
Abstract
Conducting materials typically exhibit either diffusive or ballistic charge transport. When electron–electron interactions dominate, a hydrodynamic regime with viscous charge flow emerges1,2,3,4,5,6,7,8,9,10,11,12,13. More stringent conditions eventually yield a quantum-critical Dirac-fluid regime, where electronic heat can flow more efficiently than charge14,15,16,17,18,19,20,21,22. However, observing and controlling the flow of electronic heat in the hydrodynamic regime at room temperature has so far remained elusive. Here we observe heat transport in graphene in the diffusive and hydrodynamic regimes, and report a controllable transition to the Dirac-fluid regime at room temperature, using carrier temperature and carrier density as control knobs. We introduce the technique of spatiotemporal thermoelectric microscopy with femtosecond temporal and nanometre spatial resolution, which allows for tracking electronic heat spreading. In the diffusive regime, we find a thermal diffusivity of roughly 2,000 cm2 s−1, consistent with charge transport. Moreover, within the hydrodynamic time window before momentum relaxation, we observe heat spreading corresponding to a giant diffusivity up to 70,000 cm2 s−1, indicative of a Dirac fluid. Our results offer the possibility of further exploration of these interesting physical phenomena and their potential applications in nanoscale thermal management.
Similar content being viewed by others
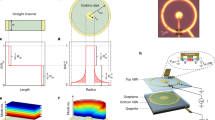

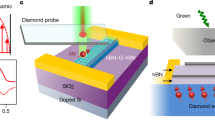
Main
During the last few years, signatures of viscous charge flow in the so-called Fermi-liquid hydrodynamic regime were observed in two-dimensional (2D) electron systems, especially graphene, using electrical device measurements7,8,9,11,12 and scanning probe microscopy10,13,22. A second hydrodynamic regime, which has no analogue in classical fluids, can occur very close to the Dirac point. When the Fermi temperature (TF = EF/kB, where EF is the Fermi energy and kB is the Boltzmann constant) becomes small compared to the electron temperature Te, the system becomes a quantum-critical fluid3,6,14,15,17. In this Dirac-fluid regime, the non-relativistic description of the viscous fluid is replaced by its ultra-relativistic counterpart, which accounts for the presence of both particles and holes, as well as for their linear energy dispersion. In line with theoretical predictions in this regime15, electrical measurements at cryogenic temperatures indicated a deviation from the Wiedemann–Franz law19 and from the Mott relation20, and a terahertz-probe study revealed the quantum-critical carrier scattering rate21.
Here, we follow electronic heat flow in the diffusive and hydrodynamic regimes at room temperature, and demonstrate a controlled Fermi-liquid to Dirac-fluid crossover, with a strongly enhanced thermal diffusivity close to the Dirac point. These observations are enabled by ultrafast spatiotemporal thermoelectric microscopy, a technique inspired by all-optical spatiotemporal diffusivity measurements23,24,25, with the crucial difference that the observable is the thermoelectric current, which is directly, and exclusively, sensitive to electronic heat26. We use a hexagonal boron nitride (hBN)-encapsulated graphene device that is both a Hall bar for electrical measurements and a split-gate thermoelectric detector (Fig. 1a). Since we use ultrashort laser pulses, with an approximate instrument response time (ΔtIRF where IRF means instrument response function) of 200 fs, to generate electronic heat, we are able to examine the system before momentum relaxation occurs, as we measure a momentum relaxation time, τmr, around 350 fs (Extended Data Fig. 1). In this temporal regime before momentum is relaxed, we enter the hydrodynamic window, because the electron–electron scattering time τee is <100 fs (ref. 27), that is τee < ΔτIRF < τmr. This is a different approach compared to most previous studies, where hydrodynamic effects were observed by using small system dimensions L to eliminate effects of momentum relaxation, that is vFτee < L < vFτmr (refs. 7,8,9,10,11,12,13,19,22) (vF = 106 m s−1 is the Fermi velocity). Our approach furthermore exploits elevated carrier temperatures, which greatly increases the accessibility of the Dirac-fluid regime, as for increasing carrier temperatures the crossover occurs increasingly far away from the Dirac point14,17 (Fig. 1b). As we will show, during the hydrodynamic window substantially more efficient heat spreading occurs in the Dirac-fluid regime than in the Fermi-liquid regime and in the diffusive regime (Fig. 1c,d).

a, Concept of the experiment, where a graphene Hall bar and thermoelectric device is illuminated by two femtosecond heat-generating pulses with a relative temporal offset ∆t and a symmetric spatial offset ∆x with respect to the pn junction where electronic heat generates a thermoelectric current. The junction is created by applying +∆U to one gate and −∆U to the other. We isolate the differential thermoelectric current corresponding to light-induced electronic heat from both pulses that has travelled to the junction, where the heat adds up in a non-linear fashion. b, Phase diagram of the Dirac-fluid regime, calculated following ref. 14. For increasing Te, the Dirac-fluid regime occurs increasingly far away from the Dirac point. c,d, Illustration of light-triggered spreading of electronic heat in the Fermi-liquid regime (c) and Dirac-fluid regime (d). In both cases, for Δt > τmr, diffusive transport dominates (straight blue lines), while in the hydrodynamic window, with Δt < τmr, extremely efficient heat transport occurs in the Dirac-fluid regime (wavy red lines). e,f, Sketch of the spatial broadening of the heat spots for low (e) and high (f) diffusivity, D, indicating more interacting heat at the junction region, hence higher ∆ITE signal for higher D.
Our technique works by using two ultrafast laser pulses that produce localized spots of electronic heat within tens of femtoseconds27. These spots are characterized by an increased carrier temperature Te > Tl, with Tl the lattice temperature (300 K). The degree of spatial spreading of these electronic heat spots as a function of time is governed by the diffusivity D. We control the relative spatial and temporal displacement, Δx and Δt, of the two pulses with sub-100-nm spatial precision and roughly 200 fs temporal resolution. Each laser pulse is incident on opposite sides of a pn junction at a distance ∆x/2 from the junction. This pn junction is created by applying opposite voltages ±ΔU with respect to the Dirac point voltages to the two backgates that form a split-gate structure. The two photo-generated electronic heat spots spread out spatially and part of the heat can reach the pn junction after a certain amount of time, generating a thermoelectric current at the junction through the Seebeck gradient26. The small region of the pn junction thus serves as a local probe of the electron temperature. While each of the heat spots can create thermoelectric current independently, we obtain spatiotemporal information by examining exclusively the signal that corresponds to heat generated by one of the pulses interacting with heat generated by the other pulse—the interacting heat current, ΔITE. Since the thermoelectric photocurrent scales sublinearly with incident power, we can isolate this interacting heat current ΔITE by modulating each laser beam at a different frequency, f1 and f2, and demodulating the thermoelectric current at the difference frequency f1 − f2. As illustrated in Fig. 1e,f, the higher the diffusivity D, the more interacting heat current ΔITE remains for increasing Δx and Δt.
Figure 2a shows the measured interacting heat current ∆ITE as a function of Δx and Δt. As expected, the largest ∆ITE occurs for the largest spatiotemporal overlap at the pn junction (Δx = Δt = 0). For increasing |∆t|, we find that the normalized signal extends further spatially, indicating the occurrence of heat spreading (Fig. 2b). This spatial spread is quantified via the second moment <Δx2>, which quantifies the width of the profile at different time delays (Methods). Similar to recent all-optical spatiotemporal microscopy24,25, we obtain spatial information beyond the diffraction limit by precise spatial sampling of diffraction-limited profiles. The experimentally obtained spatial spread as a function of Δt (Fig. 2c) is very similar to the calculated results (Fig. 2d), obtained by simulating the experiment with a given diffusivity, D (Methods and Supplementary Note 1). The white lines indicate the values of the spatial spread <Δx2> for different Δt. We also compare the simulated spatial spread <Δx2> versus Δt (blue dashed line in Fig. 2e) with the theoretical expectation according to the heat diffusion equation, <Δx2> = <Δx2>focus + 2DΔt (dash-dotted line in Fig. 2e). Here, D is the same diffusivity that was used as input for the simulation, and <Δx2>focus is the minimum second moment from the two overlapping pulses (Supplementary Note 2 and Supplementary Figs. 1–4). The initial slope is the same for both the simulated heat spreading and the theoretical spreading following the heat diffusion equation.

a, The experimental spatiotemporal differential thermoelectric current ∆ITE as a function of Δx and Δt. b, Normalized profiles (norm.), showing a larger spatial spread for larger |∆t|. c,d, Experimental (c) and simulated (d) normalized ∆ITE for each ∆t, showing spatial broadening due to thermal transport as a function of ∆t. The white line indicates the spatial spread <∆x2>. e, Spatial spread <∆x2> of ∆ITE, as a function of ∆t for three different Fermi energies (symbols), with simulation results using as input the diffusivities from electrical mobility measurements (purple solid lines), with offset due to ultrafast heat spreading around time zero. Simulation (blue dashed line) and theoretical heat equation (black dash-dotted line) results with the same input diffusivity and no ultrafast spreading around time zero. Heat spreading with ultrahigh diffusivity in the Dirac-fluid regime (red line), which lasts for a few hundred fs, explains the time zero offset.
We first discuss the experimental results in the diffusive regime, where |Δt| > τmr. For three different gate voltage combinations, corresponding to Fermi energies between 75 and 190 meV (TF = 900 − 2,200 K), we extract the spatial spread as a function of time delay (symbols in Fig. 2e), and compare it with the results from simulations (solid lines). For these simulations, we have used the diffusivity values that we obtain directly from electrical measurements of charge mobility μ on the same device (Extended Data Fig. 1), and the relation between mobility and diffusivity: D = μEF/2e (Methods). We find excellent agreement, if we account for short-lived ultrafast heat spreading around Δt = 0, which leads to a larger-than-expected initial spread at time zero <Δx2>min, as we will explain below. The agreement between the measured heat spread for |Δt| > τmr and the one calculated using the measured charge mobilities shows that electronic heat and charge flow together, as expected in the diffusive regime. Furthermore, it confirms that our technique is a reliable method for obtaining thermal diffusivities in a quantitative manner.
We now turn to the non-diffusive regime, by exploring the behaviour in the hydrodynamic window, where |Δt| < τmr. The experimentally obtained spatial spreads start at a minimum value <Δx2>min larger than 2 μm2, rather than starting at an expected <Δx2>focus = 0.56 μm2. A second device of hBN-encapsulated graphene with similar mobility reproduces this larger-than-expected spatial spread at time zero (Supplementary Note 3 and Extended Data Fig. 2). We exclude the possibility of an experimental artefact such as an underestimation of the laser spot size, since we repeated the measurements while scanning through the laser focus, and measured the focus size (Supplementary Figs. 1–4). Furthermore, we observe that the offset depends on the Fermi energy, while keeping all other experimental parameters fixed. Finally, we measured a third device with a lower charge mobility and shorter hydrodynamic time window: τmr < 100 fs, which is smaller than ΔtIRF. This device exhibits systematically less heat spreading around time zero (Supplementary Note 4 and Extended Data Fig. 3), consistent with its smaller hydrodynamic time window. We therefore attribute the large experimentally observed minimum <Δx2>min in Fig. 2e to ultrafast initial heat spreading that occurs before momentum relaxation takes place, Δt ≲ 350 fs (see the schematic illustration of spatiotemporal heat spreading in Fig. 1d). The dynamics of this initial heat spreading are washed out by the finite time resolution ΔtIRF, and manifests as a large minimum <Δx2>min at time zero. The observed initial spatial spread suggests a thermal diffusivity of D = (<Δx2>min − <Δx2>focus)/2ΔtIRF ≅ 70,000 cm2 s−1 for the lowest measured EF of 75 meV. Simulations of heat spreading with an input diffusivity of 100,000 cm2 s−1 are indeed consistent with the experimentally observed spread in the hydrodynamic window (the red line in Fig. 2e).
We attribute this observation of highly efficient initial heat spreading to the presence of the quantum-critical electron-hole plasma. We can exclude that the observed initial spreading is the result of ballistic transport, as we calculate that the ballistic contribution to initial heat spreading would give only <Δx2>ball = 0.68 μm2 (Supplementary Note 2 and Extended Data Fig. 4). Besides, ballistic transport has a very weak dependence (<10%) on carrier density in this range, as the Fermi velocity does not change appreciably for the Fermi energies considered here28. The reason for the high diffusivity in the Dirac-fluid regime is that the hot electrons and hot holes that coexist in this regime move in the same direction under a thermal gradient, with inter-particle scattering events conserving total momentum19. We note that typical transport measurements probe the sum of the momentum-conserving thermal resistivity (due to carrier–carrier scattering) and the momentum-non-conserving thermal resistivity (due to carrier–impurity and carrier–phonon scattering), where the latter dominates at room temperature. The ability of our technique to interrogate the system during the 350 fs before any momentum-non-conserving scattering occurs, means that this contribution to the overall resistivity is negligible. Therefore, we probe exclusively the momentum-conserving thermal conductivity, which diverges towards the Dirac point and towards infinite electron temperature.
To provide further evidence of hydrodynamic heat transport, we demonstrate the ability to control the crossover between the Fermi-liquid and quantum-critical Dirac-fluid regimes via the ratio Te/TF, by independently varying Te via the incident laser power and TF via the applied gate voltage. A larger ratio results in less Coulomb screening and correspondingly stronger hydrodynamic effects due to electron–electron interactions. If Te is larger than TF, electrons and holes coexist and the Dirac-fluid regime becomes accessible (Fig. 1b). We perform spatial scans in the hydrodynamic window at a temporal delay of Δt = 0, in a geometry with one laser pulse impinging on the junction, while scanning the other pulse across (x axis) and along (y axis) the junction region. Figure 3a–d shows four representative spatial ∆ITE maps with varying Te/TF, yet similar signal magnitudes. Clearly, the signal is broader for larger Te/TF, indicating faster thermal transport. We repeat these measurements for a range of Te and TF values and quantify the initial heat spreading using Gaussian functions, with widths σx and σy, to describe ∆ITE at Δt = 0 as a function of Δx or Δy (Fig. 3e,f and Supplementary Fig. 5). As expected for a crossover from the diffusive Fermi-liquid regime to the hydrodynamic Dirac-fluid regime, both spatial spreads σx and σy increase substantially for increasing ratio Te/TF. These spreads correspond to a diffusivity up to 40,000 cm2 s−1 (Methods), similar to the 70,000 cm2 s−1 we found earlier.

a–d, Time-zero spatial maps of ∆ITE for low optical power P and high gate voltage ∆U with an np junction (a) and with a pn junction (d); and for high optical power and low gate voltage with an np junction (b), and with a pn junction (c). For larger ratio Te/TF (that is, larger P/∆U) the spatial extent is clearly larger. e,f, Time zero Gaussian widths for spatial scans with one pulse on the junction and the second one scanning across (e) and along (f) the graphene pn junction, as a function of P and ∆U. The strong dependence on Te/TF demonstrates our ability to transition under control into the Dirac-fluid regime with strongly increased thermal diffusivity. g,h, Calculation of the thermal diffusivity following refs. 6,18 with only electron–electron interactions (g) and only long-range Coulomb scattering (h). The contours in g are the calculated time zero spreads \(\sigma _{{{{\mathrm{calc}}}}}^2\) (Methods).
We compare our experimental results to Boltzmann transport calculations following refs. 6,18, including carrier interactions and long-range impurity scattering. We model impurities as Thomas–Fermi screened Coulomb scatterers of density 0.24 × 1012 cm−2. Figure 3g shows the calculated thermal diffusivity D as a function of TF and Te, when considering only the hydrodynamic term due to electron–electron interactions, relevant in the hydrodynamic window where |Δt| < τmr. A higher electron temperature, or lower Fermi temperature, leads to strongly increased diffusivity, signalling a crossover from the Fermi-liquid to the Dirac-fluid regime. This is the same qualitative trend as for the experimental data taken at Δt = 0 in Fig. 3e–f, where a larger initial width originates from a larger diffusivity, thus supporting our interpretation of a hydrodynamic crossover.
A more quantitative comparison shows that the calculated D in the diffusive regime is around 2,000 cm2 s−1 (Fig. 3h), in quantitative agreement with the experiment in the diffusive regime. The obtained thermal diffusivity in the hydrodynamic window close to the Dirac point reaches values above 100,000 cm2 s−1, even higher than our experimental estimates of 35,000–70,000 cm2 s−1. Using the calculated diffusivities, we estimate the spatial spread at time zero σcalc (Methods), as shown in Fig. 3g. These are similar to the experimentally obtained ones, thus confirming our conclusion of highly efficient heat spreading in the Dirac-fluid regime at room temperature, with a diffusivity that is almost two orders of magnitude larger than in the diffusive regime. We note that the theoretical calculations predict that even higher diffusivities are attainable.
Finally, we discuss the three-dimensional (3D) thermal conductivity, to assess the ability to transport useful amounts of heat. We find roughly 100 W m−1 K−1 in the diffusive regime (Methods), in agreement with ab initio calculations29. In the Dirac-fluid regime, with an electron temperature of roughly 1,000 K, we obtain a thermal conductivity of 18,000–40,000 W m−1 K−1. This is in agreement with ref. 15, where values up to 100,000 W m−1 K−1 were predicted theoretically for large Te/TF. The thermal conductivity we obtain is about three orders of magnitude larger than the one obtained in the Dirac-fluid regime at cryogenic temperatures19. Our results show that in the Dirac-fluid window the electronic contribution to heat transport can be much larger than the phononic contribution with a conductivity of >2,000 W m−1 K−1 (ref. 30), which is already exceptionally high and can also be enhanced hydrodynamically, as shown recently31. Thus, the Dirac electron-hole plasma can contribute strongly to thermal transport, extracting heat from hot spots much faster than predicted by classical limits.
In conclusion, our results show that the—until recently unreachable—physical phenomena associated with the Dirac fluid do not only offer an exciting playground for interesting physical phenomena, but also hold great promise for applications, for example in thermal management of nanoscale devices. We note that the quantum-critical behaviour can be switched on and off using a modest gate voltage and in systems prepared by standard fabrication techniques. Finally, we believe that the optoelectronic technique we have introduced—with the potential of increased spatial accuracy and temporal resolution—will be a valuable tool to reach a better understanding of the thermal behaviour of a broad range of quantum materials, with great promise for new technological applications.
Methods
Fabrication of split-gate thermoelectric device
The split-gate device with Hall geometry consists of exfoliated, single layer graphene encapsulated by hBN, prepared using standard exfoliation and dry transfer techniques. The hBN-graphene-hBN stack is placed on a predefined split-gate structure made of graphene, grown by chemical vapour deposition, where the gap between the two gates is roughly 100 nm, created via electron-beam lithography and reactive ion etching (RIE). The top hBN and graphene are etched into a Hall bar shape with laser lithography and RIE, keeping the split-gate intact and not etching completely through the bottom hBN. Finally, the Ti/Au side contacts are created by a further step of lithography, RIE and metal evaporation. The fabrication steps are shown in Supplementary Fig. 6.
Spatiotemporal thermoelectric current microscopy setup
Our setup enables us to follow electronic heat spreading in space and time, because we use the thermoelectric signal generated by electronic heat interacting at a fixed location (the pn junction), while we vary the spatial displacement of our two laser pulses with respect to this junction and vary the temporal delay between the two ultrashort pulses. This means that we are following in space and time the diffusion of light-induced electronic heat from the location of light incidence to the pn junction. It is the thermoelectric effect at the pn junction, governed by the Seebeck coefficient, that generates our observable signal, the thermoelectric current. We note that although the value of the Seebeck coefficient itself changes when changing EF, and when entering the hydrodynamic regime20, this only affects the magnitude of the thermoelectric current, rather than how electronic heat is diffusing outside the pn junction, which is what we are following with our spatiotemporal technique.
A sketch of the setup is shown in Supplementary Fig. 7. A Ti:sapphire oscillator (886-nm centre wavelength, 76-MHz repetition rate), is split into two beam paths. Both beams are modulated with optical choppers, at frequencies f1 = 741 and f2 = 529 Hz. The relative time delay between the two pulses is controlled by a mechanical delay line. The spatial offset of one beam with respect to the other is controlled with a mirror galvanometer, while the position of the sample with respect to the beams is controlled with a piezo scanning stage. The beams are focused onto the sample with a ×40 0.6 numerical aperture objective lens. We collect the thermoelectric (TE) photocurrent between the source and drain contacts through the graphene sheet on either side of the junction via lock-in amplification. By demodulating the current signal at the difference frequency of the two modulation frequencies, f2 − f1 = 211.7 Hz, we isolate the signal caused by the interaction of both heating sources, which we call the interacting heat current ∆ITE. The temporal resolution of the setup of 200 fs is determined in the sample plane of the microscope (Supplementary Fig. 8). The spatial resolution defined by our spot sizes is below 1 μm, whereas the accuracy with which we can observe electronic heat spreading is determined by the signal-to-noise ratio, and is estimated to be below 100 nm.
Estimating Fermi temperature controlled by gate voltage
During photocurrent measurements, the gate voltage Ux is applied to one side (x is ‘A’) or the other side (x is ‘B’) side of the split gate. We always apply a symmetric voltage around the experimentally determined Dirac point voltage \(U_{{{\mathrm{x}}}}^{{{{\mathrm{DP}}}}}\): \(U_{{{\mathrm{A}}}} = U_{{{\mathrm{A}}}}^{{{{\mathrm{DP}}}}} + {\Delta} U\) and \(U_{{{\mathrm{B}}}} = U_{{{\mathrm{B}}}}^{{{{\mathrm{DP}}}}} - {\Delta} U\). The gate electrode and the graphene form a capacitor with the dielectric hexagonal boron nitride (hBN), with a thickness of thBN = 70 nm, and a relative permittivity of \({\it{\epsilon }}_{{{{\mathrm{hBN}}}}} = 3.56\). The carrier density n is calculated via \(n = \frac{{{\it{\epsilon }}_0{\it{\epsilon }}_{{{{\mathrm{hBN}}}}}}}{{e\,t_{{{{\mathrm{hBN}}}}}}}{\Delta} U\), where \(\epsilon_{0}\) is the vacuum permittivity. We calculate the Fermi energy EF and the Fermi temperature TF via \(E_{{{\mathrm{F}}}}^2\) = πħ2\(v_{{{\mathrm{F}}}}^2 \cdot n\) and \(T_{{{\mathrm{F}}}} = \frac{{E_{{{\mathrm{F}}}}}}{{k_{{{\mathrm{B}}}}}},\) where kB is the Boltzmann constant.
Estimating carrier temperature controlled by laser power
The thermoelectric photovoltage is assumed to be proportional to the time-averaged increase of the electronic temperature Te above the ambient temperature T0, as in ref. 32. The sublinear dependence of the thermoelectric current ITE on optical power for the device under study here for illumination with a single pulsed laser (λ = 886 nm) is shown in Supplementary Fig. 9. With a linear temperature scaling of the electronic heat capacity for graphene away from the Dirac point, Ce(T) = γT, we integrate the heat energy per unit area dQ = CedT, that is, \(\mathop {\smallint }\limits_{Q_0}^{Q_0 + {\Delta} Q} {{{\mathrm{d}}}}Q = \mathop {\smallint }\limits_{T_0}^{T_e} \gamma T{{{\mathrm{d}}}}T\). With the incident power P proportional to the absorbed heat energy per unit area ∆Q, we find that the peak Te as a function of the laser power P scales as in ref. 32, \(T_e = \root {2} \of {{T_0^2 + bP}}\). Here, the parameter b is defined via bP = 2∆Q/γ, and is used to convert incident power to peak electron temperature (Supplementary Fig. 9).
Simulation of the experiment
A detailed description of the simulation can be found in Supplementary Note 1 and Supplementary Fig. 10. In brief, we calculate the spatiotemporal evolution of electronic heat generated by the two optical pulses in the graphene sheet via the heat equation with a finite difference method. We define Gaussian heating pulses and calculate their temperature rise via the experimentally measured non-linear power scaling. We extract the differential TE current contribution as a function of ∆x and ∆t by the difference of the heating at the pn junction region in the presence of both pulses with respect to simulations with only one pulse at a time, analogous to the experimental difference-frequency demodulation.
Quantifying the spatial spread
The following analysis is conducted both on the experimental and the simulated data of ∆ITE(∆x, ∆t) for ‘symmetric experiments’ with optical pulses incident at a distance ∆x on each side of the pn-junction (Figs. 1 and 2). For each ∆t of the datasets ∆ITE(∆x, ∆t) we calculate the width of the signal via the second moment, which for an ideal Gaussian profile is equal to the squared Gaussian width σ2. The second moment is calculated from the pixels ∆xi (i = 1,…, N) via
We note that the minimum second moment at the focus <∆x2>focus of 0.56 µm2 comes from simulating the symmetric experiment, using as input the measured Gaussian beam width at the focus σfocus2 = 0.14 µm2 (Supplementary Note 2). For the ‘asymmetric experiments’ with one optical pulse always incident on the pn junction (data for Fig. 3), we always consider the spatial profile only at time zero. Here we find that Gaussian fits with a background give the most reliable results. The entire set of data is shown in Supplementary Fig. 5. For each dataset ∆ITE(∆x) or ∆ITE(∆y) taken at ∆t = 0, we perform Gaussian fits using the function \({{{\mathrm{f}}}}({\Delta} x) = a\exp \left( { - \frac{{{\Delta} x^2}}{{2\sigma ^2}}} \right) + b\), where the Gaussian squared width σ2 indicates the thermal spreading. Here, the minimum simulated Gaussian widths are (σx2)focus = 0.34 µm2 and (σy2)focus = 0.44 µm2 (Supplementary Note 2). The experimentally obtained widths from this dataset as function of gate voltage and optical power are also shown in Supplementary Fig. 5, showing an increase with power, that is a larger Te, and an increase towards the Dirac point, that is a smaller TF. We estimate the theoretical Gaussian widths in Fig. 3g using \(\sigma _{{{{\mathrm{calc}}}}}^2\) = (σx2)focus + 2D ΔtIRF, where D are the calculated diffusivities.
Electrical measurements
We characterize our device electrically with four-probe measurements (Extended Data Fig. 1), finding a charge mobility μ of 30,000–50,000 cm2 Vs−1, depending on carrier density. The measured mobilities correspond to a momentum relaxation time 𝜏mr of 300–500 fs. These relaxation times are longer than the temporal resolution (the IRF) of our measurement technique, ΔtIRF ≅ 200 fs, thus allowing us to probe our system before and after momentum relaxation occurs, that is in the non-diffusive and diffusive regime. We use these measured charge mobilities to calculate the expected thermal diffusivity via the Einstein relation33,34 \(\mu _{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}} = \frac{e}{{n_{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}}}}\frac{{\partial n_{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}}}}{{\partial E_{{{\mathrm{F}}}}}}D_{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}}\), where e is the elementary charge, EF is the Fermi energy and ne/h is the electron/hole carrier density. For highly doped graphene (\(E_{{{\mathrm{F}}}} \gg k_{{{\mathrm{B}}}}T\)) the carrier density expression \(n_{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}} = \frac{{E_{{{\mathrm{F}}}}^2}}{{\pi \hbar ^2v_{{{\mathrm{F}}}}^2}}\), leads to the simple relation:\(D_{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}} = \frac{{E_{{{\mathrm{F}}}}}}{{2e}}\mu _{{{{\mathrm{e}}}}/{{{\mathrm{h}}}}}\). We note that we obtain the identical result by calculating D from the ratio of the 2D thermal conductivity κe,2D and the electronic heat capacity Ce and using the Wiedemann–Franz law: \(\kappa _{{{{\mathrm{e}}}},2{{{\mathrm{D}}}}}/\sigma = \pi ^2/3\cdot (k_{{{\mathrm{B}}}}/e)^2T_{{{\mathrm{e}}}}\), where kB is the Boltzmann constant and e the elementary charge, together with the conductivity σ = neμ and the following heat capacity for graphene (valid for Te < TF): \(C_{{{\mathrm{e}}}} = \frac{{2\pi \varepsilon _{{{\mathrm{F}}}}k_{{{\mathrm{B}}}}^2T_{{{\mathrm{e}}}}}}{{3\hbar ^2v_{{{\mathrm{F}}}}^2}}\). Given the measured mobilities, we expect thermal diffusivities around 2,000 cm2 s−1 for our sample.
Thermal diffusivity and conductivity of the Dirac fluid
We estimate the enhanced thermal diffusivity of the Dirac fluid by comparing the measured width at time zero <Δx2>min to the expected width <Δx2>focus explained above, via D = (<Δx2>min − <Δx2>focus)/2ΔtIRF. We find values up to 74,000 cm2 s−1 for the symmetric scan (Fig. 2), and 29,000 and 39,000 cm2 s−1 for the x and y directions of the asymmetric scan (Fig. 3), respectively, where <Δx2> is replaced with (σx2) and (σy2), respectively. The same calculation for a second device (Supplementary Note 3 and Extended Data Fig. 2) gives a diffusivity of 100,000 cm2 s−1. The 3D thermal conductivity κ3D of the Dirac fluid is calculated from the diffusivity D and the electronic heat capacity Ce, via κ3D = DCe/d, where d is the thickness of graphene, 0.3 nm. For the Dirac fluid, we have Te > TF, and therefore use the ‘undoped’ electronic heat capacity35 \(\frac{{18\,\zeta (3)}}{{\pi (\hbar v_{{{\mathrm{F}}}})^2}}k_{{{\mathrm{B}}}}^3T_{{{\mathrm{e}}}}^2\), where \(\zeta \left( 3 \right) \approx 1.202\). With the above estimate D = 35,000–70,000 cm2 s−1 and Te = 1,000 K, we obtain the 3D thermal conductivity κ3D = 18,000–40,000 W mK−1. This corresponds to a 2D κ2D > 5 μW K−1. This value is orders of magnitude larger than the value found in ref. 19. The reason for this is that our electron temperature is more than ten times higher, and therefore the electronic heat capacity is >100× higher. Furthermore, we reach a Te/TF > 3, while their maximum Te/TF was around two, which means that we are further in the Dirac-fluid regime with its diverging thermal diffusivity.
Dirac-fluid crossover temperature
Following the treatment in ref. 14, we find the crossover temperature from Fermi liquid to Dirac fluid, as a function of Fermi temperature as
where \(\lambda = {{{\mathrm{e}}}}^2/16\epsilon _0{\it{\epsilon }}_{{{\mathrm{r}}}}v_{{{\mathrm{F}}}}\hbar \approx 0.55/{\it{\epsilon }}_{{{\mathrm{r}}}}\) for graphene with the dielectric environment \({\it{\epsilon }}_{{{\mathrm{r}}}} \approx 3.56\) for hBN. The temperature \(T_0 = \frac{{2\hbar v_{{{\mathrm{F}}}}\sqrt \pi }}{{3^{3/4}k_{{{\mathrm{B}}}}a_0}} \approx 8.4\times 10^4\,{{{\mathrm{K}}}}\), with the inter-atomic distance \(a_0 = 1.42\times 10^{ - 10}\,{{{\mathrm{m}}}}\). The resulting crossover temperature is shown in Fig. 1b. We note that the relatively high refractive index of the hBN encapsulant makes the Dirac fluid more easily accessible, as it lowers the crossover temperature compared to vacuum, by a factor of about two for the range of TF values studied here.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Polini, M. & Geim, A. K. Viscous electron fluids. Phys. Today 73, 28 (2020).
Müller, M., Schmalian, J. & Fritz, L. Graphene: a nearly perfect fluid. Phys. Rev. Lett. 103, 025301 (2009).
Foster, M. S. & Aleiner, I. L. Slow imbalance relaxation and thermoelectric transport in graphene. Phys. Rev. B. 79, 085415 (2009).
Narozhny, B. N., Gornyi, I. V., Titov, M., Schütt, M. & Mirlin, A. D. Hydrodynamics in graphene: linear-response transport. Phys. Rev. B. 91, 035414 (2015).
Levitov, L. & Falkovich, G. Electron viscosity, current vortices and negative nonlocal resistance in graphene. Nat. Phys. 12, 672–676 (2016).
Zarenia, M., Principi, A. & Vignale, G. Disorder-enabled hydrodynamics of charge and heat transport in monolayer graphene. 2D Mater. 6, 035024 (2019).
Moll, P. J. W., Kushwaha, P., Nandi, N., Schmidt, B. & Mackenzie, A. P. Evidence for hydrodynamic electron flow in PdCoO2. Science 351, 1061–1064 (2016).
Bandurin, D. A. et al. Negative local resistance caused by viscous electron backflow in graphene. Science 351, 1055–1058 (2016).
Krishna Kumar, R. et al. Superballistic flow of viscous electron fluid through graphene constrictions. Nat. Phys. 13, 1182–1185 (2017).
Braem, B. A. et al. Scanning gate microscopy in a viscous electron fluid. Phys. Rev. B. 98, 241304 (2018).
Gooth, J. et al. Thermal and electrical signatures of a hydrodynamic electron fluid in tungsten diphosphide. Nat. Commun. 9, 4093 (2018).
Berdyugin, A. I. et al. Measuring hall viscosity of graphene’s electron fluid. Science 364, 162–165 (2019).
Sulpizio, J. A. et al. Visualizing Poiseuille flow of hydrodynamic electrons. Nature 576, 75–79 (2019).
Sheehy, D. E. & Schmalian, J. Quantum critical scaling in graphene. Phys. Rev. Lett. 99, 226803 (2007).
Xie, H. Y. & Foster, M. S. Transport coefficients of graphene: interplay of impurity scattering, Coulomb interaction, and optical phonons. Phys. Rev. B. 93, 195103 (2016).
Lucas, A., Crossno, J., Fong, K. C., Kim, P. & Sachdev, S. Transport in inhomogeneous quantum critical fluids and in the Dirac fluid in graphene. Phys. Rev. B. 93, 075426 (2016).
Lucas, A. & Fong, K. C. Hydrodynamics of electrons in graphene. J. Phys. Condens. Matter 30, 053001 (2018).
Zarenia, M., Smith, T. B., Principi, A. & Vignale, G. Breakdown of the Wiedemann–Franz law in AB-stacked bilayer graphene. Phys. Rev. B 99, 161407 (2019).
Crossno, J. et al. Observation of the Dirac fluid and the breakdown of the Wiedemann–Franz law in graphene. Science 351, 1058–1061 (2016).
Ghahari, F. et al. Enhanced thermoelectric power in graphene: violation of the Mott relation by inelastic scattering. Phys. Rev. Lett. 116, 136802 (2016).
Gallagher, P. et al. Quantum-critical conductivity of the Dirac fluid in graphene. Science 364, 158–162 (2019).
Ku, M. J. H. et al. Imaging viscous flow of the Dirac fluid in graphene. Nature 583, 537–541 (2020).
Ruzicka, B. A. et al. Hot carrier diffusion in graphene. Phys. Rev. B. 82, 195414 (2010).
Zhu, T., Snaider, J. M., Yuan, L. & Huang, L. Ultrafast dynamic microscopy of carrier and exciton transport. Annu. Rev. Phys. Chem. 70, 219–244 (2019).
Block, A. et al. Tracking ultrafast hot-electron diffusion in space and time by ultrafast thermomodulation microscopy. Sci. Adv. 5, eaav8965 (2019).
Gabor, N. M. et al. Hot carrier-assisted intrinsic photoresponse in graphene. Science 334, 648–652 (2011).
Brida, D. et al. Ultrafast collinear scattering and carrier multiplication in graphene. Nat. Commun. 4, 1987 (2013).
Stauber, T. et al. Interacting electrons in graphene: Fermi velocity renormalization and optical response. Phys. Rev. Lett. 118, 266801 (2017).
Kim, T. Y., Park, C. H. & Marzari, N. The electronic thermal conductivity of graphene. Nano Lett. 16, 2439–2443 (2016).
Balandin, A. A. et al. Superior thermal conductivity of single-layer graphene. Nano Lett. 8, 902–907 (2008).
Machida, Y., Matsumoto, N., Isono, T. & Behnia, K. Phonon hydrodynamics and ultrahigh–room-temperature thermal conductivity in thin graphite. Science 367, 309–312 (2020).
Tielrooij, K. J. et al. Out-of-plane heat transfer in van der Waals stacks through electron-hyperbolic phonon coupling. Nat. Nanotechnol. 13, 41–46 (2018).
Zebrev, G. I. I. in Physics and Applications of Graphene—Theory (ed. Mikhailov, S.) 475–498 (IntechOpen, 2011).
Rengel, R. & Martín, M. J. Diffusion coefficient, correlation function, and power spectral density of velocity fluctuations in monolayer graphene. J. Appl. Phys. 114, 143702 (2013).
Lui, C. H., Mak, K. F., Shan, J. & Heinz, T. F. Ultrafast photoluminescence from graphene. Phys. Rev. Lett. 105, 127404 (2010).
Acknowledgements
We thank M. Polini and P. Piskunow for fruitful discussions, and H. Agarwal and K. Soundarapandian for help with sample fabrication. We acknowledge the following funding sources: European Union’s Horizon 2020 research and innovation programme under grant nos. 804349 (K.-J.T.), 873028 (A.P.), 785219 (F.H.L.K. and S.R.), 881603 (F.H.L.K. and S.R.) and 670949 (N.F.v.H.); Spanish MCIU/AEI under grant nos. RYC-2017-22330 (K.-J.T.), PID2019-111673GB-I00 (K.-J.T.), BES-2016-078727 (M.L.), RTI2018-099957-J-I00 (M.L.) and PGC2018-096875-B-I00 (M.L. and N.F.v.H.); the Government of Catalonia under grant nos. SGR1656 (F.H.L.K.) and 2017SGR1369 (N.F.v.H.) and the CERCA program (ICN2 and ICFO); Spanish MINECO under grant nos. SEV-2017-0706 (ICN2) and CEX-2019-000910-S (ICFO); the International PhD fellowship program ‘la Caixa’ (A.B.); Leverhulme Trust grant no. RPG-2019-363 (A.P.); the Elemental Strategy Initiative conducted by the MEXT, Japan, grant no. JPMXP0112101001 (K.W. and T.T.), JSPS KAKENHI grant no. JP20H00354 (K.W. and T.T.) and the CREST (grant no. JPMJCR15F3) and JST (K.W. and T.T.); Fundació Privada Cellex (ICFO) and Fundació Mir-Puig (ICFO).
Author information
Authors and Affiliations
Contributions
K.-J.T. conceived and supervised the project. A.B. developed the experimental setup under supervision of M.L. and N.F.v.H. and input from K.-J.T. N.C.H.H. performed sample fabrication, with material input from K.W. and T.T., under the supervision of F.H.L.K. and K.-J.T. A.B. performed the measurements under the supervision of K.-J.T. A.P. performed the theoretical hydrodynamic transport calculations. A.B. and K.-J.T. interpreted and analysed the data, with input from M.L., A.W.C. and N.F.v.H. A.B. developed the model that simulated the experiment with input from K.-J.T. A.W.C. and S.R. developed the ballistic transport model. A.B. and K.-J.T. wrote the paper, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Nanotechnology thanks Giulio Cerullo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Electrical mobility measurement.
Momentum relaxation time from four-probe measurements and corresponding (calculated) heat diffusivity (solid line). Four-probe measurements were performed by applying 1 V to a MΩ series resistor, such that a current of 1 μA flows between the two outer contacts of the device (see inset). We then measure the voltage drop between two lateral contacts of the graphene device. This yields the sheet conductance σ as a function of gate voltage. We then use σ = neμ, in order to extract the mobility µ and use \(\tau _{mr} = \frac{{\mu E_{{{\mathrm{F}}}}}}{{ev_{{{\mathrm{F}}}}^2}}\) to obtain the momentum relaxation time. Here, n is the carrier density, EF is the Fermi energy, vF ≈ 106 m/s is the Fermi velocity, and e is the elementary charge. We measured up to EF = 150 meV, and extrapolated the data to higher Fermi energies. The three symbols indicate the Fermi energy and predicted diffusivity corresponding to the diffusivity measurements in Fig. 2e.
Extended Data Fig. 2 Spatiotemporal results from a second device.
a, Asymmetric spatiotemporal ∆ITE maps for three different gate voltages. b, Extracted width as a function of ∆t. A lower Fermi level leads to a higher time-zero width, in accordance with hydrodynamic transport, as presented for the main device in the manuscript. (c) ∆ITE maps, taken at ∆t = 0, as a function of beam offset (∆x, ∆y), as well as sample height (z). (d) extracted line profiles for the two dimensions. e, Resulting signal width σ2 for both dimensions as extracted from Gaussian fits at each z-position. The same measurements are presented in Supplementary Fig. 4 for the main device. These experiments were performed with two beams of wavelength 443 nm and 886 nm, respectively.
Extended Data Fig. 3 Spatiotemporal results from a third device.
a, Microscope image of the device. This sample has split-gates made from graphite, with a gap that is 200 nm. It then has a 30 nm thick layer of SiO2, and then we transferred a graphene flake grown by chemical vapour deposition (CVD) on top of the split-gate structure using an hBN flake. b, Gate-dependent current measurement, which gives an estimated mobility of this half-encapsulated CVD graphene sample of around 8,500 cm2/Vs (solid lines). c-d, Asymmetric spatiotemporal ∆ITE maps at time zero for different gate voltages and laser powers without (c) and with normalization (d). e-f, Comparison between first and third device. Time zero Gaussian widths for spatial scans with one pulse on the junction and the second one scanning across (e) and along (f) the graphene pn-junction, as a function of power and gate voltage, for both the first device (hBN-encapsulated with high mobility, presented in the main text, blue-purple colours, ‘hBN’) and the third device (on SiO2 with low mobility, yellow-red colors, ‘SiO2’). The low-mobility sample with shorter hydrodynamic time window shows systematically less heat spreading around time zero, in agreement with our picture of hydrodynamic heat spreading during the hydrodynamic time window.
Extended Data Fig. 4 Ballistic spreading simulation.
a, Time-dependent output distributions for quantum mechanical calculations for a single electron (top row) and an ensemble of independent electrons (bottom row). b, Resulting width σball2 for ballistic transport for the Monte Carlo method with varying Fermi velocities, as well as the quantum calculation for an ensemble of independent electrons. Both calculations essentially agree and the spread within 0.25 ps leads to final widths of below 0.25 µm2 for realistic values of the Fermi velocity.
Supplementary information
Supplementary Information
Supplementary Notes 1–4 and Figs. 1–10.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Block, A., Principi, A., Hesp, N.C.H. et al. Observation of giant and tunable thermal diffusivity of a Dirac fluid at room temperature. Nat. Nanotechnol. 16, 1195–1200 (2021). https://doi.org/10.1038/s41565-021-00957-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-021-00957-6
This article is cited by
-
Light-driven nanoscale vectorial currents
Nature (2024)
-
Ultrafast light-based logic with graphene
Nature Materials (2023)
-
Giant magnetoresistance of Dirac plasma in high-mobility graphene
Nature (2023)
-
Two-dimensional Dirac plasmon-polaritons in graphene, 3D topological insulator and hybrid systems
Light: Science & Applications (2022)
-
Hydrodynamic approach to two-dimensional electron systems
La Rivista del Nuovo Cimento (2022)
