
The outbreak of respiratory disease, COVID-19 caused by SARS-CoV-2 has now been spread globally and the number of new infections is rising every moment. There are no specific medications that are currently available to combat the disease. The spike receptor of SARS-CoV-2 facilitates the viral entry into a host cell and initiation of infection. Targeting the viral entry at the initial stage has a better advantage than inhibiting it in later stages of the viral life cycle. This study deals with identification of the potential natural molecule or its derivatives from MolPort Databank as SARS-CoV-2 spike receptor inhibitors using structure-based virtual screening followed by molecular dynamics simulation. On the basis of ADME properties, docking score, MMGBSAbinding energy, 150 ns molecular docking studies, and final molecular dynamics analysis, two natural compounds – 3 (MolPort-002-535-004) docking score –9.10 kcal mol-1 and 4 (MolPort-005-910-183) docking score –8.5 kcal mol-1, are selected as potential in-silico spike receptor inhibitors. Both hits are commercially available and can be further used for in-vitro and in-vivo studies. Findings of this study can facilitate rational drug design against SARS-CoV-2 spike receptor.
Similar content being viewed by others

INTRODUCTION
The outburst of SARS-CoV-2 infection has created an emergency situation globally. Starting from the epicenter of COVID-19, Wuhan city of China, the infection rapidly spread worldwide. Coronavirus (CoVs) species are enveloped positive-sense single strand-largest RNA viruses belonging to the Coronaviridae family of the order Nidovirales, which are divided into four genera (α, β, γ, and δ) [1]. In the last two decades, two highly pathogenic human coronaviruses (HCoVs) namely, Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) [2] and Middle East Respiratory Syndrome coronavirus (MERS-CoV) [3] appeared in years 2002 and 2012, respectively. The two viruses originated from animal reservoirs, had caused global epidemics with fatality rate of which were 9.5 and 34%, respectively [4]. The third pathogenic coronavirus namely SARS-CoV-2 was identified to be the reason for a typical pneumonia infection outbreak [5 – 9]. Due to the high dissemination capacity of SARS-CoV-2, it has created a pandemic situation to public health throughout the world and potentially brings a challenge to the scientific community to control the spread of the virus. According to Worldometer’s COVID-19 data, as of 16 August 2020, about 21.6286 million confirmed cases of COVID-19 infection with 0.7691 million death cases were reported over 215 countries [10]. Due to the absence of any effective treatment and medication against SARS-CoV-2 infection and the high dissemination rate of the infection worldwide, prompted the WHO to declare the SARS-CoV-2 outbreak as a global public health emergency of international concern [11, 12]. This urgent situation is pressing the world scientific community to respond with the development of novel vaccine or small molecule therapeutics for SARS-CoV-2 [13, 14]. In the emergency situation drug repurposing may be a short-term and non-specific solution to treat COVID-19 patients [15], therefore the development of more target oriented natural inhibitors is the need of the hour.
Depending on the target, therapeutics against COVID-19 disease can be divided into two categories: one is acting on the human immune system or host cells, and the other is on SARS-CoV-2 life cycle. The therapeutics acting on the coronavirus includes inhibiting the viral RNA through interacting with the genetic material of the virus. Preventing the viral replication process by interacting with its crucial life cycle enzymes or by resisting the viral spike receptor binding to human ACE-2 or inhibiting the virus’s self-assembly process through acting on some structural proteins of the virus [16]. Depending upon the protein sequence alignment analyses of SARS-CoV-2, it has proven that, the genomic RNA of SARS-CoV-2 contains 29,736 nucleotides. The envelope and nucleocapsid proteins of SARS-CoV-2 are two evolutionarily conserved regions, having the sequence identities of 96% and 89.6%, respectively, compared to SARS-CoV [17]. Whereas, the spike protein of SARS-CoV-2 exhibited 77% sequence similarities with SARS-CoV [12]. The spike is a transmembrane glycoprotein that promotes the host cell attachment and virus-cell membrane fusion during virus infection. SARS-CoV-2 utilizes the homotrimeric glycoprotein of each spike, which consists of two monomeric subunits (S1 sub-unit and S2 sub-unit), to bind with the host cell receptor.
Recently, Lan, et al. [18] showed that SARS-CoV-2 viral spike S1 sub-unit receptor binding domain (RBD) is able to utilize host receptor angiotensin-converting enzyme 2 (ACE2) as an entry receptor in ACE2-expressing cells, suggesting potential drug targets for therapeutic development. In addition, the viral entry requires, the spike proteins priming through cellular proteases of the host cell, which promotes spike (S) protein cleavage at the S1/S2 site and facilitates the viral fusion with the host cell [19 – 22]. Past cryo-electron microscopy investigations of the SARS-CoV-2 spike protein interaction with the host cell ACE2 receptor have indicated that receptor attachment initiates the separation of the viral S1 sub-unit with ACE2 receptor. This emphasizes the S2 sub-unit to form an increasingly steady post-fusion state that is basic for membrane fusion [19]. Furthermore, cryo-EM structure of the spike protein and biophysical assays reveal that the SARS-CoV-2 spike binds with ACE2 in higher affinity than SARS-CoV [23]. The receptor binding domain (RBD) of the S1 sub-unit stabilizes membrane attached S2 sub-unit of the virus [18, 24]. A potential ACE2 inhibitor arbidol is suggested for the treatment COVID-19 infection, but due to the low selectivity and a number of side effects, the ACE2 receptor inhibitor may not be the potential therapeutic against COVID-19 infection. It was also revealed in recent studies that the virus can enter through the transmembrane cellular serine protease (TMPRSS2) receptor of the host cell and facilitates the viral pathogenesis in the host body [22 – 24]. Therefore, the spike receptor binding domain (RBD) of SARS-CoV-2 virus can be a potential target for the treatment and development of new drugs against COVID-19 viral pathogenesis.
Li, et al. [25] identified various traditional herbal medicines as potential ant-viral against SARS-CoV infection although the mechanism of anti-viral activities is not clearly understood. There are evidences that plants also exhibit anti-viral activities against other coronavirus species [26]. The mechanism of action is mainly through the inhibition of the viral replication process and prevention of viral entry into the host cell. Baicalin and baicalein are two natural flavones showing potential in vitro inhibitory activity against SARS-CoV-2 main protease [27]. Baicalein is available from Oroxylum indicum, the fruit of which is used as vegetable in North East India [26]. Similarly, docking study of seventeen organosulfur compounds available in garlic essential oil showed that these compounds may exhibit good binding affinity with human ACE2 and SARS-CoV-2 main protease. The in-silico study suggests that the garlic oil may be an important natural antivirus source that can prevent entry of SARS-CoV-2 into the human cell [18].
In the present study, a total of 1,20,000 natural compounds were initially selected from the MolPort database and, depending upon their pharmacokinetic properties, about 50,000 of compounds were filtered through pkCSM open source server. A virtual screening based molecular docking of selective ligands against the receptor binding domain (RBD) of SARS-CoV-2 was performed. Depending upon the binding interaction with binding threshold value > -8.00 kcal mol-1, top 4 molecules were selected. To analyze the stability of the protein-ligand complex and backbone, NVT and NPT ensemble 50 ns molecular dynamics (MDs) simulations were performed for each molecule and the RMSD (root mean square deviation) and RMSF (root mean square fluctuation) values of protein-ligand complex and backbone were reported. Most of the works related to SARS-CoV-2 drug design are re-purposing the existing drugs or in-silico identification of natural compounds. But in our study, the selected molecules are natural products or natural product derivatives which are commercially available. Therefore, scientific community can easily explore their biological activity in wet laboratory.
MATERIALS AND METHODS
Data Set
The x-ray crystal structure of the recently discovered SARS-CoV-2 spike receptor (PDB ID: 6M0J, Resolution: 2.45Å) [19] was retrieved from RCSB Protein Data Bank (www.rcsbpdb.org) in PDB format. The MolPort Database [https://www.molport.com] which contains 1.12 lakh commercially available natural compounds and related derivatives were used for virtual screening.
Protein Preparation
The protein was imported in Auto Dock Tools 1.5.6 and removed chain A, water molecules, and hetero atoms [28] and saved it as pdb format. This E chain of protein was used for Computed Atlas of Surface Topography of proteins (CASTp 3.0) server [29] and identified the binding site amino acid residues (338, 339, 342, 343, 367, 368, 371, 373, and 374), volume and area of the active site pocket were 60.837 Å3 and 73.389 Å2, respectively. This server uses the alpha shape method to determine the aforementioned topographic features for the input PDB file. The binding site information generated by CASTp 3.0 server was used to generate the grid box [30].
Preparation of Protein for Vina
The previously saved pdb format of the protein was imported and polar hydrogens were added to the protein. Then necessary charges like, Gasteiger and Kollman charges were added. The protein was assigned to Auto Dock Tools 1.5.6 and was exported as PDBQT format. Depending upon the CASTp 3.0 binding site information the grid box was prepared and saved as a text file. The grid box dimension centre was of X, Y and Z set to –36.022, 9.043 and 29.693, respectively.
Preparation of Ligands
A total of 120,000 natural compounds or natural compound derivatives were retrieved from Mol-Port Database. The drug-likeness property of the molecules depends upon the absorption, distribution, metabolism, excretion and toxicological (ADMET) and pharmacodynamics properties. Based on the Lipinski’s rule of five, these molecules were filtered, depending upon the absorption, distribution, metabolism, excretion and toxicological properties. The ADMET properties of these compounds were predicted by open source server pkCSM [31] and 50,000 numbers of hits were filtered out for further studies. These molecules were converted to PDBQT format by using a script written program of Samadani, et al. [32]. The force field was used during ligand preparation was Gaff force field [33] and by applying Genetic algorithm, the best structural conformer was generated. The ligands were prepared using a steepest descent algorithm in which 1000 steps of energy minimization of the ligands were carried out.
Virtual Screening of Ligands with SARS-CoV-2 Spike-Receptor Binding Domain
The structure-based virtual screening (VS) is an important approach to identify the potential in-silico inhibitors against a particular target protein. The hits having good binding affinity towards a receptor are identified from the large database. Structure-based virtual screenings of 50,000 molecules with SARS-CoV-2 spike receptor binding domain was performed using Autodock Vina [34]. The VS work flows of the study are presented in Fig. 1. The VS was performed using the aforementioned script written program [32]. The analysis of ligand binding contribution and the semi-empirical force field were performed in AutoDock Tool. The binding poses of the ligands were generated by using Lamarckian Genetic Algorithm. Other docking parameters like, population size was set to 150, and maximum numbers of evaluations were set to 2500000. Maximum number of generations was chosen up to 27000, rate of gene mutation was set to 0.02, rate of crossover was set to 0.8 and the exhaustiveness was set to 8. The other parameters in Auto Dock Tool were set as default. Depending upon the standard-root-mean-deviation values and binding free energy, the results were clustered. The resulted protein-ligand complex was observed by UCSF Chimera 1.13 software [35] and LigPlot [36]. Based upon the binding interaction and binding score above -8.00 kcal mol-1 top hits were selected for further analysis. To determine the stability of these top four protein-ligand complexes at 50 ns NVT and NPT ensemble molecular dynamics were performed for each complex.

Virtual Screening workflow for identification of SARS-CoV-2 spike receptor inhibitors.
Molecular Dynamics Stimulation Study
The molecular dynamics (MD) simulations were carried out up to 50 ns to analyze the stability of the receptor-ligand complexes of six selected ligands using Desmond software [37, 38]. Before simulation the system was solvated in a TIP3P water model and 0.15 M NaCl was incorporated to neutralize and impersonate the physiological condition. The simulation was run for 50 ns at 315.10 K temperature [39] and 1.01325 bar pressure. The periodic boundary conditions of grid box were enforced in all directions and prepared the simulation chamber by an auto calculated orthorhombic box with buffer dimensions of 10 Å × 10 Å × 10 Å and NPT ensemble. Applying a steepest descent algorithm with 50,000 steps maximum and OPLS-2005 force field, the energy minimization and moderation of the position of the solvated system of the protein-ligand complexes were carried away. The Parrinello-Rahman coupling method was employed [40] for the equilibration of both the NVT and NPT ensemble. In case of NVT ensemble, a constant number of particles (N), volume (V), temperature (T) and coupling constant 0.1 ps for 100 ps were maintained throughout the MD simulation time. In case of NPT ensemble, a constant number of particles (N), pressure (P), temperature (T) and equal coupling constant were maintained. The pressure during MD stimulations was maintained at 1 atm using the Martyna–Tobias Klein pressure bath [41]. The Particle Mesh Ewald method [42] was used with a cut-off distance of 9.0Å to determine the electrostatic interaction. The root mean square deviation (RMSD) of the protein-ligand complex and backbone were calculated with respect to the reference structure (Rref) by using the following equation:
where M = Σi miand ri (t) describes the position of ith atom at time t, after Least Square (LS) fitting of a given molecule to the reference molecule. Root Mean Square Fluctuation (RMSF) is defined as a measure of the fluctuation between the position of ith particle and reference position and determined by the following equation:
where, T denotes the time over which one wants to calculate the average fluctuation and \( {r}_i^{ref} \) indicates the reference position of ith particle.
ADMET Predictions
The absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of the selected molecules was predicted by the open source server pkCSM-pharmacokinetics [31]. The absorption properties of these molecules were analyzed by using several physicochemical and pharmacological parameters like, solubility, human colorectal carcinogenicity, human intestinal absorption (HIA%), skin permeability, in-vivo P-glycoprotein inhibition parameter. By calculating various parameters like, Cytochrome P450 2C19, 2C9, 2D6 and 3A4 enzyme inhibition guideline, metabolic properties of the selected hits were characterized. The drug-likeness properties of these molecules were evaluated using the Lipiniski’s Rule of Five calculation, blood-brain barrier permeability and central nervous system permeability property. The toxicity assessment of the molecules were carried away by evaluating parameters like acute algae toxicity property, Ames test results, carcinogenicity bioassay analysis on mice.
Binding Free Energy Calculations
The average binding free energies (∆G bind) were calculated from the molecular dynamics trajectory. It infers important information about the affinity of the ligands towards receptor. MM-GBSA (Generalized-Born Surface Area) methods based binding energy was calculated using thermal mmpbsa.py script program. Every frames of 50ns molecular dynamics trajectory were used as an input for this script program [46]
RESULTS AND DISCUSSION
SARS-CoV-2 spike receptor binding domain plays a crucial role in viral entry into the host cell, as it directly binds with the ACE2 receptor on the host cell [19]. Recently discovered x-ray crystallographic structure of the SARS-CoV-2 spike receptor created possibility to develop drugs against the SARS-CoV-2 spike receptor binding domain to inhibit viral entry into the host cell. In step 1, the database was filtered to 50,000 compounds depending upon the ADMET properties through pkCSM open source server and filtered compounds were used for structure-based VS studies. In step 2, the VS of ADME filtered hits were performed with the binding site of the SARS-CoV-2 spike-RBD using Autodock Vina [34]. The hits with docking score greater than -8.0 were further used for MD simulations using Desmond package [38]. The interacting active site amino acid residues with interaction distance of selected hits are presented in Table 1. The docking results indicated four hits: 1 (MolPort-000-856-466), 2 (MolPort-002-535-004), 3 (MolPort-005-910-183), and 4 (MolPort-002-225-685) having nice binding affinity toward the active site of spike receptor. The docking score of compounds 1, 2, 3 and 4 was –9.1, –8.9, –8.7 and –8.5 kcal mol-1, respectively, as shown in Table 1. Among the selected compounds, the binding affinity of hit compound 1 was highest and that of 4 was the lowest.
The 2D ligand interactions of these hits were plotted by LigPlot software [36] as shown in Fig. 2. It was found that the binding energy of compound 1 was –9.1 kcal mol-1 and the interacting active site amino acid residues of the spike receptor through hydrogen bonding were CYS-336 (2.94Å, 2.83Å), VAL-362 (3.03Å), ASP-364 (2.84Å), and SER-373 (3.05Å). This compound showed hydrophobic interactions with amino acid residues LEU-335, TRP-436, PHE-342, ALA-363, VAL-367, LEU-368, SER-371, and PHE-374 of the spike receptor binding domain. The binding energy of compound 2 is –8.9 kcal mol-1 and it interacts with spike receptor binding domain amino acid residues through hydrogen bonding were SER-371 (2.98Å), SER-373 (3.15Å, 3.24Å). The hydrophobic interactions of this compound with residues were LEU-335, PHE-338, GLY-339, PHE-342, ASN-343, ASP-364, and LEU-368. The binding energy of compound 3 was –8.7 kcal mol-1 and it interacted with spike receptor binding domain residues through hydrogen bonding were CYS-336 (3.10Å, 3.01Å), GLY-339 (3.09Å), VAL-362 (3.00Å), ASP-364 (3.10Å), SER-373 (3.19Å). This compound exhibited hydrophobic interactions with LEU-335, PHE-338, PHE-342, ASN-343, ALA-363, VAL-367, LEU-368, and SER-371. The binding energy of compound 4 was –8.5 kcal mol-1 and it interacted with spike receptor binding domain through hydrogen bonding with ASP-364 (2.82Å) and hydrophobic interactions with LEU-335, CYS-336, GLY-339, PHE-338, PHE342, ASN-343, ALA-363, VAL-367, LEU-368, SER-371, SER-373, and SER-374.

LigPlot images showing hydrogen bonding and hydrophobic interactions: (1) ligand 1-6M0J interactions; (2) ligand 2-6M0J interactions; (3) ligand 3 – 6M0J interactions; (4) ligand 4-6M0J interactions. Circled residues represent hydrophobic interaction with active site residues.
All structures of selected hits with their binding energies, interacting amino acid residues, and MolPort ID, are shown in Table 2. Compound 1 showed four H-bonds and eight hydrophobic interactions with the active site of spike receptor.
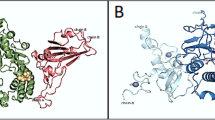
Compound 2 interacts through three H-bonds and seven hydrophobic interactions. Six H-bonds and eight hydrophobic interactions are shown by compound 3. Similarly, one H-bond and twelve hydrophobic interactions are shown by compound 4. Except compound 4, all other hits have fair number of H-bonds and all the hits exhibit good number of hydrophobic interactions. All the hits are deeply inserted into the active site of receptor and the surface diagram of SARS-CoV-2 spike receptor with ligands 1, 2, 3 and 4 are shown in Fig. 3.

Surface diagrams of SARS-CoV-2 spike receptor and ligands 1, 2, 3 and 4 (shown in yellow stick).
In this work, 50-ns MD simulations were performed for apo-protein and selected ligand-spike receptor (6M0J) complexes to analyze stability the of receptor-ligand complex. The calculated root mean square deviation (RMSD), from first to last stimulated trajectories provided the structural and conformational information of the protein-ligand complexes. The average RMSD of apo-protein backbone was ~2.90Å (Table 2) which was stable throughout MD simulations run. Average RMSD of the spike receptor (6M0J)-ligand complexes of 1, 2, 3 and 4 were ~2.97Å, ~3.25Å, ~2.61Å and ~1.96Å and ~2.35Å, respectively, are shown in Fig. 4 and Table 2. The RMSD of receptor-ligand complexes of molecules 3 and 4 were compared with apo-protein, i.e. after binding in the active site these ligands showed a small change in the receptor backbone during the simulation time. The RMSD of ~2.94Å was observed after the ~67 ns in the trajectory of molecule 1-6m0j complex system probably due the presence of higher degree of rotatable bonds or unable to attain the structural flexibility inside the binding cavity of the spike protein. For the rest of the stimulation time the complex system exhibit an average RMSD of ~3.16Å. The molecule 2-6m0j complex system exhibited an undesirable deviation of ~2.99Å in RMSD at ~10.5 ns and for the entire stimulation the average RMSD was observed ~3.55Å. There was a deviation observed at ~29.3 ns with a RMSF of ~3.17Å for molecule 3-6m0j complex system. The highest fluctuation of RMSD (~3.59Å) was observed at ~31.3 ns in the entire trajectory of molecule 3 – 6m0j complex system. But after the fluctuation, the system exhibited complete stability with the spike protein with an average RMSD of ~3.10Å. Molecule 4 exhibited better stability with the protein with an average RMSD of ~1.96Å.

RMSD of apo-protein and protein-ligand complex in 50 ns molecular dynamics simulations. Different colors in the figure refer to apo protein (black), ligand 1-6M0J complex (red), ligand 2-6M0J complex (green), ligand 3-6M0J complex (blue), and ligand 4-6M0J complex (yellow).
The root mean square fluctuation (RMSF) values are useful for identification of any changes in backbone atoms as well as side chain atoms of protein. The average RMSF values for ligand 1, 2, 3 and 4 complex system were observed ~1.79Å, ~1.27Å, ~1.36Å and~1.01Å respectively, and the average RMSF of apo-protein was found to be ~1.47Å. The RMSF values of all the selected hits were almost lower than the apo-protein, although there was a small deviation observed in the residue range 356 – 364 amino acid (AA) for ligand 3 (Fig. 5). Lower the values of RMSF indicate that the reduced random motions and minimum fluctuations of protein backbone and side chain during the simulation run. Compound 1 showed maximum hydrophobic interactions with PHE-342, hydrogen bonding interaction with ASP-364 throughout the simulation time. The active site residues ALA-372, PHE-374, and TRP-436 showed hydrophobic interactions with the compound and ASP-364, ASN-370, SER-371, ALA-372 interacted with the ligand through water-bridge (Fig. S1). Compound 2 showed nice hydrophobic interactions with PHE-342, VAL-367, PHE-374, and TRP-436 and small fraction of hydrogen bonding interaction with SER-373, TRP-436, ASN-437 and small fraction of water bridge interactions with ASN-343, SER-373, ASN-440, and ARG-509 (Fig. S2). Compound 3 showed nice hydrogen bonding interaction with ASP-364 and small hydrogen bonding interaction with PHE-338. Other than this it also showed hydrophobic interactions with PHE-342, PHE-374, TRP-436 and small water bridge interactions with ASP-364 and SER-373 (Fig. S3). Compound 4 interacted with active site residues of spike receptor were ASP-364 through H-bonding, PHE-342, VAL-367 through hydrophobic, and ASP-364 and SER-373 through water bridge. The interaction fractions and type of interactions throughout the simulation time are shown in (Fig. S4). Compounds 3 and 4 showed stable protein-ligand complex during the 50 ns molecular dynamics simulation. Therefore, the two compounds were further studied in 150-ns MD simulations. The average RMSD values of apo-protein, 3-spike receptor complex and 4-spike receptor complex were ~2.90,~ 2.82 and ~2.16Å, respectively, for 150-ns simulation time (Fig. 6). The average RMSD of compound 3 and compound 4 are lower than the average RMSD of apo-protein. Therefore, compound 3 and 4 showed stable binding with spike receptor (Table 3). The binding energy of compound 1-, 2-, 3-, and 4-spike receptor complex predicted by prime are –52.1467, –48.8684, –59.6584, and –55.6087 kcal mol-1. The binding energy values also support the superiority of compound 3 and 4 as SARS-CoV-2 spike receptor inhibitors.

RMSF of apo-protein and protein-ligand complexes during 50 ns molecular dynamics simulations. Different colors in the figure refer to apo-protein (deep blue), ligand 1-protein complex (red), ligand 2-protein complex (yellow), ligand 3-protein complex (green), and ligand 4-protein complex (violet).

RMSD of apo-protein and protein-ligand complex in 150 ns molecular dynamics simulations. Different colors in the figure refer to apo protein (black), ligand 3-6M0J complex (blue), and ligand 4-6M0J complex (red).

Histogram of spike receptor residues interacting with MolPort-000-856-466 (interaction fractions) throughout 50 ns simulation time: (A) MolPort-000-856-466 atom interactions with the spike receptor residues; (B) bar graph showing receptor 6M0J-compound 1, H-bond interactions (green color), hydrophobic interactions (gray violet color), and water bridges (blue color).

Histogram of spike receptor residues interacting with MolPort-002-535-004 (interaction fractions) throughout 50 ns simulation time: (A) detailed MolPort-002-535-004 atom interactions with the spike receptor residues; (B) bar graph showing receptor 6M0J-compound 2, H-bond interactions (green color), hydrophobic interactions (gray violet color), and water bridges (blue color).

Histogram of spike receptor residues interacting with MolPort-005-910-183 (interaction fractions) throughout 50 ns simulation time: (A) detailed MolPort-005-910-183 atom interactions with the spike receptor residues; (B) The bar graph showing receptor 6M0J-compound 3, H-bond interactions (green color), hydrophobic interactions (gray violet color), and water bridges (blue color).

Histogram of spike receptor residues interacting with MolPort-002-225-685 (interaction fractions) throughout 50 ns simulation time: (A) detailed MolPort-002-225-685 atom interactions with the spike receptor residues; (B) bar graph showing receptor 6M0J-compound 4, H-bond interactions (green color), hydrophobic interactions (gray violet color), and water bridges (blue color).
The summary of the ADMET properties of the selected molecules are presented in Table 4. The molecular weights of selected hits 1, 2, 3 and 4 were found to be 397.427, 474.55, 373.405, and 368.385 g mol-1, respectively. All molecules have a probability to permeate through the blood brain barrier. The standard range of logBB value to permeate through the blood-brain barrier lies between the range of 0.3 to –1 [43]. The molecules 1, 2, 3 and 4 exhibited log BB values –0.497, –0.485, –0.79 and –0.565, respectively (Table 4). The maximum human intestinal absorption (HIA%) was observed for molecule 4 (HIA%- 98.8) and the others molecules 1, 2 and 3 exhibited a HIA% of 71.55%, 91.06% and 62.28%, respectively (Table 4). The aqueous solubility plays the important role in drug oral activity. Except molecule 3, all the molecules were found to be insoluble in water. The standard range of LogS for aqueous solubility indicate that LogS >0 is highly soluble; molecules of Logs value 0 to –2 are soluble; molecules with LogS value –2 to –4 are slightly soluble and LogS < –4 indicates insolubility of the molecule in aqueous phase [44]. The LogS value of molecules 1, 2 and 4 were found to be –4.124, –5.481 and –4.109, respectively (Table 4). Whereas, the molecule 3 exhibited moderate solubility in water with a LogS value of –3.759 (Table 4).All the compounds have good skin permeability (logkp < 2.5) with the logkp value –2.75, –2.73, –2.72 and –2.73 for molecules 1, 2, 3 and 4, respectively (Table 3). The above observations indicated that the molecules have good absorption properties. The Cytochrome P450 (CYP 2D6) is an important enzyme and plays the crucial role for the metabolism of more than 25 % of drugs. None of these selected molecules exhibited any inhibitory effect on this enzyme and indicated acceptable metabolic properties. The predicted acute oral toxicity (LD50) value of the selected compounds were found to be 2.538 mol kg-1, 2.239 mol kg-1, 2.856 mol kg-1, 2.559 mol kg-1and 2.519 mol kg-1 (Table 4) for molecules 1, 2, 3 and 4, respectively. The results indicated a moderate toxicity of hit molecules as per the standard value of LD50 (1 mol kg-1).
The drug-likeness properties of any molecule depends upon various physicochemical properties like numbers of hydrogen bond donors (HBD), numbers of hydrogen bond acceptors (HBA), molecular weight, number of rotatable bonds and logP value of that molecule (Lipiniski’s Rule of Five) [45]. For suitable drug like molecule, the HBD and HBA must be in the range of 5 and 10, respectively. The molecular weight must be < 500 gmol-1 and logP value should be less than 5 [45]. In the present study, all the molecules met with the standard criteria for drug-likeness properties. For the molecule 1, 2, 3 and 4 the HBD was observed 1, 2, 2 and 1, the number of HBA was estimated 5 for all these molecules and logP estimated values were found to be 3.33, 4.75, 2.33 and 4.15, respectively (Table 4). The results of physicochemical and pharmacological ADMET parameters of the selected molecules indicated that, all the potential SARS-CoV-2 spike protein inhibitors have suitable drug-likeness properties and good ADMET properties. Further in vivo and ex vivo study may be carried out to justify the predicted properties of the molecule and to observe the potentiality of these molecules against SARS-CoV-2 spike protein.
Based on the ADME properties, docking score and stability of protein-ligand complex during MD study, MMGBSA binding energy compound 3 and 4 were selected as potential spike receptor inhibitors. These two compounds showed stable protein-ligand complex throughout the 150 ns simulation time. All the molecules are commercially available and these two molecules may be used for further in vitro and in vivo studies.
References
A. B. Durojaiye, J. R. D. Clarke, G. A. Stamatiades, and C. Wang, J. Biomol. Struct. Dyn., 2020, 1 – 8 (2020).
Y. Zhou, et al., Antiviral Res., 116, 76–84(2015).
M. Prajapat, et al., Indian J. Pharmacol., 52, 56 – 65(2020).
D. Schoeman and B. C. Fielding, Virol. J., 16(1) 1 – 22 (2019).
N. H. Abd Ellah, et al., Nanomedicine, 15(21), (2020).
A. Guihot, et al., Front. Immunol., 11, 1662 (2020).
W. Tai et al, Cell. Mol. Immunol., 17, 613 – 620 (2020).
C. Liu, Q. Zhou, et al., ACS Cent. Sci., 6, 315 – 331 (2020).
S. Xiu and A. Dick, J. Med. Chem., 63, 12256 (2020).
https:// www.worldometers.info/coronavirus/https://www.worldometers.info/coronavirus/
S. A. Lauer, et al., Ann. Intern. Med., 172, 577 – 582 (2020).
T. T. Y. Lam, et al., Nature, 583, 282 – 285 (2020).
Y. Yang, et al., Virol. J., 17, 117 (2020).
S.F. Ahmed, A. A. Quadeer, and M. R. McKay, Immunol. Stud. Viruses, 12(3), 254 (2020).
A. Savarino, J. Clin. Virol, 34, 170 – 178 (2005).
M. Hoffmann, et al., Cell, 181, 271 – 280 (2020).
Y. Zhou, et al., Cell Discov., 6, 14 (2020).
J. Lan, et al., Nature, 581, 215 – 220 (2020).
B. T. P. Thuy, et al., ACS Omega, 5, 8312 – 8320 (2020).
J. Shang, et al., Nature, 581, 221 – 224 (2020).
M. Bhattacharya, et al., J. Med. Virol., 92, 618 – 631 (2020).
C. A. Devaux, J. M. Rolain, and D. Raoult, J. Microbiol. Immunol. Infect. (2020).
C. M. Yang, et al., Antiviral Res., 67, 24 – 30 (2005).
I. Astuti, Clin. Res. Rev., 14(4), 407 – 412 (2020).
S. Y. Li, et al., Antiviral Res., 67, 18 – 23 (2005).
B. Dinda, I. Silsarma, M. Dinda, and P. Rudrapaul, J. Ethnopharmacol., 161, 255 – 278 (2015).
H. Liu, et al., BioRxiv (2020).
R. Huey and G. M. Morris, J. Scripps Res. Inst., 54 – 56 (2008).
G. E. Rhizobiu, Nucleic Acids Res., 1, 13 – 14(2013).
M. Sivaramakrishnan, et al., Inform. Med. Unlocked, 19, 100331 (2020).
D. E. V. Pires, T. L. Blundell, and D. B. Ascher, J. Med. Chem., 58, 4066 – 4072 (2015).
A. Samdani and U. Vetrivel, Comput. Biol. Chem., 74, 39 – 48 (2018).
K. G. Sprenger, V. W. Jaeger, and J. Pfaendtner, J. Phys. Chem. B, 119, 5882 – 5895 (2015).
O. Trott and A. J. Olson, J. Comput. Chem., 31 (2009)
E. F. Pettersen, et al., J. Comput. Chem., 25, 1605 – 1612(2004).
W. AC, L. RA, T. JM, Protein Eng., 8 (1995).
M.I. Choudhary, M. Shaikh, A. Tul-Wahab, and A. Ur-Rahman, PLoS One, 15(7), e0235030 (2020).
K. J. Bowers, et al., J. Inst. Electr. Electron. Eng. (IEEE), 5, 43 – 43 (2007).
D. J. Evans and B. L. Holian, J. Chem. Phys., 83, 4069 – 4074 (1985).
P. Tangney, J. Chem. Phys., 124, 044111 (2006).
J. Li, A. H. W. Ngan, and P. Gumbsch, Acta Mater., 51, 5711 – 5742 (2003).
T. Darden, L. Perera, L. Li, and L. Pedersen, Structure, 7, 55 – 60 (1999).
S. Vilar, M. Chakrabarti, and S. Costanzi, J. Mol. Graph. Model., 28, 899 – 903 (2010).
J. L. McDonagh, N. Nath, L. De Ferrari, T. Van Mourik, et al., J. Chem. Inf. Model., 54, 844 – 856 (2014).
C. A. Lipinski, Adv. Drug Deliv. Rev., 101, 34 – 41 (2016).
D. K. Yadav, et al. Front Chem., 8, 1 – 14 (2020)
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sarkar, A., Sen, D., Sharma, A. et al. Structure-Based Virtual Screening and Molecular Dynamics Simulation to Identify Potential SARS-CoV-2 Spike Receptor Inhibitors from Natural Compound Database. Pharm Chem J 55, 441–453 (2021). https://doi.org/10.1007/s11094-021-02441-w
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-021-02441-w