
Abstract
Introduction
Recurrent event rates after myocardial infarction (MI) remain unacceptably high, in part because of the continued growth and destabilization of residual coronary atherosclerotic plaques, which may occur despite lipid-lowering therapy. Inflammation is an important contributor to this ongoing risk. Recent studies have shown that the broad-acting anti-inflammatory agent, colchicine, may reduce adverse cardiovascular events in patients post-MI, although the mechanistic basis for this remains unclear. Advances in endovascular arterial wall imaging have allowed detailed characterization of the burden and compositional phenotype of coronary plaque, along with its natural history and responsiveness to treatment. One such example has been the use of optical coherence tomography (OCT) to demonstrate the plaque-stabilizing effects of statins on both fibrous cap thickness and the size of lipid pools within plaque.
Methods
The Phase 2, multi-centre, double-blind colchicine for coronary plaque modification in acute coronary syndrome (COCOMO-ACS) study will evaluate the effect of colchicine 0.5 mg daily on coronary plaque features using serial OCT imaging in patients following MI. Recruitment for the trial has been completed with 64 participants with non-ST elevation MI randomized 1:1 to colchicine or placebo in addition to guideline recommended therapies, including high-intensity statins. The primary endpoint is the effect of colchicine on the minimal fibrous cap thickness of non-culprit plaque over an 18-month period.
Summary
The COCOMO-ACS study will determine whether addition of colchicine 0.5 mg daily to standard post-MI treatment has incremental benefits on high-risk features of coronary artery plaques. If confirmed, this will provide new mechanistic insights into how colchicine may confer clinical benefits in patients with atherosclerotic cardiovascular disease.
Trial Registration
ANZCTR trial registration number: ACTRN12618000809235. Date of trial registration: 11th of May 2018.
Similar content being viewed by others

Introduction
Atherosclerotic coronary artery disease (CAD) is the principal source of cardiovascular mortality, morbidity and health economic burden worldwide [1, 2]. While aggressive modification of traditional atherosclerosis risk factors has led to a substantial reduction in the incidence of myocardial infarction (MI) [3], the recurrence of major adverse cardiovascular events (MACE) post-MI remains a major concern [4]. Inflammation is independently and strongly associated with future MACE in both primary and secondary prevention settings [5,6,7,8]. Current therapies for CAD, such as lipid-lowering statins, do not adequately control plaque inflammation, meaning that new anti-atherosclerotic agents targeting inflammation are needed [9]. The positive results for the anti-interleukin-1-beta (IL-1β) monoclonal antibody, canakinumab, in the CANTOS clinical trial [10], highlighted that not only is inflammation important from a pathogenic and risk prediction perspective in CAD, but that reducing inflammation can be beneficial. More recently, low-dose colchicine, a broad acting anti-inflammatory agent, has been repurposed for the treatment of CAD. Results from the LoDoCo [11], LoDoCo 2 [12], COPS [13] and COLCOT [14] studies have demonstrated potential clinical benefit of colchicine use in patients with both stable and unstable CAD, with other Phase 3 clinical studies still ongoing [9]. Colchicine’s potential mechanism of effect in atherosclerosis is protean, and although it may primarily act via its known action on innate immune pathways, we hypothesize that it may also promote favourable plaque healing following MI, leading to the formation of a thicker plaque cap which is more resilient to rupture and thrombosis.
Coronary Atherosclerosis: an Inflammatory Process Requiring Treatment Beyond Statins
Considered a chronic inflammatory disease of blood vessels, atherosclerosis is characterized by a maladaptive immune response, triggered by subintimal lipoprotein accumulation, and resulting in the formation of lipid-rich, inflamed plaques [9]. Ultimately, systemic and local activation of both the adaptive and innate immune systems can result in either rupture or erosion of an atherosclerotic plaque, leading to thrombosis, which may manifest as unstable angina, acute MI or sudden cardiac death [15, 16]. Despite no established association between genetically elevated C-reactive protein (CRP) and coronary heart disease in genome-wide association studies [17, 18], measurement of the inflammatory biomarker, high-sensitivity CRP (hs-CRP), has been shown to independently predict future cardiovascular events in both patients with stable coronary disease [19] and those with acute coronary syndrome (ACS) [20]. After adjusting for traditional risk factors, a meta-analysis of 54 long-term prospective studies involving 160,000 participants revealed that each standard deviation increase in log-normalized hs-CRP was associated with an increase in relative risk of 1.37 (95% CI, 1.27 to 1.48) for CAD and 1.55 (95% CI, 1.37 to 1.76) for cardiovascular mortality [21]. Statins reduce hs-CRP in a partly low-density lipoprotein cholesterol (LDL-C) independent manner, and in the JUPITER study, the most favourable clinical outcomes were observed in participants who achieved both a reduction in LDL-C and hs-CRP [22]. However, the concerning frequency of recurrent cardiovascular events despite highly effective LDL-C reduction has led to concerns that residual inflammatory risk is also causal. The PROVE-IT trial showed that of the 2,099 patients with prior ACS prescribed atorvastatin 80 mg daily, 44% achieved target reductions of both LDL-C (< 1.8 mm/L) and hs-CRP (< 2 mg/L) [23, 24]. However, 29% were left with a residual inflammatory burden (hs-CRP > 2 mg/L), 13% a residual cholesterol risk (LDL-C > 1.8 mmol/L) and 14% with both, indicating that residual inflammatory risk is frequent after high-intensity statin therapy. Together, these results highlight that recurrent cardiovascular events are determined by multiple biological processes that require a tailored therapeutic approach and that future treatment for CAD must tackle residual inflammation not completely addressed by statin therapy alone.
Repurposing Potential of Colchicine in CAD
Colchicine, an orally administered, inexpensive anti-inflammatory medication used to treat gout, pericarditis and familial Mediterranean fever (FMF), has been repurposed over recent years to treat CAD. Colchicine binds to microtubule ends and inhibits cytoskeletal microtubule processes [25]. At low concentrations, it inhibits their formation, and at higher concentrations, it promotes their depolymerization [26]. Neutrophil chemotaxis, phagocytosis and protein excretion are all microtubule-dependent processes. In vitro experiments have identified that colchicine lessens endothelial selectin family-dependent adhesiveness, affecting both endothelial E-selectin and neutrophil L-selectin surface expression [27]. Additionally, it has been found to increase leukocyte cyclic adenosine monophosphate levels, inhibit IL-1 production by activated neutrophils and to downregulate tumour necrosis factor-alpha receptors in macrophages [28]. In the setting of atherosclerotic plaque containing cholesterol crystals, NLRP3 inflammasome activation is dampened in colchicine-treated neutrophils and macrophages [29]. Colchicine also suppresses neutrophil extracellular traps (NETs) by restoring cytoskeletal dynamics in patients treated with percutaneous coronary intervention (PCI) following MI [30]. Comprising strands of DNA in the form of decondensed chromatin, and liberated by activated or dying neutrophils, NETs localize at the interface of blood and the intimal surface of diseased arteries [31]. Here they form a fibrin-like base for platelet adhesion, activation and aggregation, promote the accumulation of prothrombotic molecules, including von Willebrand factor and fibrinogen, and facilitate erythrocyte adhesion, all of which contribute to thrombus formation [32].
Retrospective observational studies identified lower rates of MI and other vascular events in patients with gout and FMF treated with colchicine [33, 34]. The first prospective study to examine the impact of colchicine in cardiovascular disease was the LoDoCo study, which evaluated the effect of colchicine 0.5 mg daily in addition to antiplatelet and statin therapy in 532 patients with stable CAD [11]. After a median follow-up of 3 years, participants who had received colchicine demonstrated a significant reduction in a composite endpoint consisting of MI, cardiac arrest or non-cardioembolic stroke when compared to the no colchicine control group (hazard ratio (HR), 0.33). The LoDoCo2 study, which randomized 5,522 patients with stable CAD to colchicine 0.5 mg daily or matching placebo revealed a similar therapeutic benefit at a median follow-up of 28.6 months [12]. The colchicine-treated group had a significant reduction in the primary composite endpoint of cardiovascular death, non-procedural MI, ischemic stroke, or ischemia-driven coronary revascularization compared to placebo (HR, 0.69). The secondary endpoint composite of cardiovascular death, spontaneous MI and ischemic stroke was also significantly lowered (HR, 0.72), as were the individual endpoints of MI and revascularization.
Inflammation is especially accentuated in the coronary vascular bed following acute MI [35]. The COLCOT trial demonstrated the potential benefits of colchicine in 4,755 patients randomized within 30 days following MI to receive colchicine 0.5 mg daily or placebo in addition to standard guideline-recommended therapy [14]. The primary efficacy endpoint was a composite of cardiovascular death, resuscitated cardiac arrest, MI, stroke or urgent hospitalization for angina requiring coronary revascularization and occurred in 5.5% of the participants who received colchicine compared to 7.1% in the placebo arm (HR, 0.77). The difference in the primary endpoint was driven by a significant risk reduction in urgent hospitalization for angina requiring revascularization (HR, 0.50) and for stroke (HR, 0.26). Trends towards reduced cardiovascular death (HR, 0.83) and MI (HR, 0.91) in colchicine-treated participants did not reach statistical significance. A recent substudy of COLCOT established that there was a significant reduction in the incidence of the primary endpoint for patients in whom colchicine was initiated between days 0 and 3 compared with placebo (HR, 0.52), in contrast to patients who either started colchicine between days 4 and 7 (HR, 0.96), or from day 8 onwards (HR, 0.82) [36]. This intimated that early initiation of low-dose colchicine after MI provides maximal clinical benefit with a 48% reduction in ischemic cardiovascular events when compared to placebo and supports the strategy of early in-patient initiation of therapy.
The COPS trial randomized 795 patients with ACS, managed with either PCI or medical therapy, to receive either colchicine 0.5 mg twice daily for 1 month, then 0.5 mg daily for 11 months, or placebo [13]. At 12-month follow-up, there was no significant difference in the primary composite endpoint of all-cause mortality, ACS, ischemia-driven urgent revascularization and non-cardioembolic ischemic stroke in the colchicine group compared to placebo (6.1 versus 9.5%; P = 0.09). However, a trend towards clinical benefit following the use of colchicine was observed and may not have reached statistical significance due to the small sample size. Noteworthy, in a post hoc analysis of the composite endpoint using only cardiovascular death rather than total death, a significant reduction in events in favour of colchicine was demonstrated at 12 months (HR, 0.51).
The propensity of low-dose colchicine to cause gastrointestinal intolerance in up to 10–15% of patients [37], the increased risk of pneumonia seen in COLCOT (0.9% compared with 0.3% in placebo arm; P = 0.03) [14] and a non-significant trend toward higher mortality from non-cardiovascular causes in some studies, including a recent meta-analysis [12, 38], provide some cause for caution in colchicine’s routine use. Additional randomized controlled trial data is needed to help clarify its safety and efficacy.
Mechanistic Actions of Colchicine on Atherosclerotic Plaque
The successful repurposing of colchicine for atherosclerotic cardiovascular disease will also benefit from a better understanding of how it works. However, few studies have directly examined colchicine’s effects on atherosclerotic plaque. Published preclinical data are mostly limited to studies performed more than 20 years ago and provide conflicting evidence that colchicine may either exacerbate [39] or inhibit [40] plaque formation or have no effect [41]. Most recently, in a rabbit model of atherosclerosis, in vivo multimodality imaging using magnetic resonance imaging, 18F-Fluorodeoxyglucose-positron emission tomography, optical coherence tomography (OCT) and immunohistology demonstrated that colchicine may stabilize atherosclerotic plaque by reducing plaque inflammation and burden, without altering macrophage infiltration or atherosclerotic plaque microstructure. In this study, no difference was observed in terms of lipid plaque proportion or mean minimum fibrous cap thickness measured by OCT in either colchicine- or placebo-treated cohorts [42].
So far the only clinical evaluation of colchicine’s ability to modify coronary plaque comes from a non-randomized, single centre observational study of 80 patients post-ACS, in whom colchicine significantly reduced low attenuation plaque volume (treatment: mean 15.9 [− 40.9%] ± 17.3 mm3; control mean 6.61 [− 17.0%] ± 12.8 mm3; P = 0.008), as measured by serial coronary computed tomography angiography over 12 months [43]. This was accompanied by a reduction in hs-CRP with colchicine compared to the non-placebo control group and supports the notion that colchicine may help to stabilize plaques. Although the predominant focus on the mechanism of action of colchicine in atherosclerosis has been on its purported ability to attenuate cholesterol crystal-induced inflammasome activation [44], there is increasing evidence that other pleiotropic mechanisms may also be instrumental in providing athero-protection and plaque stabilization [45]. This is supported by a recent proteomic substudy of LoDoCo 2 [46], in which colchicine was associated with a reduction in serum levels of 11 proteins not directly involved in the NLRP3 inflammasome pathway, including strong attenuation in proteins involving neutrophil degranulation (myeloblastin, carcinoembryonic antigen-related cell adhesion molecule 8, azurocidin and myeloperoxidase). Colchicine also resulted in an increase in another 23 proteins, including intestinal fatty acid-binding protein, a biomarker of intestinal barrier dysfunction, protein-glutamine γ-glutamyltransferase 2, related to tissue repair, and both fibroblast growth factor 21 and insulin-like growth factor-binding protein 1, hypothesized to protect against atherosclerosis.
Assessment of Therapeutic Modulation of Plaque Using Endovascular Imaging
Intracoronary OCT is a catheter-based imaging technique which utilizes coherent near-infrared light, typically of a wavelength of approximately 1,300 nm, to create images of plaque atheroma in the coronary arteries from optical backscatter. Its greatest advantage is its high spatial resolution (approximately ten times higher than that of intravascular ultrasound), with an axial resolution of up to 10 μm and lateral resolution of up to 20 μm [47, 48]. This enables qualitative and quantitative analysis of the atheroma below the intimal endothelial surface, providing the ability to discriminate between fibrous, lipid-rich and calcified plaques [47, 49, 50], quantitate lipid content and macrophage burden [49, 51] and accurately measure fibrous cap thickness [50].
Lipid-rich thin-cap fibroatheromas (TCFAs) that are believed to be more prone to rupture can be identified and quantitated by OCT. The relationship between culprit site lipid-rich plaques (LRP) and cardiovascular events has been documented in patients with ACS utilizing OCT [52, 53]. A retrospective study demonstrated that OCT-detected non-culprit LRP led to a two-fold increase in non-culprit MACE (HR, 2.06), primarily driven by revascularization for recurrent ischemia [54]. In addition, OCT has been used to assess modulation of coronary atherosclerosis in response to different established therapies. Serial OCT assessment of non-culprit lesions was used in 70 patients with unstable angina and dyslipidemia in the Japanese EASY-FIT study to investigate plaque stability. Patients were randomized to either 20 mg or 5 mg of atorvastatin daily and imaging was performed at baseline and 12-month follow-up [55]. Lower LDL-C levels were obtained in the higher dose group (69 versus 78 mg/dL, P = 0.039) with OCT suggesting a more stable plaque in this group, characterized by a significant increase in fibrous cap thickness of 69% compared to 17% (P < 0.001). The increase in fibrous cap correlated with the decrease in LDL-C levels and grade of OCT measured macrophages. Similar results were seen in a single centre OCT study that examined the effects of the addition of ezetimibe 10 mg daily to treatment with fluvastatin 30 mg daily. In this study, the reduction in LDL-C was significantly larger in the group receiving combination therapy (− 34.0 ± 32.0 versus − 8.8 ± 17.4 mg/dL, P < 0.001), and while OCT demonstrated a significant increase in fibrous cap thickness after 9 months of therapy in both groups, this was greater in those receiving combination therapy [56].
These findings contribute to our understanding of the mechanistic effects of statins on plaque. As new therapies evolve in the treatment of CAD, intracoronary imaging studies will continue to complement Phase 3 outcome trials by evaluating their effectiveness at modifying the burden and composition of coronary plaques [57].
Design of the COCOMO-ACS Study
Objectives
The COCOMO-ACS study will employ serial OCT imaging to evaluate the impact of colchicine on coronary atheroma phenotype. The primary objective is to evaluate the effect of colchicine on changes in fibrous cap thickness on non-culprit plaques in patients with non-ST-elevation myocardial infarction (NSTEMI), taking maximally tolerated oral lipid-lowering therapy. Additional objectives are to evaluate the impact of colchicine on other measures of plaque phenotype and the safety and tolerability of colchicine when administered to patients in the post-ACS setting. The primary hypothesis of the study is that the addition of colchicine to high-intensity statin therapy will have a favourable effect on coronary plaque composition, with a greater increase in the minimum fibrous cap thickness compared to lipid-lowering therapy alone.
Study Design
COCOMO-ACS is an investigator-initiated, multicentre, randomized, double-blind, placebo-controlled, Phase 2 clinical trial in patients hospitalized due to NSTEMI to evaluate the effect of colchicine on non-culprit plaque phenotype as assessed by serial OCT. The COCOMO-ACS study includes patients from nine sites across Australia with no maximum limit on recruitment at each centre.
Ethical Aspects
All procedures in this study are conducted in accordance with the Declaration of Helsinki, International Conference on Harmonization, Good Clinical Practice guidelines and applicable regulatory requirements. The final protocol and informed consent forms were approved by the Royal Adelaide Hospital Human Research Ethics Committee (HREC) under the National Mutual Acceptance (NMA) system with local Research Governance Officer (RGO) approval at each site (HREC/17/RAH/366; Central Adelaide Local Health Network Reference Number R20170904). This clinical trial was registered with the Australian New Zealand Clinical Trials Registry (ACTRN12618000809235) on the 11th of May 2018.
Study Population
Randomization has now been completed for this study. Eligible participants were those aged between 18 and 82 years of age, who had a clinically indicated coronary angiogram within 72 h of presenting with NSTEMI and gave written informed consent for the study before angiography. A culprit atherosclerotic lesion was identified and managed as clinically indicated. There must have been at least one intermediate lesion in a non-culprit coronary artery, determined visually to be 20–50% stenotic, with no intention to revascularize it in the next 18 months. The non-culprit lesion was imaged by OCT at baseline angiography. OCT images were assessed within 48 h at the Atherosclerosis Imaging Core Laboratory at the South Australian Health and Medical Research Institute (SAHMRI), to ensure that they were of suitable quality and that the target vessel contained a lipid-rich plaque (fibrous cap thickness ≤ 120 μm and lipid arc > 90°). A full list of inclusion and exclusion criteria is provided in Table 1.
Randomization and Allocation Concealment
Participants who passed angiographic and OCT screening were randomized 1:1 within 10 days of their angiogram and OCT study, to colchicine 0.5 mg or placebo, administered orally daily for 18 months. Randomization was by a computer generation Web response system, and to ensure balance, patients were stratified into two groups according to prior treatment with statins. Group 1 includes patients with no statin use in the preceding 4 weeks prior to admission. Group 2 includes patients who had been on a statin at any dose for at least 4 weeks prior to the admission. The randomization schedule included blocking within each stratum with variable block size of two to four. Treatments were randomly allocated within blocks with an equal number of each treatment within each block.
Once the participants were randomized, the randomization number was given to the pharmacist, and kit numbers containing the appropriate matching allocation were dispensed to participants. All treatment packs are identical and contain either active tablets (Colgout® manufactured by Aspen Pharmacare, NSW, Australia) or matching placebo tablets consisting of PROSOLV® EASYtab SP (composition: 95.0–98.0% microcrystalline cellulose, 1.5–2.5% colloidal silicon dioxide, 0.5–2.0% sodium starch glycolate and 0.3–1.0% sodium stearyl fumarate) (manufactured by Pharmaceutical Packaging Professionals Pty Ltd trading as PCI Pharma Services, Victoria, Australia). Researchers have no access to the randomization list. All researchers, study participants and adjudicators remain blinded to the treatment allocation. Only the pharmacist and a small unblinded team at SAHMRI have access to the randomization list to ensure allocation concealment throughout the study.
Standard of Care
All participants also receive currently recommended therapies after MI at the discretion of their treating clinician: dual antiplatelets, cholesterol-lowering medication including maximally tolerated dose of a statin to achieve LDL-C of < 1.8 mmol/L, with or without an ACE inhibitor/angiotensin II-receptor blocker or beta-blocker.
Study Procedures
Initial background assessments included demographics, cardiovascular risk factors, relevant medical and surgical history and laboratory data. Follow-up study visits are performed in the clinic at months 3, 6 and 12 and by telephone at months 1, 9 and 15. Pill counts from study medication bottles returned by participants are used to assess adherence. Imaging of the target artery by OCT was performed at the time of screening visit coronary angiography and will then be repeated at month 18. A final end of study visit will be performed at 18 months to complete the safety evaluation. The final OCT originally planned to be performed at 12 months has been extended to 18 months in the updated protocol due to restrictions on elective surgery schedules due to the COVID-19 pandemic. The study flowchart is presented in Fig. 1.

COCOMO-ACS study design
Image Analysis
All imaging performed is anonymized, digitally stored and transferred to the SAHMRI Atherosclerosis Imaging Core Laboratory for analysis. Screening OCT imaging was assessed within 48 h for image quality and to determine that the patient met all eligible imaging inclusion criteria. Paired baseline and final OCT images will be analysed at the end of the study, with measurements of plaque phenotype. The segment selected for analysis will be defined by proximal and distal side branches and by the presence of at least one image containing a fibrous cap thickness ≤ 120 μm and one image with a lipid arc > 90°. The same segment will undergo measurements by analysts who are blinded to the treatment status of the patient. Cross-sectional images spaced 0.2 mm apart will be selected for analysis. For each image, where plaque is present, measurement of minimum fibrous cap thickness, mean fibrous cap thickness, lipid arc and the lipid length will be performed (Fig. 2). Each image will be graded for the presence of macrophage infiltration, microchannels and calcium accumulation [58]. The presence of TCFA, plaque rupture and thrombus will also be recorded.

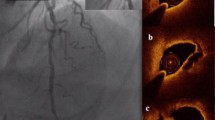
OCT plaque measurements. Example of coronary angiogram image in AP Cranial projection, identifying non-culprit plaques in the left anterior descending artery, with corresponding cross-sectional OCT image from the segment denoted by red arrow, highlighting the thin fibrous cap, lipid pool and lipid arc.
Sample Collection
Blood samples are being collected at baseline, 3-, 6-, 12- and 18-month visits. Clinical laboratory testing is performed by the local laboratory for complete blood picture, biochemistry, creatinine and estimated glomerular filtration rate (eGFR), liver function tests, thyroid function tests, lipids (LDL-C, high-density lipoprotein cholesterol (HDL-C), triglycerides, non-HDL-C), hs-CRP, creatine kinase, troponin, HbA1c and vitamin B12 level. Plasma, serum and peripheral blood mononuclear cells are also prepared from fasting blood collected at baseline and final study visit, stored at – 80 °C or in liquid nitrogen and transferred to SAHMRI at the end of the study, for exploratory analysis of inflammatory cytokines and other biomarkers.
Study Endpoints
The primary endpoint of the study is the percentage change in minimum FCT in a non-culprit plaque from baseline to 18 months. The secondary endpoints include absolute change in plaque minimum FCT, absolute and percentage change in plaque mean FCT, change in lipid pool size and absolute change in plaque macrophage content. All endpoints, including exploratory endpoints, are listed in Table 2.
Statistical Analysis
Sample size estimation was based around studies that have demonstrated a mean % change in minimum FCT of 65 ± 35% with statins, which corresponds here to the expected change in the placebo group [55, 59,60,61,62,63,64]. We assume that colchicine will result in a segment-specific 50% relative increase of the expected change in minimum FCT (97 ± 35%). With a two-sided alpha of 0.05 and power of 90%, 52 participants (26 per group) are required to meet these expectations. Allowing for approximately 20% loss in follow-up due to missing data or patient withdrawal, we randomized a total of 64 participants (32 per group) with recruitment completed in December of 2020.
We will perform intention-to-treat analysis as the primary analysis. Descriptive summary will be presented as percentage frequencies for categorical variables and as mean ± SD (or as median with interquartile range) for continuous variables by treatment group. Continuous variables will be tested for normality and appropriate non-parametric test will be used for variables not normally distributed. Group comparisons will be performed by chi-squared (or Fisher’s exact) test for categorical variables and independent samples t test for continuous variables. The % change in minimum FCT (primary endpoint) will be compared between the two groups using ANCOVA adjusting for baseline measurements. Chi-squared test or independent t test will be used for the secondary endpoints to compare the difference in changes between groups. We will explore relationships between % change in minimum FCT and % change in inflammatory biomarkers in the colchicine group using simple regression model; multivariable analysis will subsequently be used to determine independent predictors of colchicine responsiveness. Per-protocol analysis will be performed on those with treatment compliance > 80%, as a sensitivity analysis. A complete case analysis will include all patients with evaluable imaging at both time points. We will also perform a sensitivity analysis using multiple imputation to accommodate patients who were lost to follow-up. A two-sided p value of < 0.05 will be considered statistically significant. Analysis will be performed with Stata MP 14 (StataCorp, TX).
Discussion
There is compelling evidence attesting to the importance of local and systemic inflammation in all stages of atherosclerosis and its complications. Residual inflammation despite current treatment predominantly centred on statin therapy is a harbinger of recurrent cardiovascular risk. Results from recent studies have demonstrated the potential clinical benefits of colchicine in patients with established CAD, including when initiated early after MI. Further, the use of serial arterial wall imaging has provided the opportunity to characterize the effect of lipid-lowering interventions on atherosclerotic plaque in vivo.
COCOMO-ACS utilizes intracoronary OCT to extend these observations in order to determine the effect of anti-inflammatory therapy with low-dose colchicine on non-culprit, high-risk lipid-rich plaques in patients presenting with MI. Recruitment for the trial has been completed with 64 participants enrolled, and it is anticipated that statistical analysis on the entire cohort will be complete by late 2022. COCOMO-ACS findings are poised to provide unique mechanistic insights into the potential effects of colchicine on modifying coronary plaque biology. If positive, this study will contribute to the rapidly accumulating evidence that supports the use of colchicine in atherosclerotic CAD.
All Principal Investigators and Participating Centres
Peter Psaltis, Royal Adelaide Hospital; Kuljit Singh, Gold Coast University Hospital; Ajay Sinhal, Flinders Medical Centre; Sharmalar Rajendran, Lyell McEwin Hospital; Richard Alcock, Royal Perth Hospital; Peter Barlis, Northern Hospital; Sanjay Patel, Royal Prince Alfred Hospital; Dennis Wong, Monash Medical Centre; Rustem Dautov, The Prince Charles Hospital.
Data Availability
There is currently no plan to make individual participant data publicly available for this trial.
References
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–292.
Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–528.
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–25.
Jernberg T, Hasvold P, Henriksson M, Hjelm H, Thuresson M, Janzon M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. Eur Heart J. 2015;36:1163–70.
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–9.
Berk BC, Weintraub WS, Alexander RW. Elevation of C-reactive protein in “active” coronary artery disease. Am J Cardiol. 1990;65:168–72.
Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, et al. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med. 1994;331:417–24.
Ridker PM. A test in context: high-sensitivity c-reactive protein. J Am Coll Cardiol. 2016;67:712–23.
Montarello NJ, Nguyen MT, Wong DTL, Nicholls SJ, Psaltis PJ. Inflammation in coronary atherosclerosis and its therapeutic implications. Cardiovasc Drugs Ther. 2020. https://doi.org/10.1007/s10557-020-07106-6.
Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. 2017;70:2278–89.
Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61:404–10.
Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383:1838–47.
Tong DC, Quinn S, Nasis A, Hiew C, Roberts-Thomson P, Adams H, et al. Colchicine in patients with acute coronary syndrome: the Australian COPS randomized clinical trial. Circulation. 2020;142:1890–900.
Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381:2497–505.
Nguyen MT, Fernando S, Schwarz N, Tan JT, Bursill CA, Psaltis PJ. Inflammation as a therapeutic target in atherosclerosis. J Clin Med. 2019;8:1109.
Santos-Gallego CG, Picatoste B, Badimon JJ. Pathophysiology of acute coronary syndrome. Curr Atheroscler Rep. 2014;16:401.
Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123:731–8.
Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48.
Albert MA, Danielson E, Rifai N, Ridker PM, Investigators P. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64–70.
Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–65.
Emerging Risk Factors C, Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–40.
Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207.
Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504.
Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8.
Leung YY, Yao Hui LL, Kraus VB. Colchicine–update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015;45:341–50.
Fuhrman B, Koren L, Volkova N, Keidar S, Hayek T, Aviram M. Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis. 2002;164:179–85.
Cronstein BN, Molad Y, Reibman J, Balakhane E, Levin RI, Weissmann G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest. 1995;96:994–1002.
Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum. 2009;38:411–9.
Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–5.
Vaidya K, Tucker B, Kurup R, Khandkar C, Pandzic E, Barraclough J, et al. Colchicine inhibits neutrophil extracellular trap formation in patients with acute coronary syndrome after percutaneous coronary intervention. J Am Heart Assoc. 2021;10:e018993.
Doring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. 2020;126:1228–41.
Moschonas IC, Tselepis AD. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis. 2019;288:9–16.
Crittenden DB, Lehmann RA, Schneck L, Keenan RT, Shah B, Greenberg JD, et al. Colchicine use is associated with decreased prevalence of myocardial infarction in patients with gout. J Rheumatol. 2012;39:1458–64.
Langevitz P, Livneh A, Neumann L, Buskila D, Shemer J, Amolsky D, et al. Prevalence of ischemic heart disease in patients with familial Mediterranean fever. Isr Med Assoc J. 2001;3:9–12.
Martinez GJ, Robertson S, Barraclough J, Xia Q, Mallat Z, Bursill C, et al. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J Am Heart Assoc. 2015;4:e002128.
Bouabdallaoui N, Tardif JC, Waters DD, Pinto FJ, Maggioni AP, Diaz R, et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the colchicine cardiovascular outcomes trial (COLCOT). Eur Heart J. 2020;41:4092–9.
Slobodnick A, Shah B, Krasnokutsky S, Pillinger MH. Update on colchicine, 2017. Rheumatology (Oxford). 2018;57:i4–11.
Fiolet ATL, Opstal TSJ, Mosterd A, Eikelboom JW, Jolly SS, Keech AC, et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: a systematic review and meta-analysis of randomized trials. Eur Heart J. 2021;42:2765–75.
Lee WM, Morrison ES, Scott RF, Lee KT, Kroms M. Effects of methyl prednisolone and colchicine on the development of aortic atherosclerosis in swine. Atherosclerosis. 1976;25:213–24.
Wojcicki J, Hinek A, Jaworska M, Samochowiec L. The effect of colchicine on the development of experimental atherosclerosis in rabbits. Pol J Pharmacol Pharm. 1986;38:343–8.
Hollander W, Paddock J, Nagraj S, Colombo M, Kirkpatrick B. Effects of anticalcifying and antifibrobrotic drugs on pre-established atherosclerosis in the rabbit. Atherosclerosis. 1979;33:111–23.
Cecconi A, Vilchez-Tschischke JP, Mateo J, Sanchez-Gonzalez J, Espana S, Fernandez-Jimenez R, et al. Effects of colchicine on atherosclerotic plaque stabilization: a multimodality imaging study in an animal model. J Cardiovasc Transl Res. 2021;14:150–60.
Vaidya K, Arnott C, Martinez GJ, Ng B, McCormack S, Sullivan DR, et al. Colchicine therapy and plaque stabilization in patients with acute coronary syndrome: a CT coronary angiography study. JACC Cardiovasc Imaging. 2018;11:305–16.
Karasawa T, Takahashi M. The crystal-induced activation of NLRP3 inflammasomes in atherosclerosis. Inflamm Regen. 2017;37:18.
Fernando SSN, Williamson A, Toledo D, Zareh J, Di Bartolo B, Nicholls S, Psaltis P. Anti-inflammatory effects of colchicine on oxidised low-density lipoproteins and cholesterol crystal-induced macrophage activation in vitro. Heart Lung Circ. 2017;26:S69–70.
Opstal TSJ, Hoogeveen RM, Fiolet ATL, Silvis MJM, The SHK, Bax WA, et al. Colchicine attenuates inflammation beyond the inflammasome in chronic coronary artery disease: a LoDoCo2 proteomic substudy. Circulation. 2020;142:1996–8.
Jang IK, Bouma BE, Kang DH, Park SJ, Park SW, Seung KB, et al. Visualization of coronary atherosclerotic plaques in patients using optical coherence tomography: comparison with intravascular ultrasound. J Am Coll Cardiol. 2002;39:604–9.
Kawasaki M, Bouma BE, Bressner J, Houser SL, Nadkarni SK, MacNeill BD, et al. Diagnostic accuracy of optical coherence tomography and integrated backscatter intravascular ultrasound images for tissue characterization of human coronary plaques. J Am Coll Cardiol. 2006;48:81–8.
Yabushita H, Bouma BE, Houser SL, Aretz HT, Jang IK, Schlendorf KH, et al. Characterization of human atherosclerosis by optical coherence tomography. Circulation. 2002;106:1640–5.
Tearney GJ, Waxman S, Shishkov M, Vakoc BJ, Suter MJ, Freilich MI, et al. Three-dimensional coronary artery microscopy by intracoronary optical frequency domain imaging. JACC Cardiovasc Imaging. 2008;1:752–61.
Kume T, Akasaka T, Kawamoto T, Watanabe N, Toyota E, Neishi Y, et al. Assessment of coronary intima–media thickness by optical coherence tomography: comparison with intravascular ultrasound. Circ J. 2005;69:903–7.
Jang IK, Tearney GJ, MacNeill B, Takano M, Moselewski F, Iftima N, et al. In vivo characterization of coronary atherosclerotic plaque by use of optical coherence tomography. Circulation. 2005;111:1551–5.
Kubo T, Imanishi T, Takarada S, Kuroi A, Ueno S, Yamano T, et al. Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J Am Coll Cardiol. 2007;50:933–9.
Xing L, Higuma T, Wang Z, Aguirre AD, Mizuno K, Takano M, et al. Clinical significance of lipid-rich plaque detected by optical coherence tomography: a 4-year follow-up study. J Am Coll Cardiol. 2017;69:2502–13.
Komukai K, Kubo T, Kitabata H, Matsuo Y, Ozaki Y, Takarada S, et al. Effect of atorvastatin therapy on fibrous cap thickness in coronary atherosclerotic plaque as assessed by optical coherence tomography: the EASY-FIT study. J Am Coll Cardiol. 2014;64:2207–17.
Habara M, Nasu K, Terashima M, Ko E, Yokota D, Ito T, et al. Impact on optical coherence tomographic coronary findings of fluvastatin alone versus fluvastatin + ezetimibe. Am J Cardiol. 2014;113:580–7.
Nicholls SJ, Nissen SE, Prati F, Windecker S, Kataoka Y, Puri R, et al. Assessing the impact of PCSK9 inhibition on coronary plaque phenotype with optical coherence tomography: rationale and design of the randomized, placebo-controlled HUYGENS study. Cardiovasc Diagn Ther. 2021;11:120–9.
Prati F, Romagnoli E, Gatto L, La Manna A, Burzotta F, Ozaki Y, et al. Relationship between coronary plaque morphology of the left anterior descending artery and 12 months clinical outcome: the CLIMA study. Eur Heart J. 2020;41:383–91.
Kataoka Y, Puri R, Hammadah M, Duggal B, Uno K, Kapadia SR, et al. Frequency-domain optical coherence tomographic analysis of plaque microstructures at nonculprit narrowings in patients receiving potent statin therapy. Am J Cardiol. 2014;114:549–54.
Dai J, Hou J, Xing L, Jia H, Hu S, Soeda T, et al. Is age an important factor for vascular response to statin therapy? A serial optical coherence tomography and intravascular ultrasound study. Coron Artery Dis. 2017;28:209–17.
Hou J, Xing L, Jia H, Vergallo R, Soeda T, Minami Y, et al. Comparison of intensive versus moderate lipid-lowering therapy on fibrous cap and atheroma volume of coronary lipid-rich plaque using serial optical coherence tomography and intravascular ultrasound imaging. Am J Cardiol. 2016;117:800–6.
Hattori K, Ozaki Y, Ismail TF, Okumura M, Naruse H, Kan S, et al. Impact of statin therapy on plaque characteristics as assessed by serial OCT, grayscale and integrated backscatter-IVUS. JACC Cardiovasc Imaging. 2012;5:169–77.
Nishio R, Shinke T, Otake H, Nakagawa M, Nagoshi R, Inoue T, et al. Stabilizing effect of combined eicosapentaenoic acid and statin therapy on coronary thin-cap fibroatheroma. Atherosclerosis. 2014;234:114–9.
Takarada S, Imanishi T, Kubo T, Tanimoto T, Kitabata H, Nakamura N, et al. Effect of statin therapy on coronary fibrous-cap thickness in patients with acute coronary syndrome: assessment by optical coherence tomography study. Atherosclerosis. 2009;202:491–7.
Acknowledgements
We gratefully acknowledge the trial participants and all personnel involved in the COCOMO-ACS trial.
Funding
This is an investigator-initiated study with funding provided by project grants from the National Health and Medical Research Council of Australia (NHMRC ID 1127159), National Heart Foundation of Australia (NHF Vanguard Grant ID 10137) and the Sylvia and Charles Viertel Charitable Foundation (VIERCI 2017016). The study protocol was developed by an academic group led by P.J.P., who will oversee the operational conduct of the study together with SAHMRI Clinical Research. D.T.L.W. receives a Level 1 Future Leader Fellowship from the National Heart Foundation of Australia (FLF 102535). S.J.N. receives a Principal Research Fellowship from the NHMRC (ID 1111630). P.J.P. receives a Level 2 Future Leader Fellowship from the National Heart Foundation of Australia (FLF 102056) and Level 2 Career Development Fellowship from the NHMRC (CDF 1161506).
Author information
Authors and Affiliations
Contributions
Nicholas J Montarello: drafting of manuscript, final approval of version to be published. Kuljit Singh: data collection, final approval of version to be published. Ajay Sinhal: data collection, final approval of version to be published. Dennis TL Wong: data collection, final approval of version to be published. Richard Alcock: data collection, final approval of version to be published. Sharmalar Rajendran: data collection, final approval of version to be published. Rustem Dautov: data collection, final approval of version to be published. Peter Barlis: data collection, final approval of version to be published. Sanjay Patel: data collection, final approval of version to be published; Stefan M Nidorf: critical revision of manuscript, final approval of version to be published. Peter L Thompson: data collection, final approval of version to be published. Thalia Salagaras: project administration, final approval of version to be published. Julie Butters: project administration, final approval of version to be published. Nitesh Nerlekar: data analysis and interpretation, final approval of version to be published. Giuseppe Di Giovanni: data analysis and interpretation, final approval of version to be published. Juanita L Ottaway: conception and design, drafting of manuscript, critical revision of manuscript, final approval of version to be published. Stephen J Nicholls: conception and design, critical revision of manuscript, final approval of version to be published. Peter J Psaltis: conception and design, data collection, data analysis and interpretation, critical revision of manuscript, final approval of version to be published.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All procedures in this study will be conducted in accordance with the Declaration of Helsinki, International Conference on Harmonization, Good Clinical Practice guidelines and applicable regulatory requirements. The final protocol and informed consent forms were approved by the Royal Adelaide Hospital Human Research Ethics Committee (HREC) under the National Mutual Acceptance (NMA) system with local Research Governance Officer (RGO) approval at each site (HREC/17/RAH/366; Central Adelaide Local Health Network Reference Number R20170904). This clinical trial is registered with the Australian New Zealand Clinical Trials Registry (ACTRN12618000809235). Informed consent was obtained from all individual participants included in the study.
Consent for Publication.
Not applicable.
Competing Interests
K.S. has received research support and consultant fees from Abbott Vascular and Bayer. A.S. has received consultant fees and speaker honoraria from Edwards Lifesciences, Medtronic, Boston Scientific, AstraZeneca and Abbott Vascular. D.T.L.W. has received speaker honoraria from AstraZeneca, Pfizer, Bayer and Boehringer Ingelheim. P.L.T. has received research grants or honoraria from Amarin, Amgen, Aspen, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, DalCor, Merck and Pfizer. S.J.N. has received research support from AstraZeneca, Amgen, Anthera, Eli Lilly, Esperion, Novartis, Cerenis, The Medicines Company, Resverlogix, InfraReDx, Roche, Sanofi-Regeneron and Liposcience and is a consultant for AstraZeneca, Akcea, Eli Lilly, Anthera, Kowa, Omthera, Merck, Takeda, Resverlogix, Sanofi-Regeneron, CSL Behring, Esperion and Boehringer Ingelheim. P.J.P. has received research support from Abbott Vascular, consulting fees from Amgen and Esperion and speaker honoraria from AstraZeneca, Bayer, Boehringer Ingelheim, Merck Schering-Plough and Pfizer.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Montarello, N.J., Singh, K., Sinhal, A. et al. Assessing the Impact of Colchicine on Coronary Plaque Phenotype After Myocardial Infarction with Optical Coherence Tomography: Rationale and Design of the COCOMO-ACS Study. Cardiovasc Drugs Ther 36, 1175–1186 (2022). https://doi.org/10.1007/s10557-021-07240-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-021-07240-9