

Abstract
This work aims at investigating the effect that the energy transferred during particle collisions in a milling process entails on solid-gas reactions. For this purpose, the synthesis of Mg(BH4)2 from MgB2 in a pressurized hydrogen atmosphere was chosen as a model reaction. MgB2 was milled under a broad set of milling parameters (i.e. milling times and rotation regimes) and the obtained product thoroughly characterized. By proving the partial formation of Mg(BH4)2, the results of this investigation indicate that the energy transferred to the powder bed by the powder particles during milling is not negligible, in particular when the milling process is protracted for a long period.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Mechanochemistry is the branch of chemistry that studies processes that occur under mechanical energy input. Since the 1860s, the development of mechanochemical techniques increasingly influenced several technological fields among which is powder processing. These techniques found application not only in the production of fine and homogeneous powder mixtures, but also for the synthesis of advanced materials [1].
Mechanochemical processes are usually carried out via the use of high-energy mills e.g. planetary mills, roto-vibrational mills, vibration mills, attritor mills, pin mills and rolling mills.
The milling process takes place in a specifically designed milling chamber. Several different variables influence the milling outcome, e.g. the type of the mill, the material of the grinding media, the ball-to-powder ratio (BTP), the filling extent of the milling chamber, the milling atmosphere, the milling speed and the milling time etc [2].
Upon milling, in milling devices such as planetary mills, mechanical stresses are applied on a fraction of powder particles trapped between grinding media colliding with the wall of the milling chamber. When the mechanically applied stresses overcome the material yield point of the trapped material, complex sequences of local atomic rearrangement and subsequent equilibration stages lead to phenomena of cold work, fracture, and plastic deformation of the material particles. This whole sequence of events can be regarded as a modification of the atomic coordination shells, which locally generates a transient energy excess in the crystal lattice. The region of the solid where the energy excess is located moves away from equilibrium conditions generating a so-called local excited state (LES) [3]. The formation of LESs is considered to be the reason for the unusual chemical behavior observed in processes taking place under mechanochemical input (e.g. enhanced absorption of gaseous phases). In a milling process, the energy transferred as the consequence of collisions between powder particles is not considered. This is mostly due to the expected low relevance that the energy transferred through these collisions has on the overall mechanochemical process when compared to the energy transferred during the impacts of the grinding media with the powders and chamber walls.
In past decades, mechanochemical approaches have been extensively utilized in the synthesis of several hydrogen storage materials [4–24]. In addition, it is expected that mechanochemical methods hold significant unexplored potential for the development of novel materials, e.g. for 'green chemistry' [25].
Among the hydrogen storage materials that can be synthesized via mechanochemical methods, metal borohydrides, owing to their large hydrogen storage capacities, are considered as potential hydrogen storage candidates for mobile and stationary applications [26–32].
In particular, magnesium borohydride (i.e. Mg(BH4)2), owing to a gravimetric hydrogen capacity of 14.9%, a volumetric hydrogen capacity of 147 kg m−3, and an evaluated enthalpy of decomposition of about 40 kJ molH2 −1, is considered as one of the most important boron-based hydrogen storage compounds [33–37]. The uncertainty about the Mg(BH4)2 enthalpy of decomposition is due to the fact that, depending on the applied experimental conditions, Mg(BH4)2 might decompose following different and or multiple decomposition reaction paths, as described in equations (1)–(4)


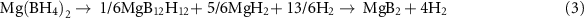

However, at high temperatures (i.e. above 500 °C) the final solid decomposition product of Mg(BH4)2 is MgB2. It must be noticed that during the decomposition process the possibility of releasing small quantities of B2H6 was also reported [5, 33, 36, 38]. In literature four different crystalline magnesium borohydride polymorphs are reported, i.e. hexagonal α (P6122) [34, 39–41], orthorhombic β (Fddd) [36, 42], cubic γ (Id-3a) [43] and trigonal ζ (P-3 m1) [44]. Depending on the utilized synthesis method, it is possible to obtain magnesium borohydride also in an amorphous state [45]. The synthesis of Mg(BH4)2 via wet chemistry methods is challenging since several possible solvent adducts can be formed (e.g. diethyl ether, toluene/heptane and amine solutions) [39]. Solvent-free processes to synthesize magnesium borohydride based on the metathesis reaction of MgCl2 with NaBH4 and LiBH4 performed under mechanochemical input were also reported [33]. The possibility to convert MgB2 into β-Mg(BH4)2 with a conversion yield of 75% was reported to be possible at 400 °C and under a hydrogen pressure of 950 bar [38]. According to the cited literature, the hydrogenation pathway of MgB2 is a multi-step process occurring at molecular scale in which the H2 dissociation at the MgB2 surface is followed by the migration of atomic hydrogen to boron sites, where the formation of stable B–H bonds leads to the generation of Mg(BH4)2. Recently, Pistidda et al reported for the first time on the possibility to form amorphous Mg(BH4)2 through high-energy ball milling (BM) of MgB2 under an atmosphere of hydrogen pressurized to 100 bar. In that work, a conversion yield of MgB2 into amorphous Mg(BH4)2 of roughly 50% was achieved after 100 h of milling under a starting pH2 of 100 bar using a BTP of 30:1 and a rotation speed of 600 rpm [5].
In this work, the synthesis of Mg(BH4)2 via the hydrogenation of MgB2 is used as a model reaction to investigate the effect that the energy transferred during particle-to-particle collisions in a milling process entails. For this reason, several different batches of MgB2 were charged into a milling chamber which was subsequently pressurized with hydrogen at 100 bar and milled (without the use of grinding media) for increasing times under a constant rotation regime (rpm) or under increasing rotation regimes for a fixed time.
2. Experimental
The MgB2 used in this work was purchased from Alfa Aesar in powder form with a purity equal to 95%. The material were mixed (no grinding media were used) under a reactive atmosphere of hydrogen by using a stainless steel pressure chamber from Evico Magnetics mounted on a Fritsch Planetary Mono Mill PULVERISETTE 6. Two MgB2 batches of 15 g were used. The first batch was the as-received MgB2: this material was divided into three parts (5 g for each test), which were then mixed for 10, 25, and 50 h at 550 rpm, respectively, under 100 bar of hydrogen. The second batch was prepared starting from the pre-milled MgB2 (10 h at 550 rpm using a BTP ratio of 10:1) divided into three parts (5 g for each test) and then mixed for 50 h at 350, 450, and 550 rpm, respectively, under 100 bar of hydrogen. For the sake of clarity, the samples are named in the following way: commercial MgB2 is reported as 'as received' and the same material after 10, 25, and 50 h of mixing at 100 bar pH2 are reported as '10 h', '25 h', and '50 h', respectively. Instead, the as-received MgB2 milled for 10 h in a Mono Mill PULVERISETTE 6 is reported as Milled and the same material after 50 h of mixing under 100 bar pH2 at 350, 450, and 550 rpm are reported as 350 rpm, 450 rpm, and 550 rpm, respectively. Powder handling and milling were carried out in dedicated glove boxes under a continuously purified argon flow (<2 ppm of O2 and H2O). Ex-situ powder x-ray diffraction analyses (PXD) were performed in-house using a Siemens D5000 x-ray diffractometer equipped with a Cu source using the Kα radiation (λ = 1.54056 Å) in the Bragg–Brentano geometry. Inside an argon filled glove box (<1 ppm of O2 and H2O), the powder was dispersed onto a silicon single crystal sample holder, airtight-sealed with a Kapton film. The microstructural parameters (i.e. crystallite size and microstrain) were investigated by fitting the diffraction patterns with the program Materials Analysis Using Diffraction (MAUD) using the Rietveld method [46]. The diffractometer instrumental function was determined by fitting with the program MAUD the diffraction pattern of a LaB6 standard reference material provided by the National Institute of Standardss & Technology. The material thermal stability was investigated in the range of temperature between 20 and 500 °C via differential thermal analysis (DTA) using a NetzschSTA409 machine. The DTA analyses were performed using a constant argon flow of 50 ml min−1 and heating rates of 1, 3, and 5 °C min−1 in open Al2O3 crucibles. For each DTA analysis, about 30 mg of material was charged in the crucibles. The evolution of H2 and B2H6 from the samples heated in the DTA equipment at a heating rate of 3 °C min−1 were monitored using a HPR20 Benchtop Gas Analysis System from Hiden Analytical.
The volumetric analysis of all the investigated samples was performed using a Sievert´s type apparatus (Hera, Quebec, Canada). The measurements were performed at a pH2 of 1 bar, heating the material from about 90 °C up to 450 °C (heating rate of 3 °C min−1) and then keeping it isothermally at 450 °C for several minutes. The material morphology was characterized by scanning electron microscopy (SEM), using an EvoMA10 microscope (Zeiss, Germany) equipped with a LaB6 filament. To avoid possible oxygen and moisture contaminations, an in-house built sample holder was used. The samples were charged into the sample holder in an argon-filled glove box and then transported to the SEM.
The composition of the gaseous phase released during the desorption reaction of the investigated samples was analyzed using a Hiden Analytical HAL 201 Mass-Spectrometer, which is coupled with a Netzsch STA 409C Differential Thermal Analysis (DTA-MS). About 5 mg of each sample was placed in a Al2O3 crucible that was heated from room temperature up to 500 °C and then cooled down to room temperature in the DTA apparatus, with a heating rate of 3 °C min−1. The measurements were performed under a continuous Ar flow of 50 ml min−1.
The solid-state nuclear magnetic resonance (SSNMR) investigations were carried out using a Bruker Avance or Avance II 400 MHz spectrometers operating at 400.2 and 128.3 MHz for 1H and 11B nuclei, respectively. The investigated samples were packed in a glove box under Ar atmosphere into cylindrical zirconia rotors with a 4 mm o.d. and an 80 μl volume. The 11B direct excitation magic-angle spinning (MAS NMR) spectra of figure 5 were acquired using a spinning speed of 12 kHz in a dry nitrogen gas flow. The 1H-11B cross-polarization (CPMAS) spectrum of the 550 rpm sample (shown in figure 6) was acquired at room temperature at a spinning speed of 10 kHz, using a ramp cross-polarization pulse sequence with a 90° 1H pulse of 3.80 μs, a contact time of 1 ms, an optimized recycle delay of 3.2 s and a number of scans of 32750. A two-pulse phase modulation decoupling scheme was used, with a radiofrequency field of 69.4 kHz. The 11B chemical shift scale was calibrated through the 11B signal of external standard NaBH4 (at −42.0 ppm with respect to BF3·O(CH2CH3)2). The spinning sidebands are indicated by the asterisk symbol '*'. The boron distribution in the samples 350 rpm, 450 rpm, and 550 rpm (table 2) was determined using the software MestReNova integrating the area enclosed in the center and spinning sidebands of the 11B spectra of figure 5(B). The first-principles calculation of NMR parameters was carried out with the gauge-including projector augmented-wave (GIPAW) [47] and the projector augmented-wave (PAW) method as implemented in the Vienna ab initio simulation package [48]. Exchange and correlation effects were described using the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) functional [49]. The plane wave expansion includes all plane waves within a kinetic energy cut-off of 600 eV. The sampling of the Brillouin zone was performed using a Monkhorst-Pack scheme with a dense k-mesh corresponding to a k-spacing of 0.2 Å. The structures were optimized prior to GIPAW calculations until the forces on each atom were less than 0.01 eV Å−1. GIPAW calculations yielded the principal components of the absolute shielding tensor (σ). The isotropic chemical shift (δiso) could then be determined by comparing a model system computed at the same level to the known experimental shift (δiso e) to obtain the reference isotropic shielding (δiso r). In this study, δiso r is set as −101.39 ppm to fit the δiso e of Mg(BH4)2 and MgB2.
3. Results
The starting materials, and the same after mixing in hydrogen atmosphere, were characterized by means of ex-situ PXD. The acquired diffraction patterns are displayed in figure 1. The patterns of as received and of the same material after mixing for increasing time under hydrogen atmosphere (10 h, 25 h, and 50 h) are shown in the insert A. The patterns of the ball-milled material (Milled) and of the same one after milling (without grinding media) in a hydrogen atmosphere under different rotation regimes (350 rpm, 450 rpm and 550 rpm) are shown in the insert B. All the specimens appear to be composed mostly of crystalline MgB2 plus a small amount of magnesium. Despite the apparently identical composition, for the MgB2 phase it is possible to perceive a progressive increment of the value of full width at half maximum of the material treated under a hydrogen atmosphere. This effect is more marked for the material that underwent BM prior to the hydrogen treatment (figure 1(B)). Considering that the diffraction measurements were acquired all at the same temperature (room temperature) and using the same instrumental setup, the broadness of diffraction peaks can be related only to the size of the MgB2 crystallites and to its lattice strains. In order to quantify variations of the crystallite size and lattice strain, the Rietveld refinement of all the diffraction patterns was carried out. The refinement results are summarized in table 1.

Figure 1. Ex-situ PXD patterns of: (A) as received, 10 h, 25 h and 50 h; (B) milled, 350 rpm, 450 rpm, and 550 rpm; x = Mg and q = MgB2.
Download figure:
Standard image High-resolution imageTable 1. MgB2 crystallite size and microstrain obtained by Rietveld refinement of the PXD patterns reported in figure 1.
| Sample name | Crystallite size (Å) | Microstrain |
|---|---|---|
| As received | 830 ± 40 | 1.0 × 10−3 |
| 10 h | 630 ± 30 | 6.5 × 10−4 |
| 25 h | 630 ± 30 | 6.7 × 10−4 |
| 50 h | 530 ± 25 | 4.8 × 10−4 |
| Milled | 170 ± 25 | 2.5 × 10−3 |
| 350 rpm | 230 ± 10 | 1.5 × 10−3 |
| 450 rpm | 160 ± 10 | 2.2 × 10−3 |
| 550 rpm | 110 ± 5 | 3.6 × 10−3 |
The Rietveld refinement of the as received pattern sets the composition of the starting material to 97 wt% MgB2 and 3 wt% Mg. In these specimens the crystallite size of MgB2 is 830 ± 40 Å and the microstrain is 1.0 × 10−3. Compared to the starting material, the value of the crystallite size of the 10 h sample decreases to 630 ± 30 Å. This value remains unchanged also for the pattern of 25 h. The microstrain decreases to 6.5 × 10−4 for the pattern of 10 h and to 6.7 × 10−4 for the pattern of 25 h. Finally, for the pattern of 50 h the MgB2 crystallite size decreases to 530 ± 25 Å while the microstrain increases to 4.8 × 10−3. As expected, the BM of the material sensibly altered the microstructural properties of MgB2. In fact, the crystallite size of MgB2 in the Milled pattern is 170 ± 25 Å, and the microstrain equals 2.5 × 10−3. These values first increase to 230 ± 10 Å and 1.5 × 10−3 for the pattern of 350 rpm and then decrease to 160 ± 10 Å and 2.2 × 10−3 for the pattern of 450 rpm and 110 ± 5 Å and 3.6 × 10−3 for pattern of 550 rpm.
To study the morphological features of the starting materials and the changes which they supposedly underwent during mixing, all the investigated specimens were characterized by the SEM technique, and the results are summarized in figure 2.

Figure 2. SEM micrograph of: as received, milled, 10 h (A, A1), 25 h (B, B1), and 50 h (C, C1); 350 rpm (D, D1), 450 rpm (E, E1), and 550 rpm (F, F1).
Download figure:
Standard image High-resolution imageThe SEM micrograph of the as received material indicates that most of the MgB2 particles have a size that ranges between 70 and 200 μm whereas the Milled material appears to be constituted mostly from particles of size between 100 and 140 μm. In both cases, smaller fractions of MgB2 particles are observed, i.e. between 1 and 10 μm for the as received material and between 30 and 50 μm for the Milled material. The milling of the as-received MgB2 at 550 rpm for increasing time (10 h, 25 h, and 50 h) leaves the average particle size of the main portion of the sample almost unchanged; however, the formation of an increasing portion of particles of size between 1 and 20 μm is observed. The micrographs acquired for the material milled for 50 h under different rotation regimes (350 rpm, 450 rpm, and 550 rpm) indicate that, in all three cases, the initial average particle size is reduced to 50–70 μm.
In order to understand if the applied mechanical treatments led to the hydrogenation of a fraction of the starting MgB2, the obtained specimens were further characterized by means of volumetric technique. The acquired results are summarized in figure 3. The volumetric curves of the as-received material after milling at 550 rpm for increasing time under hydrogen atmosphere are shown in the insert A. The volumetric curves obtained for the material milled under a hydrogen atmosphere under different rotation regimes (350 rpm, 450 rpm, and 550 rpm) are shown in the insert B. These analyses clearly show that, upon milling, the material absorbed quantities of hydrogen which appears to be related to the magnitude of the applied mechanical process. Upon heating, all the samples of figure 3(A) start to release hydrogen at about 200 °C and reach a maximum rate of release at about 350 °C. This process appears to be complete shortly after reaching the isothermal period at 450 °C. Overall, the desorption process appears to take place in a single step. The amount of released hydrogen is 0.11 wt%, 0.18 wt% and 0.26 wt% for the 10 h, 25 h and 50 h samples, respectively. The volumetric curves of figure 3(B), similarly to those of figure 3(A), show that the hydrogen release takes place in a single step. The sample 350 rpm starts to release hydrogen at about 250 °C and its dehydrogenation is complete when the temperature reaches 450 °C. The overall amount of released hydrogen is 0.7 wt%. The 450 rpm sample starts releasing hydrogen at about 250 °C until an amount of desorbed hydrogen equal to 1.4 wt% is achieved shortly after reaching the isothermal period at 450 °C. The 550 rpm sample starts releasing hydrogen at about 200 °C and reaches a maximum amount of desorbed hydrogen equal to 2.1 wt% after 100 min at 450 °C.

Figure 3. Volumetric analyses of the products of (A) 10 h, 25 h, 50 h, and (B) 350 rpm, 450 rpm, 550 rpm. The measurements were performed at a pH2 of 1 bar, heating the material from about 90 °C up to 450 °C with a heating rate of 3 °C min−1 and then keeping it isothermally at 450 °C for several minutes.
Download figure:
Standard image High-resolution imageIn order to ensure that the observed capacities are due to the release of hydrogen only (boron-containing hydrides might decompose releasing B2H6), the composition of the gaseous phase released upon heating was investigated by means of an MS device (figure 4). The monitored gases were H2 and B2H6. The results of the MS characterizations confirm that the only gas released upon heating is hydrogen. At this point, it is clear that at least a portion of the samples undergoes hydrogenation upon mechanical treatment. Moreover, since no peak belonging to hydrogenated products is visible in the diffraction patterns of figure 1 the hydride phase/phases must be in an amorphous or nanocrystalline state. Both possibilities are likely. In fact, in our previous work [5] the hydrogenated phases formed via BM under hydrogen atmosphere were also not visible in the diffraction patterns; moreover, the amorphization process and/or the reduction of the crystallite size to the nano-range size might be also a direct consequence of the hydrogenation process.
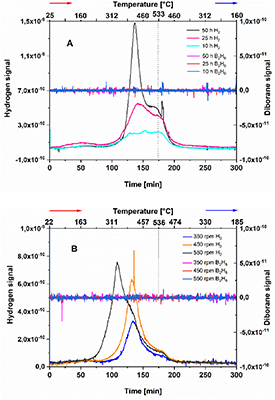
Figure 4. MS signals for H2 and B2H6 measured for samples 10 h, 25, 50 h (A) and 350 rpm, 450 rpm, 550 rpm (B).
Download figure:
Standard image High-resolution imageAiming to thoroughly characterize the composition of the mechanically treated specimens, the MAS NMR technique was used. Indeed, it is well known that SSNMR provides complementary information with respect to diffraction techniques [50] on several types of materials, independently on their form, either crystalline, nano-/microcrystalline or amorphous [51]. The results of this analysis are summarized in figure 5. In the 11B MAS NMR spectra aquired for the 10 h, 25 h, and 50 h samples (figure 5(A)) the main resonance of MgB2 at δ(11B) = 99.3 ppm is visible. It is interesting to notice that in the spectra of 25 h and 50 h a not well-resolved peak starts to be visible at δ(11B) = −38.4 ppm. This resonance is most likely related to the presence of a small fraction of Mg(BH4)2. Unfortunately, due to the low quality of the Mg(BH4)2 band a quantification of the B-distribution between the phases contained in the sample could not be performed. The spectra reported in figure 5(B) clearly show the presence of unreacted MgB2 at δ(11B) = 99.3 ppm, and compared to the spectra of figure 5(A) a better-defined peak at δ(11B) = −38.4 ppm increases with the increasing milling rotation regime. These results undoubtedly confirm the formation of Mg(BH4)2. Spectra acquired at different spinning speeds, 10 and 12 kHz (see figure S1 in the supplementary information (available online at stacks.iop.org/JPENERGY/3/044001/mmedia)), allow an unambiguous assignment of the spinning sidebands of MgB2. This highlighted the presence of an additional peak at 3.5 ppm associated with a new B-containing phase which, if acquired at 12 kHz, resulted overlapped with one of the spinning sidebands of the MgB2 resonance. This signal becomes more pronounced with the increasing of the frequency of rotation in the following order: 350 rpm < 450 rpm < 550 rpm. This explains the increase of the intensity of the associated spinning sidebands (at about +200, −80 and −170 ppm). Since at this moment the nature of this phase is unknown we indicate it as B??? For the spectra displayed in figure 5(B) an attempt to calculate the B-distribution between MgB2, Mg(BH4)2, and B??? was carried out. The possibility that some B2O3 might be present in the analyzed samples was considered; however, after a careful analysis of the spectra we established the quantity of this phase to be neglectable.
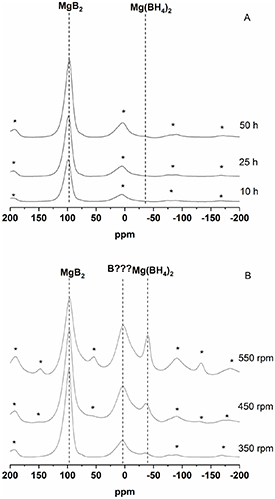
Figure 5. Solid-state 11B MAS NMR spectra acquired using a spinning speed of 12 kHz for (A) 10 h, 25 h, 50 h, and (B) 350 rpm, 450 rpm, 550 rpm. Spinning sidebands are indicated with an asterisk. Spinning sidebands are indicated with an asterisk. The spinning sidebands associated with the MgB2 signal are at about +200, +5, −80 and −170 ppm; the spinning sidebands associated with the B??? signal are at about +200, −80 and −170 ppm; the spinning sidebands associated with the Mg(BH4)2 signal are at about +150, +60 and −130 ppm.
Download figure:
Standard image High-resolution imageFrom the integration of the area enclosed in the center and spinning sidebands of the spectra of figure 5(B) we estimated the following boron distribution (table 2).
Table 2. B atoms distribution in MgB2, B and Mg(BH4)2 for the samples 350 rpm, 450 rpm and 550 rpm.
| Sample | % B atoms in MgB2 | % B atoms in B??? | % B atoms in Mg(BH4)2 |
|---|---|---|---|
| 350 rpm | 90 | 7.7 | 2.8 |
| 450 rpm | 76.8 | 16.5 | 6.7 |
| 550 rpm | 67.6 | 23.3 | 9.1 |
4. Discussion
The results reported in the previous section indicate that the energy transferred during particle-to-particle collisions in a milling process cannot be neglected. In fact, for the chosen model reaction, the transferred energy not only allowed to refine the material microstructure (figures 1 and 2) but it also allowed its partial hydrogenation (figures 3–5). It must be noticed that, despite the relatively favorable enthalpy of hydrogenation (ΔHf ∼ −40 kJ molH2 −1), due to kinetic constrains the hydrogenation of MgB2 under static conditions occurs only under high temperature and high pressure conditions [38, 52, 53].
As reported in the literature, a significant number of gas-solid reactions, typically performed under extreme conditions, can be promoted at room temperature via mechanochemical route by exploiting BM techniques: reactive BM consists in repeated single impact events under reactive atmospheres, such as hydrogen, carbon monoxide and dioxide, which induce structural and chemical transformations by lowering the activation energy barrier necessary to trigger the reaction [54–56]. However, the formation of MgBH4 here described and performed without any grinding media, was quite unexpected. As emerged from the SEM analyses reported in figure 2 and the volumetric analyses of figure 3, the particle reactivity seems to be connected to their dimension. For the hydrogenation processes performed under identical rotation regime, extent of time, and consequently milling chamber temperature (lower than 60 °C), the reduced MgB2 particle dimensions allowed achieving a larger degree of hydrogenation i.e. 2.1 wt% for 550 rpm and 0.26 wt% for 50 h. Interestingly, for the material milled for the same extent of time (e.g. 350 rpm, 450 rpm, and 550 rpm) a clear correlation between the increased number of rpm and the yield of the conversion is seen (table 2). Therefore, not only a mechanical input appears to be necessary to initiate the hydrogenation reaction, but also the magnitude of the energy transferred during the collisions appears to play an important role in the conversion of MgB2 into Mg(BH4)2. To further investigate this aspect, it is plausible to estimate the collision energy for each particle, assuming that their motion during rotation can be approximately treated as the balls in a mechanochemical reactor. For this purpose, it is then possible to exploit the model proposed by Burgio et al [57, 58]. According to this work, the total energy (ΔE*), released by one particle in a mechanochemical reactor, can be calculated by the equation (5):
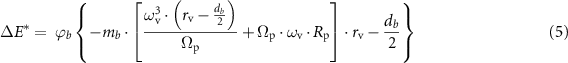
where  is the degree of milling which can be approximated to 1 due to the small diameter of particles, mb
the mass of each particle, i.e. 2.32 × 10−9 kg considering a starting particle dimension of 120 μm, a density of 2.57 g cm−3, and a spherical geometry of the particles, Ωp the rotation velocity of the plate (rad s−1), ωv the rotation velocity of chamber (i.e. 36633 rad s−1, 47100 rad s−1, and 57566 rad s−1 for the sample 350 pm, 450 rpm and 550 rpm, respectively), rv the chamber radius (i.e. 0.0322 m), Rp the plate radius (0,0611 m) and dp the particle diameter (1.2 × 10−4 m). The resulting total energy which each particle dissipated is reported in table 3.
is the degree of milling which can be approximated to 1 due to the small diameter of particles, mb
the mass of each particle, i.e. 2.32 × 10−9 kg considering a starting particle dimension of 120 μm, a density of 2.57 g cm−3, and a spherical geometry of the particles, Ωp the rotation velocity of the plate (rad s−1), ωv the rotation velocity of chamber (i.e. 36633 rad s−1, 47100 rad s−1, and 57566 rad s−1 for the sample 350 pm, 450 rpm and 550 rpm, respectively), rv the chamber radius (i.e. 0.0322 m), Rp the plate radius (0,0611 m) and dp the particle diameter (1.2 × 10−4 m). The resulting total energy which each particle dissipated is reported in table 3.
Table 3. Total energy which each particle of 350 rpm, 450 rpm, and 550 rpm dissipates.
| Sample name | Total energy per particle |
|---|---|
| 350 rpm | 1.39 × 10−8 J |
| 450 rpm | 2.30 × 10−8 J |
| 550 rpm | 3.44 × 10−8 J |
These energy values are 5–6 orders of magnitude smaller than those typically reported for gas–solid reactions activated by mechanochemical input in planetary apparatus [15, 59]. On the other hand, it seems enough to impart the threshold energy level to trigger mechanochemical transformation. Starting from this parameter, it is then possible to additionally determine the velocity possessed by each particle during the mixing treatment, which is estimated to be equal to 3.46 m s−1, 4.45 m s−1, and 5.44 m s−1 for 350 rpm, 450 rpm, and 550 rpm, respectively. The particle velocity represents an important parameter that can be exploited for upscaling the gas–solid reaction, for example in a fluidized bed reactor dedicated to the sustainable synthesis, at room temperature, of complex borohydrides. However, despite this first attempt, a deep characterization of the dynamics of the mixing process combined with the determination of a more accurate threshold energy and shear contributions should be carried out.
Further analyzing the 11B MAS NMR results reported in figure 5 it appears clear that in all the investigated specimens the milling treatment led to the partial conversion of MgB2 into Mg(BH4)2. However, the additional signal at δ(11B) = 3.5 ppm revealed the formation of the unidentified phase B???. In order to better understand the nature of this phase and if it contains hydrogen a 1H-11B CPMAS analysis of the 550 rpm sample was carried out. This analysis is aimed at revealing the presence of 11B nuclei dipolarly coupled with 1H nuclei, i.e. 11B nuclei that present 1H nuclei in their chemical environment (close in space). In other words, all signals displayed in the spectra are associated to H-containing phases. The result of this analysis is reported in figure 6.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
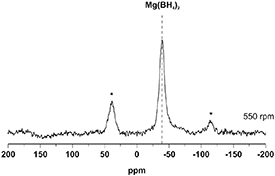
Figure 6. Solid-state 1H-11B CPMAS NMR spectrum of the 550 rpm sample, acquired using a spinning speed of 10 kHz.
Download figure:
Standard image High-resolution image{kind=link}
The 1H-11B CPMAS spectrum of 550 rpm shows only one resonance, at −38.4 ppm, which appeared also in the direct 11B measurements of figure 5 and is due to the formation of Mg(BH4)2. Thus, the signal visible in the direct 11B spectra at 3.5 ppm (figure 5) corresponds to 11B domains that do not incorporate hydrogen atoms. Differently from the previously published works on the hydrogenation of MgB2 [5, 38] no MgB12H12 was detected. This might be a direct consequence of the mild energy transferred to the system which prevents the formed Mg(BH4)2 from decomposing following the reaction path reported in equation (3).
To shed further light on the nature of phase B???, first-principles calculation of the NMR shielding tensors was utilized. 11B NMR parameters for a chosen variety of boron coordination environments were simulated in periodic calculation of bulk systems utilizing density functional theory. The obtained results (table 4) indicate that the unidentified peak lies in the scope of boron icosahedral clusters. All optimized structures used for the NMR calculations and their relative coordinates are reported in figure S2 and table S1, respectively in the supplementary information. Mg–B compounds can be ruled out seeing as their computed δiso values lie out of the range of the signal in question, which is localized around 3 ppm. As table 4 shows, structures containing B12 clusters as building units give chemical shifts in the expected range, which is in agreement with work by others [60, 61]. The structures of both α- and β-rhombohedral boron phases are closely related and contain B12 subunits. Since both allotropes could potentially form under the experimental conditions, they are both likely candidates for the origin of the unidentified signal in the spectrum, although the disordered β-phase is marginally more stable [62]. The lack of diffraction peaks associated to the presence of this boron phase in the PXD analyses of figure 1 might be justified by the existence of this phase in a nanostructured state.
Table 4. GIPAW NMR isotropic chemical shifts (δiso, given in ppm) for symmetry-inequivalent 11B atoms in selected boron-containing systems calculated with the PBE functional.
| Compound | Calculated δiso, |
|---|---|
| MgB2 | B-1: 96.40 (97.2) a |
| Mg(BH4)2 | B-1: −40.0 (−40.0) a |
| B12 icosahedra | B-1: 10.54, B-2: 7.93 (5.6) b |
| B12 cuboctahedra | B-1: 38.50 |
| B36 quasi-planar | B-1: 59.76, B-2: −8.93, B-3: 21.99 B-4: 27.77, B-5: −9.07, B-6: −12.19 |
| B28 icosahedra | B-1: 9.20, B-2: 10.70, B-3: 4.43, B-4: −5.30, B-5: 6.83 |
| MgB7 | B-1: −31.34, B-2: 17.79, B-3: 39.88, B-4: 1.83, B-5: 13.73 |
| MgB4 | B-1: −4.06, B-2: −51.06 |
| MgB3 | B-1: 26.15, B-2: −38.75 |
a Experimental chemical shift from this work. b Computed chemical shift from [60].
When comparing the % of B atoms contained in Mg(BH4)2 (the only B-phase containing hydrogen among the products) in each sample with the amount of hydrogen released during the volumetric measurements (figure 3) it appears as if the values reported in table 2 for the % B atoms in Mg(BH4)2 are slightly underestimated. Although we do not possess a piece of direct evidence, this inconsistency might be justified by the presence of nanocrystalline MgH2 among the reaction products. In fact, the NMR analyses of figures 5 and 6 clearly indicated that during milling an increasing amount of boron is freed into the system due to an incomplete conversion of MgB2 into Mg(BH4)2. Consequently, an increasing amount of Mg should be also present among the reaction products. Considering that in the ex-situ PXD analysis of figure 1 the diffraction peaks of Mg (present already in the as-received material) do not increase in intensity, the additional Mg formed during the milling processes might be in a nanocrystalline state. Thus, it might at least partially react with hydrogen to form MgH2. In fact, it was reported that nanocrystalline Mg can react with hydrogen to form MgH2 already at room temperature [63].
It is important to mention that so far the impact of the energy transferred during particle-to-particle collisions in mechanochemical activated/sustained processes involving powdery materials has been always considered to be irrelevant. In this work, we prove that this energy contribution is not negligible, and to the best of our knowledge, an attempt to quantify it was made for the first time.
5. Conclusion
In this work, the effect of the energy transferred through particle-to-particle collisions during the milling process of MgB2 under a hydrogen atmosphere was studied. The obtained pieces of evidence demonstrated that this energy allowed the partial hydrogenation of the starting MgB2 to form Mg(BH4)2. Owing to a series of MAS NMR measurements and numerical simulations it has been possible to find out that consequently to the mechanochemical treatment a fraction of the starting MgB2 is converted in α- or β-rhombohedral boron. Although, it has not been possible to irrefutably prove it, the formation of Mg and/or MgH2 is also expected. Taking advantage of the kinematic model developed by Burgio et al, an attempt to estimate the collision energy for each particle was made for the first time, to the best of the authors' knowledge. The results of this calculation revealed that for the applied measurement conditions and powder features, the energy transferred during a single particle-to-particle impact is 5–6 order of magnitude smaller than the typical energy transferred during ball-to-powder impact during a similar milling process carried out in the presence of grinding media. This work opens a new frontier on the study of the synthesis of material under low-energy mechanochemical input in reactive atmospheres.
Acknowledgments
The authors of this work are grateful to Dr Nils Bergemann and Dr Pau Nolis for the support in performing part of the presented analyses.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.