate to 150 °C for 30 min in order to separate the low temperature ammonia physisorption step, then up to 600 °C with the same ramp for 1 h. The NH3 concentration in the effluent gas flow was monitored by thermal conductivity detector (TCD) after passing through a dry-ice trap.
FT-IR spectra of adsorbed pyridine (Py) were measured on a Nicolet Compact 640 spectrometer by self-supported wafer technique. Pellets (10 × 20 mm) were pressed from the samples, placed into a special IR cell with in situ heat treatment facility, heated up to 250 °C in high vacuum (10–6 mbar) with a rate of 10 °C/min and dehydrated for 1 h. Following 30 min contact with Py (6 mbar) at 100 °C, the sample was evacuated subsequently at 100, 200 and 300 °C, for 30 min. After each evacuation step a spectrum was recorded at IR beam temperature with a resolution of 2 cm−1. The spectra were normalized to 5 mg/cm2 weight of the wafers for comparison. The ratio of Brønsted/Lewis acid sites were calculated by dividing the corresponding band areas of the 200 °C Py desorption spectrum (L:1450 cm−1, B:1545 cm−1).
The SEM images were recorded on a FEI/ThermoFisher Apreo S scanning electron microscope.
The in-house-made flow reactor is made up of the following parts: JASCO PU-2087 Plus HPLC pump, Jetstream 2 HPLC Peltier Column Thermostat, FC-204 Fraction Collector. The pre-heater coil is a 275 cm long rolled up steel pipe with an internal diameter of 1.016 mm. The column for the drying agent (4 Å molsieve) has a length of 150 mm, and an internal diameter of 4.6 mm. The column for the catalyst is 30 mm long and has an internal diameter of 4.6 mm. Both columns are made of stainless steel and are equipped with stainless steel frits at their ends.
4.2 Preparation of PTA/SiO2 Composites
Silica was suspended in an aqueous solution of PTA and the resulting mixture was stirred for 12 h at room temperature. (In the case of 50 wt% PTA/SiO2 1–1 g of the reagents were mixed in 8 mL of doubly distilled water.) After being stirred, water was evaporated at 60 °C under reduced pressure. The remaining solid was then dried at 80 °C for 2 h under reduced pressure (1.5 mbar) and for an additional 10 h at 150 °C in same vacuum.
4.3 Catalytic Alkylation in Batch Conditions
In a flame dried Schlenk tube toluene (15 mmol) and 1-octene (1.5 mmol) were mixed and 23 mg of supported catalyst was added. The reaction mixtures were stirred for the given time and at the given temperature. In the case of the recycling experiments, the solid catalyst was left to settle and cool and the reaction mixture was carefully removed by a syringe. Then a fresh mixture of toluene (15 mmol) and 1-octene (1.5 mmol) was added to the catalyst and the reaction was initiated by stirring at 70 °C. All manipulations were performed under argon.
4.4 Catalytic Alkylation in Flow Conditions
The 4 Å molecular sieve and the catalyst were filled into the corresponding stainless steel columns. The columns containing the catalyst could be filled uniformly. The flow system was flushed with absolute toluene. The reaction mixture (feed) was prepared. The inlet tube was changed from the toluene to the feed and the continuous feeding of the toluene/1-octene mixture was started.
4.5 Determination of Product Composition
The composition of the product was determined with GC using an Agilent 7820A, equipped with an injector at 250 °C and a FID detector at 250 °C. N2 was used as carrier gas. Conversion of octene was calculated as the following: the sum of the corrected peak area of the products (Amonooctyl-toluene + 2Adioctyl-toluene) was divided by the sum of the corrected peak areas of the products (Amonooctyl-toluene + 2Adioctyl-toluene) and the peak area of the remaining alkene (Aoctene). (The sole products of the reaction are mono- and dioctyl-toluenes, no other products formed in the experiments.)
The definition of E-factor:
In the catalytic reactions mono- and dioctyl-toluene formed as mixtures of regioisomers. Additionally, in the case of uncompleted reactions the significant isomerization of the 1-octene starting material could be observed. These isomers are linear chain regioisomers (i.e. 2-, 3- or 4-octene) as was proved by 1H NMR spectroscopy. This corroborates the fact that only linear chain monooctyl-toluenes formed in the reactions. Based on the 1H NMR study of the product, 3 × 3 isomers could be identified corresponding to the 2-, 3- and 4-octyl derivatives and the o-, m- and p-positions on the aromatic ring, no 1-aryl-octane isomers could be detected. The exact ratio of the product monooctyl-toluene isomers could not be determined due to the significant signal overlap in the 1H NMR spectrum.
References
Weissermel K, Arpe H-J (2003) Industrial organic chemistry. Wiley-VCH, Weinheim
Caeiro G, Carvalho RH, Wang X, Lemos MANDA, Lemos F, Guisnet M, Ramôa Ribeiro F (2006) Activation of C2–C4 alkanes over acid and bifunctional zeolite catalysts. J Mol Catal A Chem 255(1–2):131–158. https://doi.org/10.1016/j.molcata.2006.03.068
Alotaibi A, Bayahia H, Kozhevnikova EF, Kozhevnikov IV (2015) Selective alkylation of benzene with propane over bifunctional Pt-heteropoly acid catalyst. ACS Catal 5(9):5512–5518. https://doi.org/10.1021/acscatal.5b01102
Ymamoto H, Ishihara K (eds) (2008) Acid catalysis in organic synthesis. Wiley-VCH, Weinheim
Macquarrie DJ (2009) Industrial Friedel–Crafts chemistry. In: Bandini M, Umani-Ronchi A (eds) Catalytic asymmetric Friedel–Crafts alkylations. Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim. https://doi.org/10.1002/9783527626977.ch8
Hancsók J, Visnyei O, Holló A, Leveles L, Thernesz A, Varga G, Valyon J (2019) Alternative diesel fuels with high hydrogen content in their molecular structures. Renew Energy 142:239–248. https://doi.org/10.1016/j.renene.2019.04.105
Srivastava SP, Hancsók J (2014) Fuels and fuel-additives. Wiley, Hoboken
Wilson K, Clark JH (2000) Solid acids and their use as environmentally friendly catalysts in organic synthesis. Pure Appl Chem 72(7):1313–1319. https://doi.org/10.1351/pac200072071313
Scheibel JJ (2004) The evolution of anionic surfactant technology to meet the requirements of the laundry detergent industry. J Surfactants Deterg 7(4):319–328. https://doi.org/10.1007/s11743-004-0317-7
Dasgupta S, Török B (2008) Environmentally benign contemporary Friedel–Crafts chemistry by solid acids. Curr Org Synth 5(4):321–342. https://doi.org/10.2174/157017908786241572
Kozhevnikov IV (1998) Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem Rev 98(1):171–198. https://doi.org/10.1021/cr960400y
Misono M, Ono I, Koyano G, Aoshima A (2000) Heteropolyacids. Versatile green catalysts usable in a variatey of reaction media. Pure Appl Chem 72(7):1305–1311. https://doi.org/10.1351/pac200072071305
Okuhara T, Misono M (1994) Oxide catalysts in solid state chemistry. In: King RB (ed) Encyclopedia of inorganic chemistry. Wiley, Chichester
Almohalla M, Rodríguez-Ramos I, Guerrero-Ruiz A (2017) Comparative study of three heteropolyacids supported on carbon materials as catalysts for ethylene production from bioethanol. Catal Sci Technol 7:1892–1901. https://doi.org/10.1039/C7CY00155J
Alcañiz-Monge J, Trautwein G, Marco-Lozar JP (2013) Biodiesel production by acid catalysis with heteropolyacids supported on activated carbon fibers. Appl Catal A Gen 468:432–441. https://doi.org/10.1016/j.apcata.2013.09.006
Ferreira P, Fonseca M, Ramos AM, Vital J, Castanheiro JE (2011) Acetylation of glycerol over heteropolyacids supported on activated carbon. Catal Commun 12(7):573–576. https://doi.org/10.1016/j.catcom.2010.11.022
Micek-Ilnicka A, Bielańska E, Lityńska-Dobrzyńska L, Bielański A (2012) Carbon nanotubes, silica and titania supported heteropolyacid H3PW12O40 as the catalyst for ethanol conversion. Appl Catal A Gen 421–422:91–98
Peng L, Gao X, Liu Y, Zhang J, He L (2021) Coupled transfer hydrogenation and alcoholysis of furfural to yield alkyl levulinate over multifunctional zirconia-zeolite-supported heteropoly acid. Energy Fuels 35(5):4182–4190. https://doi.org/10.1021/acs.energyfuels.0c04222
Sert E, Atalay FS (2012) Esterification of acrylic acid with different alcohols catalyzed by zirconia supported tungstophosphoric acid. Ind Eng Chem Res 51(19):6666–6671. https://doi.org/10.1021/ie202609f
Quereshi S, Ahmad E, Pant KK, Dutta S (2019) Synthesis and characterization of zirconia supported silicotungstic acid for ethyl levulinate production. Ind Eng Chem Res 58(35):16045–16054. https://doi.org/10.1021/acs.iecr.9b01659
Viswanadham B, Vishwanathan V, Chary KVR, Satyanarayana Y (2021) Catalytic dehydration of glycerol to acrolein over mesoporous MCM-41 supported heteropolyacid catalysts. J Porous Mater. https://doi.org/10.1007/s10934-021-01070-8
Ma T, Ding J, Shao R, Xu W, Yun Z (2017) Dehydration of glycerol to acrolein over Wells-Dawson and Keggin type phosphotungstic acids supported on MCM-41 catalysts. Chem Eng J 316:797–806. https://doi.org/10.1016/j.cej.2017.02.018
Kocaman E, Akarçay Ö, Bağlar N, Çelebi S, Uzun A (2018) Isobutene oligomerization on MCM-41-supported tungstophosphoric acid. Mol Catal 457:41–50. https://doi.org/10.1016/j.mcat.2018.07.013
Sheng X, Kong J, Zhou Y, Zhang Y, Zhang Z, Zhou S (2014) Direct synthesis, characterization and catalytic application of SBA-15 mesoporous silica with heteropolyacid incorporated into their framework. Microporous Mesoporous Mater 187:7–13. https://doi.org/10.1016/j.micromeso.2013.12.007
Pinto T, Arquillière P, Niccolai GP, Lefebvre F, Dufaud V (2015) The comparison of two classes of bifunctional SBA-15 supported platinum–heteropolyacid catalysts for the isomerization of n-hexane. New J Chem 39:5300–5308. https://doi.org/10.1039/C5NJ00453E
Pinto T, Dufaud V, Lefebvre F (2014) Isomerization of n-hexane on heteropolyacids supported on SBA-15. 1. Monofunctional impregnated catalysts. Appl Catal A Gen 483:103–109. https://doi.org/10.1016/j.apcata.2014.07.003
Tropecêlo AI, Casimiro MH, Fonseca M, Ramos AM, Vital J, Castanheiro JE (2010) Esterification of free fatty acids to biodiesel over heteropolyacids immobilized on mesoporous silica. Appl Catal A Gen 390(1–2):183–189. https://doi.org/10.1016/j.apcata.2010.10.007
Moffat JB, Kasztelan S (1988) The oxidation of methane on heteropolyoxometalates II. Nature and stability of the supported species. J Catal 109(1):206–211
Yanga Y, Lv G, Guo W, Zhang L (2018) Synthesis of mesoporous silica-included heteropolyacids materials and the utilization for the alkylation of phenol with cyclohexene. Microporous Mesoporous Mater 261:214–219. https://doi.org/10.1016/j.micromeso.2017.11.018
Pezzotta C, Fleury G, Soetens M, Van der Perre S, Denayer JFM, Riant O, Gaigneaux EM (2018) Improving the selectivity to 4-tert-butylresorcinol by adjusting the surface chemistry of heteropolyacid-based alkylation catalysts. J Catal 359:198–211. https://doi.org/10.1016/j.jcat.2018.01.010
Yu FL, Wang QY, Yuan B, Xie CX, Yu ST (2017) Alkylation desulfurization of FCC gasoline over organic-inorganic heteropoly acid catalyst. Chem Eng J 309:298–304. https://doi.org/10.1016/j.cej.2016.10.003
Zhang P, Yang B, Ma H, Wu Z (2021) Graphene modified porous organic polymer supported phosphotungstic acid catalyst for alkylation desulfurization. Fuel 293:120438. https://doi.org/10.1016/j.fuel.2021.120438
Sartori G, Maggi R (2011) Update 1 of: use of solid catalysts in Friedel−Crafts acylation reactions. Chem Rev 111(5):PR181–PR214. https://doi.org/10.1021/cr100375z
Ammar M, Jiang S, Ji S (2016) Heteropoly acid encapsulated into zeolite imidazolate framework (ZIF-67) cage as an efficient heterogeneous catalyst for Friedel–Crafts acylation. J Solid State Chem 233:303–310. https://doi.org/10.1016/j.jssc.2015.11.014
Fang Z, He W, Tu T, Lv N, Qiu C, Li X, Zhu N, Wan L, Guo K (2018) An efficient and green pathway for continuous Friedel–Crafts acylation over α-Fe2O3 and CaCO3 nanoparticles prepared in the microreactors. Chem Eng J 331:443–449. https://doi.org/10.1016/j.cej.2017.09.001
Costa VV, da Silva Rocha KA, Kozhevnikov IV, Gusevskaya EV (2010) Isomerization of styrene oxide to phenylacetaldehyde over supported phosphotungstic heteropoly acid. Appl Catal A Gen 383(1–2):217–220. https://doi.org/10.1016/j.apcata.2010.06.005
Cotta RF, Martins RA, Pereira MM, da Silva Roch KA, Kozhevnikova EF, Kozhevnikov IV, Gusevskaya EV (2019) Heteropoly acid catalysis for the isomerization of biomass-derived limonene oxide and kinetic separation of the trans-isomer in green solvents. Appl Catal A Gen 584:117173. https://doi.org/10.1016/j.apcata.2019.117173
Song D, An S, Sun Y, Guo Y (2016) Efficient conversion of levulinic acid or furfuryl alcohol into alkyl levulinates catalyzed by heteropoly acid and ZrO2 bifunctionalized organosilica nanotubes. J Catal 333:184–199. https://doi.org/10.1016/j.jcat.2015.10.018
Da Silva MJ, Teixeira MG (2017) An unexpected behavior of H3PMo12O40 heteropolyacid catalyst on the biphasic hydrolysis of vegetable oils. RSC Adv 7:8192–8199. https://doi.org/10.1039/C6RA27287H
Liu W, Qi W, Guo X, Su D (2015) Heteropoly acid/nitrogen functionalized onion-like carbon hybrid catalyst for ester hydrolysis reactions. Chem Asian J 11(4):491–497. https://doi.org/10.1002/asia.201500944
Hosseini H, Masteri-Farahani M (2019) Surface functionalization of magnetite nanoparticles with sulfonic acid and heteropoly acid: efficient magnetically recoverable solid acid catalysts. Chem Asian J 14(7):1076–1083. https://doi.org/10.1002/asia.201801810
Huang F, Su Y, Tao Y, Sun W, Wang W (2018) Preparation of 5-hydroxymethylfurfural from glucose catalyzed by silica-supported phosphotungstic acid heterogeneous catalyst. Fuel 226:417–422. https://doi.org/10.1016/j.fuel.2018.03.193
Li J, Yang Z, Hu G, Zhao J (2020) Heteropolyacid supported MOF fibers for oxidative desulfurization of fuel. Chem Eng J 388:124325. https://doi.org/10.1016/j.cej.2020.124325
Kanno M, Yasukawa T, Ninomiya W, Ooyachi K, Kamiya Y (2010) Catalytic oxidation of methacrolein to methacrylic acid over silica-supported 11-molybdo-1-vanadophosphoric acid with different heteropolyacid loadings. J Catal 273(1):1–8. https://doi.org/10.1016/j.jcat.2010.04.014
Li J, Yang Z, Li S, Jin Q, Zhao J (2020) Review on oxidative desulfurization of fuel by supported heteropolyacid catalysts. J Ind Eng Chem 82:1–16. https://doi.org/10.1016/j.jiec.2019.10.020
Lefebvre F (2017) Acid catalysis by heteropolyacids: transformations of alkanes. Curr Catal 6(2):77–89. https://doi.org/10.2174/2211544706666170203123816
Izumi Y, Natsume N, Takamine H, Tamaoki I, Urabe K (1989) Silica-supported heteropoly acid catalyst for liquid-phase Friedel–Crafts reactions. Bull Chem Soc J 62(7):2159–2162. https://doi.org/10.1246/bcsj.62.2159
Zhu H-O, Wang J, Zeng C-Y, Zhao D-Y (2003) Novel SBA-15 supported heteropoly acid catalysts for benzene alkylation with 1-dodecene. Stud Surf Sci Catal 146:661–664. https://doi.org/10.1016/S0167-2991(03)80470-6
Ren Y, Yue B, Gu M, He H (2010) Progress of the application of mesoporous silica-supported heteropolyacids in heterogeneous catalysis and preparation of nanostructured metal oxides. Materials 3:764–785. https://doi.org/10.3390/ma3020764
Liu Y, Xu L, Xu B, Li Z, Jia L, Guo W (2009) Toluene alkylation with 1-octene over supported heteropoly acids on MCM-41 catalysts. J Mol Catal A Chem 297(2):86–92. https://doi.org/10.1016/j.molcata.2008.09.007
Devassy BM, Lefebvre F, Bohringer W, Fletcher J, Halligudi SB (2005) Synthesis of linear alkyl benzenes over zirconia-supported 12-molybdophosphoric acid catalysts. J Mol Catal A Chem 236:162–167. https://doi.org/10.1016/j.molcata.2005.03.033
Zhang J, Zhu Z, Li C, Wen L, Min E (2003) Characterization and kinetic investigation of tungstophosphoric acid supported on SiO2 for alkylation of benzene with 1-dodecene to synthesize linear alkylbenzene. J Mol Catal A Chem 198:359–367. https://doi.org/10.1016/S1381-1169(03)00004-9
Zhang J, Chen B, Li C, Zhu Z, Wen L, Min E (2003) Kinetics of benzene alkylation with 1-dodecene over a supported tungstophosphoric acid catalyst. Appl Catal A Gen 249:27–34. https://doi.org/10.1016/S0926-860X(03)00206-0
Hung C-T, Liu L-L, Wang J-J, Wu P-H, Wang C-B, Tsai T-C, Liu S-B (2019) Acidity and alkylation activity of 12-tungstophosphoric acid supported onionic liquid-functionalized SBA-15. Catal Today 327:10–18. https://doi.org/10.1016/j.cattod.2018.07.041
Blasco T, Corma A, Martínez A, Martínez-Escolano P (1998) Supported heteropolyacid (HPW) catalysts for the continuous alkylation of isobutane with 2-butene: the benefit of using MCM-41 with larger pore diameters. J Catal 177(2):303–313. https://doi.org/10.1006/jcat.1998.2105
Kuang W, Rives A, Fournier M, Hubaut R (2003) Structure and reactivity of silica-supported 12-tungstophosphoric acid. Appl Catal A Gen 250:221–229. https://doi.org/10.1016/S0926-860X(03)00239-4
Kozhevnikov IV, Kloetstra KR, Sinnema A, Zandbergen HW, van Bekkum H (1996) Study of catalysts comprising heteropoly acid H3PW12O40 supported on MCM-41 molecular sieve and amorphous silica. J Mol Catal A Chem 114:287–298. https://doi.org/10.1016/S1381-1169(96)00328-7
Newman AD, Brown DR, Siril P, Lee AF, Wilson K (2006) Structural studies of high dispersion H3PW12O40/SiO2 solid acid catalysts. Phys Chem Chem Phys 8:2893–2902. https://doi.org/10.1039/B603979K
Mastikhin VM, Kulikov SM, Nosov AV, Kozhevnikov IV, Mudrakovsky IL, Timofeeva MN (1990) 1H and 31P MAS NMR studies of solid heteropolyacids and H3PW12O40 supported on SiO2. J Mol Catal 60:65–70. https://doi.org/10.1016/0304-5102(90)85068-S
Lefebvre F (1992) 31P MAS NMR study of H3PW12O40 supported on silica: formation of (≡SiOH2+)(H2PW12O40−). J Chem Soc Chem Commun. https://doi.org/10.1039/C39920000756
Bronnimann CE, Chuang I-S, Hawkins BL, Maciel GE (1987) Dehydration of silica-aluminas monitored by high-resolution solid-state proton NMR. J Am Chem Soc 109(5):1562–1564. https://doi.org/10.1021/ja00239a043
Bronnimann CE, Zeigler RC, Maciel GE (1988) Proton NMR study of dehydration of the silica gel surface. J Am Chem Soc 110(7):2023–2026. https://doi.org/10.1021/ja00215a001
Haukka S, Lakomaa EL, Root A (1993) An IR and NMR study of the chemisorption of titanium tetrachloride on silica. J Phys Chem 97(19):5085–5094. https://doi.org/10.1021/j100121a040
Ghambari-Siahkali A, Philippou A, Dwyer J, Anderson MW (2000) The acidity and catalytic activity of heteropoly acid on MCM-41 investigated by MAS NMR, FTIR and catalytic tests. Appl Catal A Gen 192(1):57–69. https://doi.org/10.1016/S0926-860X(99)00333-6
Pezotta C, Marakati VS, Gaigneaux EM (2020) Role of Lewis and Brønsted acid sites in resorcinol tert-butylation over heteroplyacid-based catalysts. Catal Sci Technol 10:7984–7997. https://doi.org/10.1039/D0CY01030H
Thomas A, Dablemont C, Basset J-M, Lefebvre F (2005) Comparison of H3PW12O40 and H4SiW12O40 heteropolyacids supported on silica by 1H MAS NMR. C R Chim 8:1969–1974. https://doi.org/10.1016/j.crci.2005.06.006
Wang J-J, Chuang Y-Y, Hsu H-Y, Tsai T-C (2017) Toward industrial catalysis of zeolite for linear alkylbenzene synthesis: a mini review. Catal Today 298:109–116. https://doi.org/10.1016/j.cattod.2017.05.046
Lovás P, Horňáček M, Hudec P, Jorík V (2014) Preparation of an active and regenerable catalyst for liquid-phasealkylation of toluene with 1-decene. Appl Catal A Gen 475:341–346. https://doi.org/10.1016/j.apcata.2014.01.047
Acknowledgements
We thank Mr Béla Édes for skillful assistance in analytical measurements and synthetic experiments. The research was supported by the project GINOP-2.3.2-15-2016-00053.
Funding
Open access funding provided by University of Pannonia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Császár, Z., Juzsakova, T., Jakab, M. et al. Continuous Flow Friedel–Crafts Alkylation Catalyzed by Silica Supported Phosphotungstic Acid: An Environmentally Benign Process. Top Catal (2021). https://doi.org/10.1007/s11244-021-01497-y
Accepted:
Published:
DOI: https://doi.org/10.1007/s11244-021-01497-y