
Abstract
The first iodide cluster of molybdenum with the metal cage in the form of a tetragonal pyramid (Bu4N)[Mo5OI13]·THF (I) and cocrystallizate (Bu4N){[Mo5OI13]0.9[Mo6I14]·THF (II) are synthesized for the first time by heating a LiI–I2–Mo mixture in a temperature range of 300–400°С followed by the extraction of the product. Complexes I and II are studied by X-ray diffraction analysis (CIF files CCDC nos. 2063029 (I) and 2063030 (II)). The molybdenum atoms in [Mo5OI13]– form a square pyramid with Mo–Mo distances of 2.67 Å between the basal molybdenum atoms and the Mo–Mo distances equal to 2.72 Å between the apical and basal molybdenum atoms. The oxygen atom is coordinated to the pyramid base (Mo–μ4-O 2.10 Å). The cluster anion [Mo5OI13]– can be presented as the octahedral cluster anion [Mo6I14]2– in which the position of the {MoI}– fragment (d0, 6e) is occupied by the isoelectronic oxygen atom (s2p4, 6e). In the structure of compound II, the [Mo5OI13]– and [Mo6I14]2– cluster anions occupy close atomic positions.
Similar content being viewed by others
INTRODUCTION
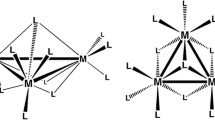
The formation of stable cluster groups with metal–metal bonds of various multiplicity is a characteristic feature of lower molybdenum and tungsten halides [1]. The clusters containing the cluster cores {M6X8}4+ (M = Mo, W; X = Cl, Br, I) are most studied [2]. Active interest in them is presently evoked by the ability of these clusters to exhibit bright red phosphorescence, which can find use in diverse areas [3]. As a rule, the structures of metal cages of the clusters of lower nuclearity (3–5 metal atoms) can formally be removed from the octahedron by the removal of one (square pyramid), two (“butterfly” or square), and three (triangle) vertices (Fig. 1), although the tetrahedral clusters are also known [4, 5]. The pentanuclear clusters with the square pyramid structure are of interest as possible precursors of heterometallic octahedral clusters of the {M5M'X8}4+ type (M' is heteroatom), especially in the case of X = I, since these are the iodide cluster complexes of molybdenum [Mo6I8L6]z (L are terminal ligands, and z is the charge of the coordination sphere) that demonstrate the highest quantum yields and lifetimes [3]. It can be expected that the replacement of one of the molybdenum atom by a heterometal would increase the range of quantitative characteristics of phosphorescence (maximum of emission wavelength, quantum yield, and lifetime). The pentanuclear iodide clusters both in the composition of binary phases [5] and as anionic complexes [W5I13]–/2– and [W5(µ4-C)I13]– [6] were described for tungsten, while only the chloride and bromide clusters [Mo5X13]2– (X = Cl, Br) were synthesized for molybdenum [7–10].

Quasi-homological cluster series from triangular to octahedral clusters.
The first example of the molybdenum iodide cluster (Bu4N)[Mo5OI13]·THF with the metal cage structure as a square pyramid was obtained and studied by X-ray diffraction analysis (XRD) in this work.
EXPERIMENTAL
Organic solvents (tetrahydrofuran (THF), acetonitrile, and ethanol) were purified using standard procedures, and hexane (special purity grade) and diethyl ether were used as received. The starting reagents (Mo, I2, LiI, and Bu4NI) with the purity not lower than analytical grade were purchased from commercial sources. High-temperature syntheses were carried out in evacuated and sealed glass ampules. After the ampules were opened, all manipulations were carried out in air. Anhydrous LiI was stored in a dry box.
IR spectra were recorded on VERTEX 80 (600–100 cm–1) and SCIMITAR FTS 2000 (4000–400 cm–1) spectrometers. Raman spectra were recorded on a LabRAM HR Evolution spectrometer (Horiba, Japan) at the 632.8 nm line of a He–Ne laser. Analysis by energy dispersive X-ray spectroscopy (EDXA) was performed on a Hitachi Tabletop Microscope TM-3000 spectrometer equipped with a QUANTAX 70 system. Elemental analysis was conducted on a Euro EA 3000 CHNS analyzer at the Analytical Laboratory of the Nikolaev Institute of Inorganic Chemistry (Siberian Branch, Russian Academy of Sciences).
Mass spectrometric data were obtained on a 6130 Quadrupole MS, 1260 infinity LC liquid chromatograph (Agilent) combined with a mass spectrometer (LC-MS). The LC-MS analysis was carried out in a range of (m/z) 350–3000 for both positively and negatively charged ions in the scan mode. Electrospray was used as the ionization source. A gaseous nitrogen flow served as a drying agent (350°С, flow rate 7 L/min, pressure on the sprayer (nitrogen) 60 psi, and voltage on the capillary 4000 V). In order to retain weakly bound forms in the mass spectra, the voltage on the fragmentor was zero in all experiments. A solution of the studied compound (5 μL) in deuterated acetonitrile with a concentration of ~10–4 g/mL was injected into the mobile phase (special purity grade acetonitrile) with a flow rate of 0.4 mL/min, sprayed, and ionized. The experimental peaks were compared with the calculated ones, including those by the isotope distribution. The Molecular Weight Calculator by Matthew Monroe program was used for the calculations.
Synthesis of (Bu4N)[Mo5OI13]·THF (I). A mixture of the Mo powder (1.403 g, 0.014 mol), I2 (4 g, 0.016 mol), and LiI (0.767 g, 0.0057 mol) in a dry box was placed in a glass ampule, which was evacuated, sealed, and placed in a muffle furnace, which was heated to 400°С for 4 h. This temperature was maintained for 48 h. After the ampule was cooled to room temperature, the melt was taken out and triturated with a Bu4NI powder (6 g, 0.016 mol), and the resulting mixture was heated to 150°С in a glass ampule for 24 h. The cooled melt was washed with ethanol to remove Bu4NI excess and dissolved in acetone. The residue was extracted with THF, combined with the acetone extract, and evaporated. The solid product was washed with ethanol and diethyl ether on a porous glass filter. The weight of the washed and dried (with an air flow on the glass filter) chocolate-colored powder was 3.330 g. The product contained a mixture of (Bu4N)2[Mo6I14] and (Bu4N)[Mo5OI13], which were separated by the extraction of the octahedral cluster with 100 mL of CH3CN. Upon this procedure, (Bu4N)2[Mo6I14] (1.00 g, 0.35 mmol) got into the solution, and (Bu4N)[Mo5OI13] (2.33 g, 0.97 mmol) remained in the insoluble precipitate, which was highly soluble in THF. The full separation was achieved by double recrystallization from THF with the slow saturation of the solution with hexane vapors to form solvate I. The yield of the pure phase was 1.86 g (27%).
For C20H44NO2I13Mo5 | |||
Anal. calcd., % | C, 9.7 | H, 1.8 | N, 0.6 |
Found, % | C, 9.7 | H, 1.8 | N, 0.6 |
IR (4000–400 cm–1; KBr), ν, cm–1: 2954 s, 2926 s, 2867 m, 1716 w, 1520 w, 1467 s, 1378 m, 1328 w, 1281 w, 1256 w, 1165 m, 1131 w, 1060 m, 1029 w, 991 w, 883 m, 800 w, 735 m, 525 w, 445 s. IR (600–100 cm–1; KBr), ν, cm–1: 442.2 w, 200.5 m, 152.4 w, 121.5 m. Raman spectrum (ν, cm–1): 14.9 s, 23.2 m, 45.8 m, 55.1 m, 99.2 w, 107.6 m, 115.5 m, 122.7 m, 129.3 m, 154.9 s, 208.4 w. Electrospray MS (MeCN, m/z): average 2145.3 ([Mo5OI13]–: calculated 2145.3). EDXA: atomic ratio Mo : I = 5.0 : 13.0.
Single crystals suitable for XRD were obtained by the diffusion of hexane vapors to a solution of (Bu4N)[Mo5OI13] in THF. The single crystals of the cocrystallizate (Bu4N){[Mo5OI13]0.9[Mo6I14]0.1}·THF (II) were obtained by the saturation of a mixture of the clusters in THF with hexane vapors.
XRD of the samples of compounds I and II was carried out at 150 K on a Bruker D8 Venture diffractometer (Center for Collective Use of the Nikolaev Institute of Inorganic Chemistry (Siberian Branch, Russian Academy of Sciences)) equipped with a CMOS PHOTON III detector and an IµS 3.0 microfocus source (MoKα, Montel focusing mirrors). The data were reduced using the APEX3 program package [11]. The structures were solved and refined using the SHELXT [12] and SHELXL [13] programs and the Olex2 software shell [14]. The studied sample of compound I was a joint of two main crystalline domains. The data with allowance for the both domains were not reduced correctly because of a too high difference in intensities of the low- and high-angle reflections. Data reduction for the main domain and the subsequent introduction of the twinning matrix did not allow us to completely avoid the presence of the artifact residual electron density, which resulted in a relatively high final R factor. The structure of compound II exhibited the disordering of the O and {MoI}– fragments of two different clusters. The Bu4N+ atoms should also occupy different positions according to different steric effects of the clusters, but the disordering of Bu4N+ was not introduced because of the low population of alternative atomic positions and the absence of the corresponding pronounced peaks. The crystallographic data and experimental and structure refinement details for compounds I and II are given in Table 1.
The XRD data were deposited with the Cambridge Crystallographic Data Centre (CIF files ССDC nos. 2063029 (I) and 2063030 (II); http://www.ccdc.cam. ac.uk/conts/retrieving.html) and are available at request of the authors.
RESULTS AND DISCUSSION
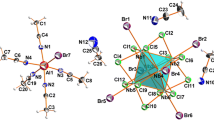
The oxoiodide cluster complex (Bu4N)[Mo5OI13]· THF (I) was synthesized in two stages. At the first stage, a mixture of Mo, I, and LiI was heated at 400°С for 48 h in the ratio assuming the formation of the cluster salt of the pyramidal Li2Mo5I13 cluster, which is presumably formed by analogy to the known [W5I13]2– salts [6]. At the second stage, the cation was replaced by tetrabutylammonium by heating the reaction product with Bu4NI at the melting point of the latter (150°С) for 24 h. The final product turned out to be the oxoiodide cluster isolated from a THF solution as solvate I after double recrystallization. The cluster anion of compound I is shown in Fig. 2. In addition, (Bu4N)2Mo6I14 was isolated, which assumes the formation of Mo6I12 as one of the products of the high-temperature reaction (it becomes the single product when the reaction occurs at temperatures higher than 500°С). It can be assumed that the second product is [Mo5I13]2–, which is oxidized to [Mo5OI13]2– upon extraction in air.

Structure of the cluster anion [Mo5OI13]– (atomic shift ellipsoids of 50% probability). One of two crystallographically nonequivalent anions is shown.
A temperature of 400°С was chosen on the basis of an assumption that the formation of the pyramidal iodide cluster of Mo should be expected at relatively low temperatures in a range of 300–500°С, while the octahedral {Mo6I8}4+ clusters will be formed at the temperatures higher than 500°С. The preliminarily experiments showed that at 300°С Mo reacted with iodine to an insignificant extent (a minor amount of I2 remains in the ampule), although compound I can be isolated as particular single crystals under these conditions. The temperature 400°С seems to be optimal, and the fraction of (Bu4N)2[Mo6I14] will increase with increasing temperature. Under the chosen synthesis conditions, the amount of formed (Bu4N)[Mo5OI13] is by ~2 times higher (by weight) than that of (Bu4N)2[Mo6I14]. On the one hand, no formation of molybdenum iodide clusters of lower nuclearity was observed at Т > 300°С. On the other hand, it is known that the tetranuclear molybdenum cluster [Mo4OI12]2– will be the final product if another molybdenum source (Mo(CO)6) would be used and the reaction with iodine would be carried out at a substantially lower temperature (110°С) in diglyme [15]. An increase in nuclearity in the series [Mo4OI12]2––[Mo5OI13]––[Mo6I14]2– is parallel with a decrease in the oxidation state of molybdenum (+3.0, +2.8, and +2.0, respectively). A correlation between the degree of nuclearity of the cluster cores (or the number of metal atoms in the metal cage of the cluster cores) and synthesis temperatures is observed for the syntheses of the iodide clusters of W and can be described as follows: (a) the clusters of higher nuclearity are formed at higher temperatures and (b) the clusters of lower nuclearity can be transformed into the clusters of higher nuclearity with increasing temperature [5, 6].
The (Bu4N)2[Mo6I14] cluster is readily separable from the mixture due to a substantial difference in solubility of tetrabutylammonium cluster salts in acetonitrile. The (Bu4N)[Mo5OI13] cluster is poorly soluble in acetonitrile but is highly soluble in THF, the phase of compound I can be obtained in the pure state after recrystallization from THF, and the purity of the phase can be monitored by mass spectrometry and elemental analysis. If the phase is insufficiently purified from (Bu4N)2[Mo6I14], the mass spectrum of an acetonitrile solution of the product exhibits the signal with the center at 1176.5 (from [Mo6I14]2– anions) along with the signal with the center at 2145.3 (m/z) (from [Mo5OI13]– cluster anions) (Fig. 3).

Found (designated by dark gray) and calculated (light gray) isotopic distributions for the [Mo5OI13]– particles in an acetonitrile solution of compound I.
The oxoiodide pyramidal [Mo5OI13]– and octahedral anionic [Mo6I14]2– clusters can form the cocrystallizate (Bu4N){[Mo5OI13]0.9[Mo6I14]0.1}·THF (II), the structure of which manifests the disordering of the oxygen atom and {MoI}– fragment (in the base of the square pyramid of the {Mo5} cluster cage) (Fig. 4).

Structure of the cluster anion in cocrystallizate II (atomic shift ellipsoids of 50% probability).
According to the XRD data, the molybdenum atoms in compound I (Fig. 2) form a square pyramid with average Mo–Mo distances of 2.67 Å between the basal atoms and Mo–Mo distances (2.72 Å) between the basal and apical atoms. The oxygen atom is coordinated to the pyramid base, and the average Mo–O distance is 2.10 Å. The cluster anion [Mo5OI13]– can be presented as the octahedral cluster anion [Mo6I14]2– with the detached {MoI}– vertex and the oxygen atom coordinated instead. The Mo–Mo distances in the [Mo5OI13]– anion are identical to the Mo–Mo distances in the {Mo6I8}4+ cluster cores (2.67–2.68 Å) [16, 17]. This indicates that the replacement of the {MoI}– fragment (d0, 6e) by the isoelectronic oxygen atom (s2p4, 6e) does not substantially affect the bonding cluster skeletal orbitals responsible for the formation of metal–metal bonds. In the oxoiodide cluster (Bu4N)2[Mo4OI12] with the “butterfly” structure, the Mo–Mo distances are not equivalent and range from 2.65 Å (shortest bond in the “butterfly” structure {Mo4}) to 2.73 Å (other Mo–Mo bonds), and the Mo–O distances also differ: 2.07 and 2.15 Å [15]. The comparable (with those found in I) metal–metal distances belong to the tungsten clusters (W–W distance in [PrN]2[W5I13] is 2.64 Å [9], while in [W6I14]2– this distance is somewhat longer: 2.67 Å [6]). Moreover, it is interesting that (Bu4N)[Mo5OI13]·THF and (Bu4N)[W5(C)I13]·THF are isostructural, the average W–W distance (2.69 Å) in [W5(C)I13]– coincides with the average Mo–Mo distance in compound I (and is also comparable with the W–W distance in [W6I14]2–), and the average W–C distance (2.16 Å) is somewhat longer (by 0.06 Å) than the Mo–O distance in compound I.
Since the sizes and shapes of the [Mo6I14]2– and [Mo5OI13]– anions are close, it is not surprising that cocrystallizate II is formed in which all crystallographic positions of the atoms of these clusters coincide in pairs except for the O atom and {MoI}– fragment, and the occupancy of the positions of the latter is 90 and 10, respectively.
The analogy in the structures of the octahedral closo-clusters [M6Х14]2– and their derivatives, nido-analogues [Mo5Х13]–/2–, [Mo5OI13]–, and [W5CI13]– (M = Mo, W; X = Cl, Br, I), is explained well if their electronic states are considered according to the scheme of MO filling proposed by Johnston and Mingos [4]. The scheme assumes the presence of 12 bonding orbitals М–М (24 cluster skeletal electrons, 84 valence electrons). The removal of one vertex results in the formation of the pyramidal nido-cluster [Mo5Х13]– having only nine these molecular orbitals (MOs) (18 skeletal electrons, 8 valence electrons). Three MOs of the previous octahedral cluster become antibonding toward the М–М bonding, although one electron can occupy the lowest-lying molecular orbital of these MOs. These orbitals are shown in Fig. 5. Evidently, the inverse coordination to the square face of the {MX} metal fragments, which are isoelectronic to {MoI}– (e.g., {NbI}2–) [18], recovers the scheme of binding with 12 bonding MOs. In the case of nonmetal (O or C), only two orbitals (е set) are appropriate in symmetry for the interaction with р orbitals (e.g., px and py), which additionally provides two bonding orbitals, the capacity of which is four electrons. This does not change the number of skeletal electrons (18) but totally gives 72 cluster valence electrons if μ4-С and μ4-О are considered as donors of four electrons. Unlike C and O, {MoI}– and {NbI}– (formally d6) use the d orbitals, the symmetry properties of which assume binding with both the е and b1 orbitals using all the six electrons. To conclude, it can be assumed that the nido-clusters [Mo5Х13]– would form stable closo-clusters upon the addition of the metal fragments {MX} with the d6 electronic configuration or capped ligands capable of donating four electrons. In the first case, the pentanuclear cluster would act as a unique cluster ligand (nucleophile), whereas the second case is the polynuclear complex (electrophile) exhibiting the dual nature like the well-known cluster boron hydrides [19].

Localized MOs at the base of the pyramid [Mo5Х13]–.
REFERENCES
Sokolov, M.N., Naumov, N.G., Samoylov, P.P., and Fedin, V.P., Comprehensive Inorganic Chemistry II, Oxford: Elsevier, 2013, vol. 2.
Welch, E.G. and Long, J.R., Progress Inorg. Chem., 2005, vol. 54, p. 1.
Mikhaylov, M.A. and Sokolov, M.N., Eur. J. Inorg. Chem., 2019, p. 39.
Johnston, R.L. and Mingos, D.M.P., Inorg. Chem., 1986, vol. 25, p. 1661.
Ströbele, M. and Meyer, H.-J., Dalton Trans., 2019, vol. 48, p. 154.
Franolic, J.D., Long, J.R., and Holm, R.H., J. Am. Chem. Soc., 1995, vol. 117, p. 8139.
Jödden, K. and Schäfer, H., Z. Anorg. Allg. Chem., 1977, vol. 430, p. 5.
Jödden, K., von Schnering, H.G., and Schäfer, H., A-ngew. Chem., 1975, vol. 87, p. 5945.
Ahmed, E., Ahrens, E., Heise, M., and Ruck, M., Z. Anorg. Allg. Chem., 2011, vol. 637, p. 961.
Zietlow, T.C. and Gray, H.B., Inorg. Chem., 1986, vol. 25, p. 631.
Bruker Apex3 Software Suite: Apex3, SADABS-2016/2 and SAINT (version 2018.7-2), Madison: Bruker AXS Inc., 2017.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. 71, p. 3.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Mikhaylov, M., Abramov, P., Novozhilov, I., and Sokolov, M., Z. Anorg. Allg. Chem., 2018, vol. 644, p. 438.
Bruckner, P., Preetz, W., and Punjer, M., Z. Anorg. Allg. Chem., 1997, vol. 623, p. 8.
Kirakci, K., Cordier, S., Roisnel, T., et al., Z. Kristallogr. New Cryst. Struct., 2005, vol. 220, p. 116.
Artemkina, S.B., Tarasenko, M.S., Virovets, A.V., and Naumov, N.G., Russ J. Coord. Chem., 2012, vol. 38, no. 4, p. 257.
Sivaev, I.B., Russ. J. Inorg. Chem., 2020, vol. 65, no. 12, p. 1854. https://doi.org/10.1134/S0036023620120165
ACKNOWLEDGMENTS
The authors are grateful to V.S. Korenev for help in preparing illustrative materials.
Funding
This work was supported by state assignment of the Nikolaev Institute of Inorganic Chemistry (Siberian Branch, Russian Academy of Sciences) in the area of basic research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of in-terest.
OPEN ACCESSThis article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit ttp://creativecommons.org/licenses/by/4.0/.
Additional information
Dedicated to blessed memory of Prof. A.A. Pasynskii
Translated by E. Yablonskaya
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mikhaylov, M.A., Sukhikh, T.S., Sheven’, D.G. et al. First Pentanuclear Molybdenum Iodide Cluster (Bu4N)[Mo5OI13]: Synthesis and Structure. Russ J Coord Chem 47, 560–566 (2021). https://doi.org/10.1134/S1070328421080030
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328421080030