
Abstract
The construction of three-dimensional models of protein macromolecules is a serious challenge due to the possible ambiguity of solving the inverse problem of reconstructing a three-dimensional structure from a one-dimensional small-angle scattering profile. The target function of this task can have several local minima, which leads to the dependence of the solution on the initial values of the model parameters and on the method of finding the global minimum. The problem of creating structural models is also complicated by averaging the scattering pattern over all orientations of particles in space and by the size and shape distribution of scattering objects in the case of polydispersity and/or polymorphism. In this study, the issue of ambiguity in solving inverse problems and restoring the three-dimensional structure of a protein is considered using the structure of the ectodomain of an insulin receptor-related receptor (ectoIRR) in solution as an example. The paper presents a consistent approach to solving this problem, starting from the determination of general structural parameters and ab initio reconstruction of shape to modeling by rigid bodies (using molecular tectonics), hybrid methods, and analysis of scattering profiles by singular vector decomposition.
Similar content being viewed by others

INTRODUCTION
The extensive use of synchrotron radiation sources, new types of recording devices, and the development of new approaches to processing and interpreting experimental data made the small-angle scattering (SAS) method one of the most powerful and versatile tools for studies of the supra-atomic structure of matter; it is used in the analysis of the structure of disperse systems, in molecular biology, biophysics, and other fields of science and technology [1–5].
The level of structural information contained in the SAS data is determined by the nature of the scattering objects. The theory of small-angle scattering and various approaches to solving structural problems have been developed most fruitfully for the study of monodisperse systems of identical particles. Such systems are studied, for example, in the analysis of highly purified solutions of biological macromolecules. In this case, the scattering intensity is directly related to the structure of the particles, which makes it possible to determine their shape and size, and in some cases, their internal structure, with a resolution of about 1 nm.
The general parameters of the samples under study, the so-called SAS invariants, are uniquely determined directly from the scattering curves without structural modeling [1]. The SAS invariants include the maximum particle size (Dmax), the radius of gyration (Rg), the Porod volume (Vp), and the molecular mass (MM) [1, 6]. The construction of three-dimensional models is a much more complex issue due to the poor conditionality of the inverse problem of reconstructing a three-dimensional structure from a one-dimensional scattering profile. The target function of this problem can have several local minima, which leads to the dependence of the solution on the initial values of the model parameters and on the method of finding the global minimum. The problem of creating structural models is also complicated by averaging the scattering pattern over all orientations of particles in space, and by the distribution in size and shape of scattering objects in the case of polydispersity and/or polymorphism. Thus, an unambiguous solution of the inverse scattering problem, that is, the reconstruction of the high-resolution three-dimensional structure of an object based on the experimental data of SAS is impossible in general. Nevertheless, considering the values of the invariants calculated from the experimental curves, imposing certain restrictions on the desired structure, for example, introducing the condition of particle symmetry or its homogeneity, and using the structural information obtained by complementary methods, the corridor of possible solutions can be significantly narrowed. In addition, modern methods of processing and interpreting small-angle scattering data [5, 7] enable for a relatively short time to restore the structure of the sample under study using different approaches and multiple runs of the corresponding programs; this makes it possible to compare, average, cluster, and then analyze the set of solutions obtained [8, 9]. As a result, it is possible to obtain a sufficiently detailed structure of the scattering object.
In this paper, the problem of ambiguity in solving inverse problems and reconstructing the structure from small-angle X-ray scattering (SAXS) data is considered by studying the structure of the ectodomain of an insulin receptor-related receptor (ectoIRR) in solution. The insulin receptor-related receptor (IRR) belongs to the superfamily of transmembrane receptor tyrosine kinases, which regulate various vital processes in the body by transmitting a signal about changes in environmental conditions to the intracellular space, thereby activating the cell’s response to the appeared changes. The IRR was discovered in 1989 [10], however, its structure and functions were not known for a long time. It was shown only recently for the first time that this protein is activated exclusively when the acid-base balance changes; that is, this protein does not require the addition of ligands to it to transmit a signal to the intracellular medium [11, 12]. The unusual functional properties of this receptor have attracted the attention of researchers around the world, but the structure of the full-size protein is still unknown; the conformation of its ectodomain was first determined by SAXS and atomic force microscopy at different pH only in 2019 [13]. Nevertheless, further study of the properties of ectoIRR have shown that the question of the uniqueness of the structure obtained in 2019 remains open due to the complexity of its structural organization. That is why this protein was chosen as an object for developing an optimal strategy for minimizing the ambiguity of solving inverse SAXS problems when studying the structures of complex biological macromolecules in solution.
This paper presents a consistent approach to solve this problem starting from the determination of general structural parameters and ab initio shape reconstruction to rigid body modeling (molecular tectonics) and hybrid methods.
MATERIALS AND METHODS
Preparation of recombinant ectoIRR. The recombinant ectodomain of the insulin receptor-related receptor (ectoIRR) was expressed, purified, and characterized as described in [13].
Small-angle X-ray scattering data analysis and primary data processing. A small-angle experiment with IRR was conducted at the BioSAXS P12 station at EMBL (DESY, Hamburg, Germany) [14] in conjunction with size-exclusive chromatography (SEC) to separate possible oligomers and to ensure its monodispersity [15].
Processing of the obtained experimental data and their further interpretation was carried out with the ATSAS software package [16], which is one of the most reliable and popular tools for analyzing, visualizing and modeling SAS data. All the programs and program blocks used in this study presented below are part of the latest modification of ATSAS.
The primary processing of experimental data included a series of manipulations with a set of experimental scattering curves, the thoroughness of which determined the quantity and quality of the obtained structural information. The PRIMUS multi-functional software block [17], designed to perform preliminary actions with the initial experimental data of the SAXS, provided the necessary and sufficient basis for further structural analysis.
The primary processing also included statistical analysis of a set of small-angle scattering curves for a given sample over the measurement period, their averaging and scaling. The PRIMUS software block was used to subtract the background (solvent) scattering, cross-link the data measured at different angular intervals, extrapolate to the zero concentration of the sample, calculate the invariants using the Guinier and Porod graphs, and much more.
It should be noted that in the case of using online SEC to ensure the monodispersity of the sample, the averaging of the scattering curves, as well as the subtraction of the buffer scattering were carried out with the CHROMIXS program [18].
The radius of gyration Rg was determined from the Guinier approximation [19] at very small angles, when the scattering intensity for identical non-interacting particles was defined as I(s) = I0 exp[–(sRg)2/3]. In this case, the scattering intensity I0 at zero angles was proportional to the molecular mass. The Porod volume Vp of the scattering object was calculated using the normalized Porod invariant Q [20].
To estimate the folding of the macromolecule, a graph s 2I(s) in the Kratky coordinates was used. The scattering intensity of structured, tightly folded particles showed a well-defined bell-shaped maximum on the Kratky plot [20]. Disordered, unfolded scattering objects obeyed the s–2 asymptotic, forming a statistically convoluted Gaussian chain (tangle) [21].
A modified version of the PRIMUS software block PRIMUS/QT [7] contained the “Molecular mass” tool, which gave a Bayesian estimate of the molecular mass based on several parameters [22].
The SASPLOT utility built into PRIMUS [7] enabled to evaluate the quality of difference scattering curves due to the possibility of detailed visualization of their individual sections. The determination of geometric and mass characteristics and a number of other structural parameters also required careful visual control, and although at present the primary processing of small-angle scattering data can be carried out in a fully automatic mode [23], in the case of studying complex objects and systems with polymorphism and/or polydispersity, this procedure should be performed manually, and the graphical interface of the PRIMUS complex enabled to evaluate the quality of the manipulations performed.
The SHANUM program [24] was used to estimate a useful section of the scattering curve, where high-angle points with strong noise were excluded from consideration as uninformative. The DATCLASS program [25] was used to predict the type of particle from the scattering curve (compact, elongated, flat, ring, hollow, and random chain). The ambiguity of the shape determination for specific samples was estimated using the AMBIMETER program [26], which compared the experimental scattering curve with the “ambiguity map”, calculated from 14 000 topologies, and provided a quantitative estimate for ab initio shape reconstruction. The a-score ambiguity index was the logarithm of the number of adjacent curves on the ambiguity map for a given experimental curve. The value of a-score <1.5 practically guaranteed a one-to-one reconstruction. With values of a-score >2.5, the reconstruction was plentiful; in all other cases, the reconstruction was potentially ambiguous.
Analysis of small-angle X-ray scattering data. Determination of the general shape of the particles from the SAXS data. For diluted solutions with a random orientation of monodisperse, non-interacting particles, the scattering is isotropic, and its intensity I(s) was expressed as a function of the modulus of the scattering vector s (s = |s| = 4π sin(θ)/λ), where λ was the wavelength of the incident radiation and 2θ was the scattering angle:
here, the distance distribution function p(r) was related by the Fourier transform to the scattering intensity:
Thus, the distance distribution function p(r) could be found using Eqs. (1) and (2). This function contains information about the shape and structure of the particle and enables to estimate its maximum size Dmax from the condition p(r) = 0 for r > Dmax. In practical studies, p(r) is calculated not by direct integration of the intensity (which leads to strong breakage effects), but by using the so-called indirect Fourier transform and the GNOM program of the ATSAS package [27].
Reconstructing a three-dimensional model of an object from its one-dimensional scattering curve is not a completely correct task, since multiple structures can hypothetically provide the same scattering profile. A simplification representing a low-resolution particle as a homogeneous body can reduce the ambiguity of such a reconstruction. The shape of a particle can be described as a set of finite volume elements (beads) arranged on a dense hexagonal grid. The most common approach for determining the low-resolution shape uses minimization of the target function by simulating annealing to create a compact interconnected ensemble of beads in the search volume (usually a sphere with a diameter of Dmax) that matches the experimental data. This approach was implemented in the DAMMIN program [28]. It did not require the presence of a priori structural information and, thus, the modeling was conducted ab initio.
During the modeling, the optimal model was searched, so that the deviation between the experimental data of Iexp(s) and the calculated from the model scattering profile Icalc(s) was minimized:
where c is the scaling factor, N is the number of experimental points, and σ denotes experimental errors.
To obtain structural models that meet the supraatomic resolution, the conditions of connectivity and compactness were imposed on the desired model. Therefore, the target function that was minimized in the process of restoring the structure has the form: f(X) = χ2 + αP(X), where P(X) was the penalty for looseness of the model (α > 0 was its weight, set by the user).
The DAMMIN program for ab initio modeling can be used for any monodisperse samples, and the GASBOR program was developed to reconstruct the domain structure of proteins [29]. The dummy residues in this program form a protein-like chain, folded so that the scattering from the resulting structure corresponds to the scattering from the studied protein macromolecule in solution with a resolution of about 0.5 nm.
Calculation of the SAXS curve from atomic resolution models. A more detailed interpretation of the SAXS data is possible if high-resolution atomic models of the entire macromolecule or its individual fragments are available from crystallographic data or NMR. In this case, the SAXS enables to detect the structural difference between the crystal and the solution and to determine the biologically active conformation or the oligomeric state. It should be noted that the exact construction of scattering curves in solution from the atomic coordinates of scattering objects is not a trivial task. The scattering profiles are affected by the solvent effects, in particular the presence of a hydration shell with an average density of about 10% higher than the density of the bulk solvent. Nevertheless, the calculation of the macromolecule scattering in solution and the approximation of the experimental curve can be performed by the CRYSOL program [30].
Reconstruction of the structure according to the SAXS data by the method of molecular tectonics. If the scattering particles are macromolecular complexes, an extremely important approach to the interpretation of the SAXS data is rigid body modeling, or the method of molecular tectonics [31]. Often, a complex biological molecular object cannot be crystallized, and the location of the subunits in it remains unknown; however, it is possible to crystallize and determine the structure of the individual components of the complex with atomic resolution. Then the relative position of the subunits in the complex can be obtained by molecular tectonics from the SAXS data. That is, the method is used when the structures of the individual parts that make up the complex are known. In addition, this method makes it possible to analyze the internal structural flexibility and mobility inherent in subunits in a complex or domains within the boundaries of a single macromolecule. Rigid body modeling of the spatial structure of complexes consists in moving and rotating individual elements of the structure and obtaining the minimum deviation χ2 between the model and experimental curves, which is carried out using the SASREF program [31]. The molecular tectonics modeling procedure is easily generalized to the case of an arbitrary number of rigid bodies K and can generally be described using 6(K – 1) parameters. The scattering amplitudes of individual subunits are calculated using the CRYSOL program.
Since rigid domains in protein macromolecules are often connected by flexible, disordered, mobile linkers, the analysis of the structure of such proteins is carried out using the further development of the method of molecular tectonics, which is implemented, for example, in the CORAL program [8]. The program performs hybrid modeling, using molecular tectonics to position domains and an ab initio approach to represent linkers as chains made up of dummy residues. The principle of hybrid modeling of biological objects is described below.
Hybrid methods of structure reconstruction based on SAXS data. The combination of different approaches, such as molecular tectonics and other structural methods, makes it possible to create a cluster of methods that are called hybrid. One of the most important approaches to the study of macromolecular complexes by hybrid methods is the combination of ab initio methods and molecular tectonics. Among the programs that implement this approach, we should particularly mention the EOM program. EOM implements a method for optimizing an ensemble of particles (domains and subunits) and, in addition to the ab initio function of restoring the whole form, allows analyzing the flexibility of biological macromolecules, unfolding and mobility of their individual parts relative to each other. Thus, this program is a tool for describing the distribution in shape and size of a partially or completely unfolded macromolecule in solution [32].
Models obtained by various methods were combined using the SUPCOMB program [33] to determine the difference in their spatial organization and to indicate the normalized spatial deviation (NSD). Typically, an NSD value close to 1.0 indicates that the models obtained by different methods are similar.
Analysis of polydisperse and polymorphic solutions. In the case of polydisperse and polymorphic systems (dilute solutions), the resulting scattering intensity is a linear combination of individual components and is described by the equation
where K is the number of system components, vk are the volume fractions, and Ik(s) are the scattering intensities from each component.
The OLIGOMER program was used for quantitative analysis of the volume fraction of various components in the solution (when the number of components and their scattering intensities were known) [17]. The program uses a non-negative linear least squares algorithm to minimize the discrepancy χ2 between the predicted scattering curve of the mixture and the experimental SAXS data.
One of the effective model-independent methods for estimating the number of components is the decomposition into singular vectors. When there is a set of SAXS data recorded under changing conditions, the SVDPLOT program [17] was used to calculate singular vectors and their associated singular values. The number of non-randomly oscillated singular vectors with significant singular values enables to estimate the minimum number of independent curves required to represent the entire data set, that is, the number of significant components in the mixture.
RESULTS AND DISCUSSION
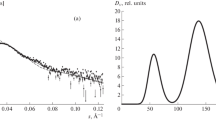
Practical application of methods for interpreting SAXS data on the example of scattering curves from ectoIRR. The experimental small-angle scattering curves of ectoIRR at pH 7.0 and 9.0 are shown in Fig. 1a. As can be seen from Fig. 1a, prior to the use of exclusive gel chromatography, the ectoIRR scattering curves were more typical for polydisperse compounds with a weakly expressed form factor. The peculiarity of these curves was also that the scattering intensity at the smallest angles was slightly lower than for the curves after chromatography. The indicated differences were revealed more clearly in the graphs in the Kratky coordinates (Fig. 1b). It can be assumed that the ectoIRR samples before passing through the chromatographic column, being polydisperse, might contain a certain amount of low-molecular impurities, which was expressed in a decrease in the scattering to the zero angle and a decrease in the amplitude of the maximum on the Kratky plot in the range of angular vectors of 0.25–0.75 nm–1. Therefore, scattering curves for purified by SEC samples were used for the structural analysis.

Experimental small-angle scattering curves (a) and Kratky plots (b) for the ectodomain of the insulin receptor-related receptor (ectoIRR) in solution at pH 7.0 and 9.0. Curves 1 (pH 7.0) and 2 (pH 9.0) reflect the measurements taken prior to the use of online chromatography; curves 3 (pH 7.0) and 4 (pH 9.0) reflect the measurements after exclusive gel chromatography.
Since IRR is activated only when the acid-base balance changes, we should expect a noticeable change in the SAXS profile when pH changes; however, the scattering curves during solution alkalization practically did not differ from each other, as we have already observed [13]; it was concluded that the structure of ectoIRR at the macro level does not change when pH changes, and conformational rearrangements are limited by the local rotation of the protein domains due to flexible linkers between them. Similar conclusions were also made for one of the IRR homologues, the ectodomain of the insulin-like growth factor receptor in [34].
To find out exactly what structural characteristics and at what is the structural level of change when ectoIRR is activated, a thorough analysis of the small-angle scattering curves was carried out in this study and the corresponding structural parameters were calculated.
Primary processing of SAXS data and general structural parameters. Since the ectoIRR chromatograms at pH 7.0 and pH 9.0 were almost identical, the only ectodomain chromatogram at pH 9.0 is presented in Fig. 2a as an example; it shows a narrow and at first glance symmetrical peak characteristic of a monodisperse substance. Several scattering curves of eluate were taken separately from the right and left sides of the peak for averaging and subsequent comparison, that is, to verify the symmetry of the chromatogram peak and to exclude the presence of additional ectoIRR fractions. Buffer scattering was subtracted from the separately averaged SAXS curves on the left and right sides of the peak (the corresponding curves are marked with black dots on the chromatogram) by the CHROMIXS program. The scattering profiles obtained in this way are shown in Fig. 2b.

(a) The chromatogram of ectoIRR at pH 9. The solid line shows the averaged scattering intensities as a function of the curve number during sample elution. Triangles show the curves from the left side of the chromatogram peak; the circles show the curves from the right side of the chromatogram peak. The points corresponding to the buffer are marked in black. (b) The averaged small-angle protein scattering curves taken from the left (1) and right (2) of the chromatogram peak.
As can be seen in Fig. 2b, the averaged ectoIRR eluate scattering curves taken for the left and right of the chromatogram peak were identical. It follows that the protein solution after the chromatographic column became monodisperse and, therefore, the scattering from it could be used for a detailed quantitative analysis of the ectoIRR structure.
According to the SHANUM program, the useful data range ends at s ≈ 2.5–3.0 nm–1 (approximately 15 Shannon channels). However, when the maximum dimensions and distance distribution functions p(r) were calculated by the GNOM program, the data range used was limited from above by 8 to 10 Shannon channels, where the width of one Shannon channel was equal to π/Dmax. This corresponds to the plateau of the Porod plot I(s)s 4, when s was in the range of approximately 1.5–1.8 nm–1 (Fig. 3). This region corresponds to the scattering by a homogeneous particle and can be used later for ab initio reconstruction of the ectoIRR shape in solution.

The Porod plots for pH 7.0 (top) and pH 9.0 (bottom) of the ectoIRR scattering curves. The vertical dashed line restricts the area of the scattering vectors corresponding to the scattering by a homogeneous particle.
The classification of the structure type using the DATCLASS program indicated the compact shape of the ectoIRR molecule domains, which was in good agreement with the shape of the scattering curves in the Kratky plot (Fig. 1b). The characteristic bell-shaped appearance of the Kratky plots for both pH 7.0 and pH 9.0 indicated that the ectoIRR mainly consisted of ordered and folded, that is, compact, domains. However, as a result of comparison with the AMBIMETER “ambiguity map”, the average value of the ambiguity parameter of 2.1 was obtained for both curves, which implies a potentially ambiguous reconstruction of the shape of the scattering object [26]. Since we have shown above that the ectoIRR solution is monodisperse at different pH, this ambiguity in the structure reconstruction could be caused by a potential polymorphism of the protein consisting of many separate domains connected by flexible linkers.
A careful determination of the integral macromolecular characteristics of scattering objects is of great importance in solving the inverse problems of SAXS. The radii of gyration calculated using the Guinier formula and other SAXS invariants calculated from the SAXS curves are shown in Table 1.
First of all, the lower values of the invariants for ectoIRR calculated from the scattering curves without the use of exclusive gel chromatography are noteworthy in Table 1. The overall data obtained indicated polydispersity, namely, low values of MM and Rg indicated the presence of low molecular mass impurities in the solution, while the increased Dmax implied the presence of a certain amount of protein aggregates. In general, this once again highlighted the need to use online SEC for detailed structural analysis of biological objects.
It was also important to choose the correct interval of angular vectors. Porod volume for ectoIRR at pH 7.0 and 9.0 (Table 1), calculated in the data range of up to 8 Shannon channels, had a clearly overestimated value, and if we used the empirical ratio MMPorod = VPorod/1.65, the molecular mass would be twice as high than the corresponding theoretical values. This value for globular proteins was obtained earlier [8]. The empirical coefficient 1.65 was determined for PDB structures that do not contain heteroatoms. In our case, the IRR molecule contains glycans, which could significantly change the ratio. In order to estimate the coefficient for this particular case, a homologous model of the insulin receptor with glycans was used, taken from the SASBDB small-angle database with the SASDHF2 code. For this model, which has MM = 251 kDa, the calculated Porod volume was of 600 nm3. Thus, the coefficient for calculating molecular masses was obtained to be of 2.4. This value was used to estimate the MM in this work in the range of angular vectors up to 15 Shannon channels. In this case, the molecular masses of ectoIRR uniquely corresponded to the dimer, which well agreed with the well-known literature data indicating that a receptor similar to the insulin receptor initially exists in the membrane as a dimer, where the corresponding subunits are connected by disulfide bridges [35]. The MM values shown in the table were in good agreement with the molecular mass calculated from the zero-angle scattering: 220 ± 20 kDa for ectoIRR after SEC.
The overestimated value of the Porod volume in the data range up to 8 Shannon channels can be explained by the heterogeneity of ectoIRR macromolecules due to the presence of glycans in their composition. The molecular mass of the ectodomain, calculated from the amino acid sequence of the protein, is approximately 190 kDa, that is, the share of glycans accounts for almost 20%, and such a scattering object cannot be considered as homogeneous. However, the initial portion of the scattering curve up to 8 Shannon channels should be used for coarse ab initio reconstruction of the ectoIRR form in solution. In this case, we used a simplification that reduced the ambiguity of the structure reconstruction by representing the shape of a low-resolution particle as a homogeneous body.
The molecular mass of the dissolved particles was also calculated using the “Molecular Mass” tool in the PRIMUS/QT program. The molecular mass calculated using this program, regardless of which data range was used (full-size or up to eight Shannon channels) was the same within the error range. The obtained value of 320 kDa with a confidence interval of (221–372) kDa corresponded to the mass of the ectoIRR dimer in the presence of glycans. In addition, it should be emphasized once again that a narrow peak on the gel chromatogram indicated the presence of only one oligomeric state, and, accordingly, only polymorphism is possible, but not polydispersity.
Ab initio modeling of the IRR shape. As a result of three-dimensional modeling by the finite volume element method with the DAMMIN program, the ectoIRR experimental data were approximated in the s range up to 8 Shannon channels, where the particle could be represented as a homogeneous body (Porod graphs, Fig. 3). This simplification for a protein such as ectoIRR containing heteroatoms is necessary to reduce the ambiguity of solving inverse problems. A typical reconstruction the shape of IRR molecule is shown in Fig. 4. The volume of the ab initio models obtained corresponded to the dimeric state of the protein (Table 1).

Ab initio reconstruction (by the DAMMIN and GASBOR programs) of the ectoIRR structure with fitting of different intervals of the scattering curves (the vertical dotted line restricts the region of about 8 Shannon channels): 1 and 4 are the experimental SAXS data for ectoIRR at pH 7.0 and 9.0, respectively; 2 and 5 are the calculated curves from models obtained by the DAMMIN program; 3 and 6 are the calculated curves from models obtained by the GASBOR program. The characteristic ab initio shapes of ectoIRR are shown on the right; the red ones were obtained using the DAMMIN program, the green ones were obtained using the GASBOR program. The inset shows the distance distribution functions p(r): 1, at pH 7.0; and 2, at pH 9.0.
Ab initio modeling by the DAMMIN and GASBOR programs gave in all cases a good approximation to the experimental scattering curves with χ2 = 1.1–1.3. The obtained shapes (Fig. 4, right), although they have the same values of the scattering invariants, were very different in their conformation structures, which confirmed the assumption of ectoIRR polymorphism in solution, arising from a large number of flexible linkers connecting individual protein domains. These shapes were present both when ectoIRR was reconstructed in solution at pH 7.0 and at pH 9.0 in the range of up to 8 Shannon channels. In the longer interval, the forms reconstructed using the DAMMIN program were loose and had no definite structure. Therefore, one of the conclusions of this section is the recommendation to use shorter ranges of the experimental profile for ab initio modeling of the structure of proteins with a large number of disordered fragments in accordance with the Shannon estimate. There is no such a restriction for the GASBOR program, since it is possible to reconstruct the domain structure of the protein with this method. Therefore, the structural models were calculated in the range of up to 15 Shannon channels. Thus, similar shapes with diverse conformations were obtained by two different methods of reconstructing the low-resolution structure. In general, it was shown at the ab initio stage of structure reconstruction based on SAXS data that the solution of the inverse problem in this case is complicated by the presence of ectoIRR polymorphism.
Approximation of the SAXS data by the atomic model of the ectodomain of the insulin receptor (ectoIR), a homolog of ectoIRR. We made an attempt to approximate the experimental SAXS data for ectoIRR by an accessible atomic model of a homologous IR protein. The ectoIR model and the corresponding approximation by the CRYSOL program are shown in Fig. 5. In this case, the entire range of experimental data was fitted, since an inhomogeneous model was used (in contrast to the ab initio modeling by the DAMMIN program). As can be seen from Fig. 5, the model did not correspond well to the experimental curve for ectoIRR apparently due to a different conformational state of the ectodomain of the insulin receptor.

Approximation of the ectoIRR scattering curve by the atomic model of the homologous ectoIR protein. Experimental SAXS data from ectoIRR (black dots) and fit by the ectoIR model (gray line). The inset shows the atomic model of the ectoIR dimer.
Molecular tectonics for quaternary structure analysis of ectoIRR. In this study, the quaternary structure of the IRR tetramer was reconstructed based on the domain structure of the homologous insulin receptor. The CORAL program modeled the mutual arrangement of individual domains in the asymmetric part of the ectoIRR molecule and the conformation of flexible linkers as chains of dummy residues connecting the corresponding domains. The symmetrically related structure of the second monomer was generated automatically. The insulin receptor ectodomain (ectoIR) also containing glycans was taken from the small-angle scattering biological data bank, SASBDB (www.sasbdb.org), the SASDHF2 access code [34], as a structural template for ectoIRR hybrid modeling. The ectoIRR dimer was represented in separate multiple runs of the CORAL program as two polypeptide chains related by a twofold axis (P2 symmetry). Each monomer was divided into the following four fragments containing residues 1–308, 312–592, 596–655, and 755–909. The successive fragments were connected by linkers from dummy residues of the appropriate length. During the modeling, the third and fourth fragments were shifted in a consorted manner (identical rotations and identical shifts) in order to preserve the structural integrity of the last two fibronectin domains at the C-end of the molecule.
Typical reconstructions are given in Fig. 6 together with the corresponding approximations of the SAXS data. As can be seen from Fig. 6, the models were not identical due to polymorphism. Repeated reconstruction resulted in many different conformations that nevertheless shared common features, and they could be divided into groups based on similarity. In general, the obtained models mainly reflected low-resolution structures of ab initio shape reconstruction, that is, structures with converged fibronectin domains (closed forms) and structures where these domains were spatially separated (open forms).

Approximation of the SAXS data by molecular tectonics for scattering from ectoIRR at pH 7.0 (1) and pH 9.0 (2). The experimental data are shown by black dots. The fits using models are given by gray lines. The typical models of the ectoIRR tetramer are shown on the right.
Despite the difference in conformations, all models, as in the case of ab initio reconstruction, showed a good approximation by the experimental scattering curves with χ2 = 1.0–1.2.
Analysis of the mobility of the IRR protein domains. The degree of mobility of the ectoIRR protein domains was evaluated using the EOM (Ensemble Optimization Method) program. As in the molecular tectonics modeling, the structure of the homologous IR protein was used, and the ectoIR monomer was divided into two domains (containing residues 1–465 and 472–910, respectively) separated by a flexible link of 7 residues. The EOM program created an ensemble of 10 000 models with randomly selected mutual domain orientations, and then, using a genetic minimization algorithm, selected from this ensemble an optimized set of models, the combination of which best approximated the small-angle data. The initial ensemble consisted of dimeric ectoIR molecules, half of which were homodimers (that is, had P2-symmetry), and the other half were heterodimers (P1 symmetry). However, with such a choice of domains, it was not possible to adequately approximate the experimental curve: apparently, the number of degrees of freedom when creating a random ensemble was insufficient. After that, each ectoIR monomer was divided into five parts (containing residues 1–290, 299–450, 459–571, 580–756, and 790–908, accordingly, and connected to each other by flexible links) and the simulation was carried out using the EOM program. In this case, the number of degrees of freedom when creating the ensemble was sufficient, and the experimental IRR data were well approximated χ2 = 2.3–2.8 (Fig. 7). At the same time, a comparison of the distribution of the gyration radii of the models of the original and optimized ensembles showed that fairly compact structures with gyration radii in the range from 4.5 to 6.0 nm were selected; the degree of domain mobility could be considered as moderate, which was confirmed by the estimates of the Rflex mobility parameters, namely, this parameter was of 87.3% for the original random ensemble, and for the optimized set of models it was of 61.6%. Thus, one of the possible states of the IRR system could be an ensemble of moderately mobile dimers with at least 3–5 flexible links separating the protein domains.

Analysis of ectoIRR disorder at pH 7 (a) and at pH 9 (b). The experimental SAXS data are shown by black dots, the fit using disordered dimers constructed by the EOM program is a solid line. The inset shows the Rg distribution in the original set (solid line) and in the optimized ensemble (dashed line).
Model-independent analysis of the minimum number of components (conformations) of the IRR protein in solution. The minimum number of independent and significant components required to describe the experimental SAXS dataset in combination with exclusive gel chromatography (SEC-SAXS) can be estimated using singular value decomposition. To do this, the set of SEC-SAXS curves can be represented as a matrix by taking the experimental curves corresponding to the chromatographic peak (curve numbers between 1600 and 1800, Fig. 2). This set is then used as input to the SVDPLOT program [17], which decomposes it into singular vectors and determines the corresponding singular values. Figure 8 shows the results of such a decomposition, where the singular values are arranged in descending order (Figs. 8a, 8b), and the corresponding singular vectors are shifted along the vertical axis relative to each other for better visualization. Figure 8c shows that the singular values reach a weakly changing plateau in the region of 10–15 components, and the first 12 singular vectors (located from top to bottom) have non-random oscillations around the horizontal line. This made it possible to conclude that there were at least 10–15 independent protein conformations in the system, which was in good agreement with the conclusion about the mobility of the ectoIRR domains of the protein when it was modeled by the ensemble optimization method (EOM), as well as the variety of shapes obtained by ab initio modeling.

Decomposition of the SEC-SAXS dataset for ectoIRR into singular vectors and determination of the corresponding singular values.
Thus, the model-independent analysis and demonstration of the presence of 10–15 protein conformations of ectoIRR in the solution confirmed the results of modeling the structure of this domain by ab initio and hybrid methods. Therefore, for a protein involved in the regulation of the acid-base balance in the body and whose structure should be determined by the pH of the solution, it was also necessary to conduct a quantitative analysis of the content of certain conformations, depending on the acidity of the medium. For this purpose, the program OLIGOMER [17] was used. To determine the volume fractions vk of each component of the mixture, the program found a linear combination of scattering intensities from each component Ik(s) in the mixture of conformations. For the calculations, the most common shapes of the protein were used, which were obtained by modeling by the hybrid method with the CORAL program. These shapes can be grouped into two main groups of closed and open conformations. As a result, it was shown that at pH 7.0 there was an open (relaxed) conformation with v1 = 0.84, and there was a fraction of the closed conformation with v2 = 0.16. At pH 9.0, in the solution there was the fraction with an open conformation v1 = 0.15, and mainly there was an active, closed conformation containing converged fibronectin domains with v2 = 0.85. The obtained approximations corresponded to the experimental data with χ2 = 2.8 for pH 7.0 and χ2 = 1.5 for pH 9.0 (Fig. 9).

The result of modeling mixtures of different conformations by the OLIGOMER program: 1 and 3 are the experimental ectoIRR SAXS curves at pH 7.0 and 9.0, respectively; 2 and 4 are the theoretical scattering from a mixture of conformations at pH 7.0 and 9.0, respectively.
CONCLUSIONS
In this paper, the issue of ambiguity in solving inverse problems and reconstructing the three-dimensional structure of the protein according to SAXS data was considered by studying the structure of the ectodomain of the insulin receptor-related receptor (ectoIRR) in solution as an example. We have shown that this protein, having many disordered flexible fragments, exists in solution as a set of conformations. It was concluded that conformational rearrangements occur not only at the local level, as previously assumed [13], but also significantly change the overall shape of the ectodomain. The data obtained correspond to the conclusions of a recently published work, where a variety of forms of a full-size receptor similar to the insulin receptor was shown by atomic force microscopy [36].
We have demonstrated that a consistent approach to the study of proteins with polymorphism, using the IRR ectodomain as an example, starting from the determination of their general structural parameters and ab initio shape reconstruction, followed by modeling with rigid bodies (using molecular tectonics) and hybrid methods, leads to a fairly complete description of the protein structure. This was especially important for the IRR ectodomain, since in previous studies we could not show a change in the protein conformation with pH changes, that is, when the receptor performs its biological functions.
The second task of our research was to demonstrate the existing possibilities of small-angle scattering using a rather complex example. We have shown that when solving the inverse problems of SAXS with the fundamental impossibility of obtaining an unambiguous structural answer, it is possible, however, to describe the conformational state of the scattering object quite fully and quantitatively. At the same time, analyzing the data of small-angle scattering, it is necessary to critically evaluate the results obtained and, if possible, use complementary methods. In our work, where we set out to show modern approaches and the possibilities of SAS, we did not use the data of other structural methods in principle, although it is their use that enables to significantly narrow the corridor of possible solutions to the inverse small-angle scattering problem.
It is also important to choose the correct range of data for the initial evaluation of general geometric parameters, such as excluded volume, radius of gyration, and maximum size; it is also important for modeling the structure by different methods. In ab initio shape modeling, only the first 8 to 10 Shannon channels should be used, and a further increase in the interval of angular vectors leads to an inadequate reconstruction of the structure with false details. On the other hand, for hybrid methods, the angular range should be as wide as possible.
The use of online exclusive gel chromatography is very important, and the reconstruction of the structure with a resolution of about 1 nm in small-angle scattering is possible only for monodisperse protein solutions. However, even for such a complex object of research as the ectodomain of the insulin receptor-related receptor, with its flexibility and polymorphism, it was possible to construct structural models that reflect its biological properties. We hope that our work is useful not only in terms of studying specifically the structure of ectoIRR, but also describes a number of significant methods of SAXS and the scenario that should be followed in the process of studying structurally intricate biological objects and complexes.
REFERENCES
Feigin L.A., Svergun D.I. 1987. Structure analysis by small-angle X-ray and neutron scattering. New York: Plenum Press.
Svergun D.I., Shtykova E.V., Volkov V.V., Feigin L.A. 2011. Small-angle X-ray scattering, synchrotron radiation, and the structure of bio- and nanosystems. Crystallography Reports. 56 (5), 725–750.
Graewert M.A., Svergun D.I. 2013. Impact and progress in small and wide angle X-ray scattering (SAXS and WAXS). Curr. Opin. Struct. Biol. 23 (5), 748–754.
Blanchet C.E., Svergun D.I. 2013. Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu. Rev. Phys. Chem. 64, 37–54.
Schroer M.A., Blanchet C.E., Gruzinov A.Y., Grawert M.A., Brennich M.E., Hajizadeh N.R., Jeffries C.M., Svergun D.I. 2018. Smaller capillaries improve the small-angle X-ray scattering signal and sample consumption for biomacromolecular solutions. J. Synchrotron Radiation. 25, 1113–1122.
Svergun D.I., Koch M.H., Timmins P.A., May R.P. 2013. Small angle X-ray and neutron scattering from solutions of biological macromolecules. London: Oxford University Press.
Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M., Svergun D.I. 2017. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50 (Pt 4), 1212–1225.
Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystal. 45 (2), 342–350.
Volkov V.V., Svergun D.I. 2003. Uniqueness of ab initio shape determination in small angle scattering. J. Appl. Crystal. 36, 860–864.
Shier P., Watt V.M. 1989. Primary structure of a putative receptor for a ligand of the insulin family. J. Biol. Chem. 264 (25), 14605–14608.
Deev I.E., Vasilenko K.P., Kurmangaliev, E. S., O. V., Popova N.V., Galagan Y.S., Burova E.B., Zozulya S.A., Nikol’skii N.N., Petrenko A.G. 2006. Effect of changes in ambient pH on phosphorylation of cellular proteins. Dokl. Biochem. Biophys. 408, 184–187.
Deyev I.E., Sohet F., Vassilenko K.P., Serova O.V., Popova N.V., Zozulya S.A., Burova E.B., Houillier P., Rzhevsky D.I., Berchatova A.A., SASashev A.N., Chugunov A.O., Efremov R.G., Nikol’sky N.N., Bertelli E., Eladari D., Petrenko A.G. 2011. Insulin receptor-related receptor as an extracellular alkali sensor. Cell Metab. 13, 679–689.
Shtykova E.V., Petoukhov M.V., Mozhaev A.A., Deyev I.E., Dadinova L.A., Loshkarev N.A., Goryashchenko A.S., Bocharov E.V., Jeffries C.M., Svergun D.I., Batishchev O.V., Petrenko A.G. 2019. The dimeric ectodomain of the alkali-sensing insulin receptor-related receptor (ectoIRR) has a droplike shape. J. Biol. Chem. 294 (47), 17790–17798.
Blanchet C.E., Spilotros A., Schwemmer F., Graewert M.A., Kikhney A., Jeffries C.M., Franke D., Mark D., Zengerle R., Cipriani F., Fiedler S., Roessle M., Svergun D.I. 2015. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystal. 48 (Pt 2), 431–443.
Jeffries C.M., Graewert M.A., Blanchet C.E., Langley D.B., Whitten A.E., Svergun D.I. 2016. Preparing monodisperse macromolecular samples for successful biological small-angle X-ray and neutron-scattering experiments. Nature Protocols. 11 (11), 2122–2153.
Manalastas-Cantos K., Konarev P.V., Hajizadeh N.R., Kikhney A.G., Petoukhov M.V., Molodenskiy D.S., Panjkovich A., Mertens H.D.T., Gruzinov A., Borges C., Jeffries C.M., Svergun D.I., Franke D. 2021. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Cryst. 54, 343–355.
Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. 2003. PRIMUS – a Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystal. 36, 1277–1282.
Panjkovich A., Svergun D.I. 2018. CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle X-ray scattering data. Bioinformatics. 34 (11), 1944–1946.
Guinier A. 1939. La diffraction des rayons X aux tres petits angles; Application a l’etude de phenomenes ultramicroscopiques. Ann. Phys. (Paris). 12, 161–237.
Glatter O., Kratky O. 1982. Small angle X-ray scattering. London: Academic Press.
Debye P. 1947. Molecular-weight determination by light scattering. J. Phys. Colloid Chem. 51 (1), 18–32.
Hajizadeh N.R., Franke D., Jeffries C.M., Svergun D.I. 2018. Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 8 (1), 7204.
Graewert M.A., Franke D., Jeffries C.M., Blanchet C.E., Ruskule D., Kuhle K., Flieger A., Schafer B., Tartsch B., Meijers R., Svergun D.I. 2015. Automated pipeline for purification, biophysical and X-Ray analysis of biomacromolecular solutions. Sci. Rep. 5, 10734.
Konarev P.V., Svergun D.I. 2015. A posteriori determination of the useful data range for small-angle scattering experiments on dilute monodisperse systems. IUCR J. 2, 352–360.
Franke D., Jeffries C.M., Svergun D.I. 2018. Machine learning methods for X-ray scattering data analysis from biomacromolecular solutions. Biophys. J. 114 (11), 2485–2492.
Petoukhov M.V., Svergun D.I. 2015. Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr. Section D. Biol. Crystallogr. 71, 1051–1058.
Svergun D.I. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503.
Svergun D.I. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76 (6), 2879–2886.
Svergun D.I., Petoukhov M.V., Koch M.H.J. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80 (6), 2946–2953.
Svergun D.I., Barberato C., Koch M.H.J. 1995. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773.
Petoukhov M.V., Svergun D.I. 2005. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 89 (2), 1237–1250.
Bernado P., Mylonas E., Petoukhov M.V., Blackledge M., Svergun D.I. 2007. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129 (17), 5656–5664.
Kozin M.B., Svergun D.I. 2001. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41.
Whitten A.E., Smith B.J., Menting J.G., Margetts M.B., McKern N.M., Lovrecz G.O., Adams T.E., Richards K., Bentley J.D., Trewhella J., Ward C.W., Lawrence M.C. 2009. Solution structure of ectodomains of the insulin receptor family: The ectodomain of the type 1 insulin-like growth factor receptor displays asymmetry of ligand binding accompanied by limited conformational change. J. Mol. Biol. 394 (5), 878–892.
De Meyts P. 2008. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 33 (8), 376–384.
Batishchev O.V., Kuzmina N.V., Mozhaev A.A., Goryashchenko A.S., Mileshina E.D., Orsa A.N., Bocharov E.V., Deyev I.E., Petrenko A.G. 2021. Activity-dependent conformational transitions of the insulin receptor-related receptor. J. Biol. Chem. https://doi.org/10.1016/j.jbc.2021.100534
ACKNOWLEDGMENTS
The authors are grateful to C.M. Jeffries and D.I. Svergun for their help in conducting a small-angle experiment at the BioSAXS station P12 (EMBL, Hamburg) and for the valuable discussion. The work was supported by the Ministry of Science and Higher Education in the framework of the State Assignment of the Federal Scientific Research Center “Crystallography and Photonics” of the Russian Academy of Sciences and by the Russian Foundation for Basic Research (projects nos. 17-00-00487 COMFI, 17-00-00488 COMFI, and 20-04-00959a).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by E. Puchkov
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petoukhov, M.V., Konarev, P.V., Volkov, V.V. et al. The Ambiguity Issue in Solving Inverse Problems of Small-Angle Scattering: A Consistent Approach Using an Insulin Receptor-Related Receptor as an Example. Methods for Interpreting SAXS Data. Biochem. Moscow Suppl. Ser. A 15, 270–283 (2021). https://doi.org/10.1134/S1990747821040097
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990747821040097