
Abstract
Lithium-ion capacitors are envisaged as promising energy-storage devices to simultaneously achieve a large energy density and high-power output at quick charge and discharge rates. However, the mismatched kinetics between capacitive cathodes and faradaic anodes still hinder their practical application for high-power purposes. To tackle this problem, the electron and ion transport of both electrodes should be substantially improved by targeted structural design and controllable chemical doping. Herein, nitrogen-enriched graphene frameworks are prepared via a large-scale and ultrafast magnesiothermic combustion synthesis using CO2 and melamine as precursors, which exhibit a crosslinked porous structure, abundant functional groups and high electrical conductivity (10524 S m−1). The material essentially delivers upgraded kinetics due to enhanced ion diffusion and electron transport. Excellent capacities of 1361 mA h g−1 and 827 mA h g−1 can be achieved at current densities of 0.1 A g−1 and 3 A g−1, respectively, demonstrating its outstanding lithium storage performance at both low and high rates. Moreover, the lithium-ion capacitor based on these nitrogen-enriched graphene frameworks displays a high energy density of 151 Wh kg−1, and still retains 86 Wh kg−1 even at an ultrahigh power output of 49 kW kg−1. This study reveals an effective pathway to achieve synergistic kinetics in carbon electrode materials for achieving high-power lithium-ion capacitors.
Similar content being viewed by others


Introduction
The rapid consumption of traditional fossil fuels promotes an ever-increasing demand worldwide for sustainable high-power energy-storage systems. Unfortunately, lithium-ion batteries (LIBs), which are ubiquitously used, afford only a low power density (<1 kW kg−1) despite their high energy density (>200 W h kg−1), while high-power supercapacitors (SCs) are restricted by a low energy density (<15 Wh kg−1)1. Recently, lithium-ion capacitors (LICs) with contrasting energy-storage mechanisms have received intense interest as a feasible solution to bridge the gap between LIBs and SCs; these LICs utilize battery-type anodes, capacitor-type cathodes, and organic electrolytes2. However, the major issue for LICs is the power capability imbalance between the cathode and anode. In detail, the capacitive cathode exhibits a high rate as the charges are swiftly transferred across the electrical double layer (EDL) near the surface of the electrode; in contrast, the anode stores energy via faradaic processes that are kinetically limited by sluggish Li+ movement within the electrode bulk3. To remedy this kinetic desynchronization, several anode materials with pseudocapacitive behavior have been vigorously investigated in the past few years, including alloy materials (Si and Sn-based compounds4,5), conversion materials (MnO6, VN7, MoS28, and Fe3O49), and intercalation materials (Li4Ti5O1210, V2O511, TiO212, Nb2O513). The large volume change of alloys or conversion anodes causes inferior rate performance and quick capacity fade, while intercalation anodes usually suffer from low Li+ storage ability14. Hence, research into novel anode materials with both a high rate and large capacity remains a large challenge.
Graphene is an allotrope of carbon with sp2-hybridized atoms in a two-dimensional sheet; and this material has been reported as an ideal Li-storing anode due to its high surface area, excellent electrical conductivity, and outstanding chemical stability15. Early studies indicated that a high capacity of 580–650 mAh g−1 can be achieved for graphene nanosheets because Li+ can be anchored on its two sides and edges16,17,18. However, later studies show that Li+ is difficult to stabilize and diffuse in pure graphene without defects, thus greatly hindering its high-rate kinetics and long-term cycling capability. Since electrochemical processes are largely dominated by the interface between the electrolyte and electrode, two mechanisms can account for the performance of graphene anodes: a surface-induced capacitive process and diffusion-controlled faradaic reaction19,20,21. This demands a high electrical conductivity to boost electron transport and a reasonable porous structure to promote ion diffusion, which cooperates to improve the reversible charge-transfer ability across the electrolyte/electrode interface22. To obtain graphene platforms with good electrical and ionic conduction for lithium-ion storage, recent work has mainly focused on the structural regulation and surface engineering of various graphene materials.
Creating reasonably porous graphene is an alternative approach to enhance its ionic conduction. Compared with pure graphene sheets without imperfections, porous and holey graphene exhibits a larger surface area and plentiful defects, which serve as electrochemically active sites to accommodate Li+ within the layers and voids of graphene. Moreover, graphene materials with well-defined porosity in the range of micro/mesopores can not only ameliorate the electrode kinetics of Li+ but also minimize the ion diffusion distance of its multiple channels. For example, Fang et al.23 proved that the electrochemical performance of porous graphene nanosheets can reach a high level of 1040 mAh g−1 at 0.1 A g−1. Using graphite oxide (GO) as a carbon precursor, Ren et al.24 fabricated a porous graphene aerogel with a superior capacity of 1100 mAh g−1 by a hydrothermal reaction, demonstrating the robustness of the porosity modification in escalating the electrochemical performance of carbon electrodes.
Another effective way to enhance the electrical conductivity of graphene is by introducing heteroatoms such as nitrogen25, sulfur26, phosphorus27, and boron28 to boost electrode wettability, the number of surface reactions and the rate of lithium storage. In particular, nitrogen doping is envisaged as an ideal method to incorporate pseudocapacitive properties into graphene for high-performance LIC anodes. The electronegativity difference between nitrogen and carbon (3.04:2.55) leads to polarized carbon atoms, providing “active sites” to influence the spin density and charge distribution of N-doped graphene (NG) nanosheets29,30. This directly catalyzes the electrochemical reactions on the graphene surface, which is usually kinetically favorable over slow ion intercalation in the electrode bulk. An NG film (9 at.%) fabricated by chemical vapor deposition (CVD) exhibits a reversible discharge capacity that is two times that of pristine graphene because of the active sites induced by N doping31. Wang et al.32 synthesized NG (~2 at.%) by the heat treatment of GO in an ammonia atmosphere, and this material demonstrated a high reversible capacity of 900 mAh g−1 (5 times higher than pristine graphene). Wu et al.28 also employed an NH3-assisted annealing method to produce NG (3 at.%) with a high reversible capacity of 872 mAh g−1 after 30 cycles at a low rate of 50 mA g−1. NG also delivers a specific capacity of 684 mAh g−1 after 500 cycles33, indicating that both high capacity and excellent cycling performance are possible through surface engineering. Wang et al.34 designed a holey NG monolith with a high volumetric capacity of 1052 mAh cm−3 by a hydrothermal process combined with annealing, further indicating the synergy of porosity regulation and N doping in enhancing the energy-storage performance of graphene materials. However, most of these N-doped graphene materials are produced by the solvothermal treatment and annealing of GO with a nitrogen precursor or by CVD using metal catalysts at high temperature, which complicate the fabrication process and usually consume large amounts of energy. A simple and feasible preparation method to manufacture graphene materials with reasonable pore structures and N doping is still urgently desired.
Combustion synthesis (CS) is a self-propagated reaction in the mode of rapid combustion waves (>20 cm s−1) after local ignition and has been employed to fabricate ceramics, refractory materials, and intermetallics35,36,37. The primary feature of the CS technique is its very short reaction time because of the fierce heat unleashed from its exothermic processes. Moreover, the products from CS usually display high purity and better crystallinity due to the self-purification effect at ultrahigh reaction temperatures (up to 5000 K)38. As a result, these inherent advantages make CS a promising alternative to fabricate novel graphene materials with tailored physical and chemical structures. However, no related research has been reported since the high reaction temperature of CS makes simultaneous surface engineering and porosity regulation of graphene materials extremely difficult. To solve this problem, we proposed a large-scale, controllable, and low-cost fabrication of N-enriched graphene frameworks (NGFs) based on a magnesiothermic CS with CO2 as the carbon source and melamine (C3H6N6) as the nitrogen source. The abundant nitrogen-containing functional groups in NGF promise good electron transport and fast surface reactions, while the mesoporous frameworks greatly improve ion diffusion through the interconnected channels within the electrode bulk. Density functional theory (DFT) calculations and electrode kinetics analysis confirm that the high doping level of pyridinic N and pyrrolic N leads to increased adsorption energy and a high carrier density. As a result, the good electronic/ionic conductivity, abundance of charge storage sites and enhanced kinetics promote NGFs as promising electrodes for high-power LICs with sizable energy storage. This work offers an effective pathway to achieve designed heteroatom doping of graphene and provides a fundamental understanding of the lithium storage process in N-doped graphene materials, showing high potential to design LICs that synergize with satisfactory energy and power performance.
Materials and methods
Synthesis of NGFs
NGFs were prepared by a magnesiothermic combustion synthesis between CO2 gas and Mg powder with melamine (C3H6N6) as the nitrogen source. Typically, 40 g of Mg powder (500 mesh), 8 g of melamine (analytical grade), and 200 g of MgO powder (300 mesh) were mixed and placed in a reaction chamber with a volume of 20 L. Then, the chamber was evacuated and rinsed with CO2 3 times to expel any oxygen. The final pressure of the chamber was 1 MPa. To start the combustion synthesis, a constant current was exerted through a tungsten coil pre-embedded in the mixture to produce sufficient heat dissipation and initiate the combustion synthesis in a self-sustaining manner. After the reaction, the resulting powder was rinsed in diluted hydrogen chloride to remove the MgO template, filtered thoroughly with deionized water and freeze-dried. The obtained graphene product was denoted as NGF-2. To prove the facile controllability of combustion synthesis in N doping, a series of experiments were further performed with 3 and 15 g of melamine without altering the reaction parameters, and the corresponding products were named NGF-1 and NGF-3, respectively. For comparison, pure graphene (NGF-0) was also prepared without the presence of melamine.
Characterization methods
The morphology of NGF was characterized by scanning electron microscopy (SEM) on a Hitachi S-4800 instrument at an accelerating voltage of 20 kV and transmission electron microscopy (TEM) on a JEOL JEM-2100F instrument at an accelerating voltage of 200 kV. A multifunctional Bruker D8 instrument was employed to record the X-ray diffraction (XRD) patterns with a monochromatic Cu Kα radiation source (λ = 1.54060 Å) at a scanning rate of 5° min−1. To analyze the chemical composition of the samples, X-ray photoelectron spectroscopy (XPS) analysis was performed with a PHI Quantear SXM instrument using an Al Kα radiation source at 1486.6 eV for excitation. Micro-Raman characterization was carried out on a confocal Horiba Jobin Yvon HR-800 instrument with an excitation wavelength of 532 nm. The Brunauer-Emmett-Teller (BET) specific surface area and pore size distribution based on density function theory (DFT) were determined from the nitrogen adsorption/desorption isotherms measured on a Micrometrics ASAP 2020 instrument at 77 K. Electrical conductivity was determined by first pressing SHSG-8 powder under 6 MPa into a platelet with a thickness of 320 μm and then measuring this platelet on a four-probe electrical conductivity instrument. Thermogravimetric analysis (TGA) was performed on a Netzsch TG 209 F3 Tarsus instrument at a heating rate of 5 K min−1 in a N2 and air atmosphere from 50 to 800 °C.
Electrode fabrication and cell assembly
The lithium storage performance of NGFs was evaluated in 2032-type coin cells. To fabricate the electrode, a slurry containing 80 wt.% active material, 10 wt.% acetylene black as the conducting additive, and 10 wt.% polyvinylidene fluoride (PVDF) in methyl-2-pyrrolidone (NMP) was uniformly coated on Cu foil (anode) or Al foil (cathode), which was then vacuum-dried at 120 °C for 12 h and punched into disks with a diameter of 13 mm. NGF half-cells with Li foil as both the counter and reference electrodes were assembled in an argon-filled glove box (MBraun), and a Celgard 2400 separator was used. The electrolyte was 1 M LiPF6 in an ethylene carbonate, diethyl carbonate and dimethyl carbonate (EC/DEC/DMC, v/v/v = 1:1:1) solution. The average mass loading of the NGF electrodes in the half-cell tests was 2 mg cm−2.
A soft-packaged all-graphene LIC full cell with NGFs as both the cathode and anode was assembled. The mass ratio of active materials (cathode/anode) was experimentally set from 1:1 to 4:1 to balance their different capacities. To eliminate irreversible side reactions during solid electrolyte layer (SEI) formation, the anode was prelithiated within the potential window of 0.03–3 V at 0.1 A g−1 for five cycles. All cells were aged at room temperature for 24 h before electrochemical measurement.
Electrochemical measurements
Cyclic voltammetry (CV), galvanostatic charge-discharge (GCD) tests and electrochemical impedance spectroscopy (EIS) were carried out using a BioLogic VMP3 electrochemical analyzer under ambient conditions. Cyclic stability tests were conducted on an Arbin MSTAT4 electrochemical station. EIS analysis was performed from 100 mHz to 100 kHz with an amplitude of 10 mV. The gravimetric specific capacitance C of the LIC (based on the total mass of both the cathode and anode) was calculated from the discharge curve using
where I stands for the constant current used in the GCD test, t is the discharge time, m is the total mass of the cathode and anode, and Vmax and Vmin are the voltages at the beginning and end of the discharge procedure, respectively. The energy density E and power density P (normalized in regard to the total mass of both the cathode and anode) were calculated by these formulae:
Theoretical computations
All computations were performed using density functional theory (DFT) and implemented in the Vienna Ab initio Simulation Package (VASP) with a plane-wave basis set39. The frozen-core all-electron projector-augmented wave (PAW) method was used for the electron-ion interaction40. The exchange and correlation terms were treated by generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional41. The cutoff energy for the plane-wave basis expansion was chosen as 550 eV. Each calculation was allowed to stop when the energy difference between two ionic steps and the lattice force were less than 1 × 10−5 eV per atom and 0.03 eV Å−1, respectively. The energy threshold of electronic self-convergence was 1 × 10−6 eV per atom. A 4 × 4 × 1 supercell with 18 carbon atoms was used to describe the layered graphene structures and simulate the interaction between isolated Li and graphene. A vacuum space of 20 Å was imposed in the lateral direction to avoid interactions between neighboring graphene sheets. To sample the Brillouin zone, a 5 × 5 × 1, 9 × 9 × 1, and 12 × 12 × 1 Γ-centered Monkhorst-Pack k-point mesh was used for the structural optimization, energy and density of state (DOS) calculations, respectively.
Results and discussion
Morphological features of NGFs
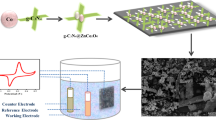
The formation of NGFs via magnesiothermic combustion synthesis is schematically illustrated in Fig. 1a. Low-cost CO2 gas was employed as the carbon source, and MgO powders were used as the hard template to guide the growth of NGFs. After brief ignition, the very large amount of heat dissipated from the reduction of CO2 by Mg powders propels the combustion synthesis of graphene in a self-sustaining manner according to the following formula:

a Schematic illustration of the fabrication of the N-doped graphene framework via combustion synthesis. b, c SEM images of mesoporous and crosslinked NGF-2. d, e TEM images of NGF-2. f–i Carbon, oxygen, and nitrogen elemental mapping images of NGF-2. j Demonstration of the 80 g NGF-2 sample showing the scalability of combustion synthesis.
Moreover, the melamine powder in the system disintegrates (decomposition temperature: 354 °C) into N-containing gases due to the high reaction temperature, which then act as a nitrogen source to promote the in situ N doping of graphene. The whole combustion reaction needs no additional energy input, and the subsequent removal of the MgO template can be easily realized by washing the product with diluted HCl. Figure 1b shows the SEM image of NGF-2, which is composed of a continuously crosslinked and porous graphene framework. The magnified picture in Fig. 1c clearly reveals well-defined graphene edges with plentiful open space between adjacent sheets. It has been reported that such an interconnected morphology would increase interlayer conductive contact and enhance charge transfer throughout the material bulk42. The representative TEM images (Fig. 1d) show an extended graphene sheet in the NGF-2 sample with an abundance of on-plane ripples and crumples. The d-spacing of the (002) plane can be measured as 0.34 nm from the high-resolution TEM image in Fig. 1e, conforming well to the interlayer distance of graphite. The elemental mapping results (Fig. 1f–i) disclose that nitrogen active sites are uniformly doped throughout the frameworks of NGF-2. Moreover, the scalable prospect of combustion synthesis can be revealed from the sample demonstration in Fig. 1j, exhibiting an attractive pathway for the facile, cost-effective and ecofriendly production of heteroatom-doped graphene materials.
To explore the morphological evolution, other NGF materials fabricated with different amounts of melamine were also prepared, and their TEM images are shown in Fig. S1 (Supporting Information). Regarding NGF-0, which was prepared without melamine (Fig. S1a), graphene presents a typical few-layered structure, indicating the successful synthesis of large graphene nanosheets by the magnesiothermic conversion of CO2. The zigzag-like and indistinct edges of NGF-0 suggest a relatively decreased crystallinity (Fig. S1b). This morphological feature varies sharply as melamine is introduced into the reaction system. In the TEM image of the NGF-1 sample (Fig. S1c, d), plentiful and obvious corrugations can be observed on the graphene sheets, and the number of graphene layers tends to increase, which provides solid proof to confirm the enhancing effect of melamine on the crystallinity of graphene. As expected, the TEM images of NGF-2 (Fig. S1e, f) and NGF-3 (Fig. S1g, h) also present sharper graphene edges and more distinct ripples on the layers, corresponding to an increased degree of crystallinity. The above TEM investigations confirm the crucial role of melamine in regulating the microstructural features of NGFs.
Physical and chemical properties of NGFs
X-ray diffraction (XRD) further determines the structure of NGFs (Fig. 2a). For graphene without nitrogen doping (NGF-0), the (002) plane located at 24° corresponds to a wider interlayer spacing of 0.36 nm compared to standard graphite (0.34 nm). This broadened peak suggests reduced crystal dimensions along the [002] direction, indicating a combination of graphitic stacking and random layer arrangement43. Notably, the weak peak related to the (100) diffraction also means a relatively low degree of graphitization in NGF-0. However, this structural property changes substantially as melamine is introduced in the reaction system. The more prominent (002) peak at 25.7° and (100) peak at 43° can be clearly observed in all the NGF samples fabricated with the aid of melamine. Moreover, the downsized full width at half maximum (FWHM) of the (002) peaks from NGF-1 to NGF-3 demonstrate a gradually enhanced crystalline structure, indicating improved electrical conductivity. These results indicate that melamine exerts a critical catalytic effect on the graphitic stacking of carbon atoms in NGF products, which is conducive to the swift transport of electrons through crystalline regions. The Raman spectra of NGFs in Fig. 2b show the typical sp2-related G band at 1578 cm−1 and disorder-induced D band at 1342 cm−1. Regarding carbon materials, the G band is a characteristic attribute of sp2-hybridized graphitic layers and the tangential vibration of the carbon atoms, while the D band is known to arise from disordered carbon atoms at the edge or defective graphitic domains44. The increase in the IG/ID ratio from 0.94 for NGF-0 to 1.15 for NGF-3 indicates an improved stacking order of carbon layers, coinciding with the SEM and TEM results.

a XRD patterns, (b) Raman spectrum, (c) XPS survey and (d) high-resolution N 1s spectrum of NGF samples.
To understand and quantify the elemental composition of NGFs, their surface characteristics were investigated by X-ray photoelectron spectroscopy (XPS). The XPS survey of NGF samples in Fig. 2c reveals that nitrogen atoms are successfully doped into the NGF products with different atomic contents of 1.3, 2.4, and 4.7 at.% for NGF-1, NGF-2 and NGF-3, respectively. The elemental compositions of the NGFs are summarized in Table S1 (Supporting Information). Generally, the chemical bonding between N and C can be divided into three categories: (i) pyrrolic N-5, (ii) pyridinic N-6 and (iii) quaternary N-Q45. N-5 represents pyrrolic N, which contributes two electrons to the conjugated system of the pentagonal ring, while N-6 denotes pyridinic N substituting a carbon atom in the C6 ring and bonds with two sp2 carbon atoms. N-Q is graphitic N located inside the graphitic plane and bonded with three sp2 carbon atoms, which is beneficial for electroconductibility due to the significant change in electron-donor characteristics34. Three characteristic peaks corresponding to N-5 (400.6 eV), N-6 (398.4 eV) and N-Q (401.1 eV) can be recognized from the deconvoluted N 1s spectrum (Fig. 2d), which implies that the N atoms are not only located at the edge of graphene layers but also doped into the basal plane. Some of the pyridinic N-6 atoms are oxidized into N-X (402.6 eV) due to the activation reaction between graphene layers and CO2 at high temperature. The percentages of the different N atoms in NGFs are listed in Table S2 (Supporting Information). Interestingly, graphitic N-Q is found to increase from 27.9% in NGF-1 to 43.2% in NGF-3, indicating that the chemical state of N atoms in graphene products can be facilely regulated by changing the amount of melamine used in the magnesiothermic combustion synthesis system. In addition, C=C (284.6 eV), C-N/C–O (285.7 eV), C-O-C (286.4 eV), C=N/C=O (288.1 eV) and O–C=O (289.9 eV) can be readily distinguished from the deconvoluted C 1 s spectra of NGFs (Fig. S2, Supporting Information). Furthermore, we employed a four-probe method to measure the electrical conductivity of NGF samples, and the results are given in Table S3 (Supporting Information). Due to the improved crystallinity and high N doping, the electrical conductivity of NGF-2 reaches 10524 S m−1, which is an 11% enhancement compared to NGF-0 without N doping (9050 S m−1). This result firmly proves the effectiveness of N doping by melamine in enhancing the electron transport capability of graphene materials via magnesiothermic combustion synthesis.
We employed nitrogen adsorption–desorption isotherms to investigate the specific surface area (SSA) and pore size distribution (PSD) of NGFs. In Fig. 3a, all the isotherms of NGFs show an apparent type IV curve with a large H3 hysteresis (according to IUPAC classification) within 0.45–0.85 P/P0, which is due to the temporary retention caused by adsorption and the delayed evaporation and desorption of liquified N2 confined in the mesopores46. The increase at higher relative pressure (0.85–0.95 P/P0) is ascribed to the unsaturated adsorption of the interlayer spaces between graphene sheets47. The SSAs of NGF-0, NGF-1, NGF-2 and NGF-3 according to the Brunauer–Emmett–Teller (BET) method are calculated to be 724, 517, 493 and 326 m2 g−1, respectively. This phenomenon is ascribed to the increased graphitic stacking order from NGF-0 to NGF-3, which has already been proven by the TEM and XRD tests. Figure 3b shows the PSD of NGFs based on density functional theory (DFT). All the samples present a predominant mesoporous structure, and the average pore size tends to increase from NGF-0 to NGF-3. These enlarged pores in NGF act as ‘ion reservoirs’ to favor ion shuttling during lithiation and buffer the concentration of electrolyte during cell formation48. The comparison of cumulative pore volume (Fig. 3c) also offers direct evidence for the average pore size change of NGFs. Thermogravimetric analysis (TGA) in air demonstrates the good thermal stability of NGF-3 (Fig. 3d), as evidenced by the minimum weight loss until reaching 520 °C. Moreover, the sample loses almost all their weight above 690 °C, verifying its extremely low ash content (~0 %) since no noble metal catalyst is used in magnesiothermic combustion synthesis and the remaining MgO template can be easily removed by acid treatment to achieve a high purity.

a N2 adsorption/desorption isotherms of NGFs, (b) pore size distribution, and (c) cumulative pore volume of NGFs based on DFT, (d) TGA profiles of NGF-2 in air from 50 to 1050 °C.
Electrochemical properties of NGFs as an LIC anode
The lithium-ion storage capacities of the NGF samples were investigated by galvanostatic charge/discharge (GCD) tests in half-cells with Li foil as both the counter and reference electrodes within the potential range of 0.03–3.0 V (vs. Li/Li+); the results are presented in Fig. 4a. All the GCD curves obtained from 0.2 to 3 A g−1 display no obvious potential plateau, which is in sharp contrast to conventional graphite with an insertion-induced flat plateau at lower than 0.2 V vs. Li/Li+. The initial coulombic efficiency of NGF-2 at 0.2 A g−1 is approximately 40%, which can be ascribed to the formation of an SEI layer on the electrode interface induced by electrolyte reduction and irreversible lithium insertion into the vicinity of residual H atoms in the carbon material49. However, after 2–3 charge/discharge cycles at the same current density, NGF-2 can achieve an excellent reversible specific capacity of 1340 mAh g−1 with a coulombic efficiency higher than 96%. The good reversibility of Li-ion storage in NGF-2 can also be revealed from its high capacity of 1361 mAh g−1 when the current rate returns to 0.2 A g−1.

a Charge and discharge profiles of NGF-2 at different current densities. b Comparison of the rate capability of different NGF electrodes. c Cyclic stability test of NGF-2 at 2 A g−1.
From the rate performance of different NGF samples in Fig. 4b, it can be found that the NGF-2 electrode displays the best Li-ion storage performance within a wide range of current densities. Specifically, it delivers high and stable reversible capacities of 1350, 1168, 1061, 985, 858, and 776 mAh g−1 at current densities of 0.2, 0.4, 0.6, 1, 2, and 3 A g−1, respectively. It is worth noting that the capacity retention at 3 A g−1 is approximately 53% compared to that at 0.2 A g−1. The superior capacity of NGF-2 can be reflected when compared with carbonaceous anodes reported recently, including prelithiated graphene (720 mAh g−1 at 0.4 A g−1)50, flash-reduced graphene oxide (660 mAh g−1 at 0.37 A g−1)51, graphene/CNT networks (503 mAh g−1 at 0.05 mAh g−1)52, carbon microbubbles (1000 mAh g−1 at 0.1 A g−1)53, graphitic carbon nanorings (1237 mAh g−1 at 0.4 A g−1)54 and N-doped carbon nanofibers (1280 mAh g−1 at 0.1 A g−1)55. A cyclic test of NGF-2 was carried out to evaluate its electrochemical stability during charge/discharge cycling. As shown in Fig. 4c, the capacity can maintain a high capacity of 825 mAh g−1 at a high current density of 2 A g−1 after 300 cycles, corresponding to an excellent capacity retention of 97.4% and a minimum capacity loss of 0.008% per cycle. Moreover, the coulombic efficiency can quickly reach nearly 100% after the first few cycles and remains unchanged throughout the cycling test. This result clearly indicates that no side reactions or irreversible faradaic deposition occurs after the successful formation of the SEI film.
Kinetic study of lithium storage in the NGFs
Intrigued by the high rate and cycling performance of NCF-2, we conducted kinetic analysis to investigate its Li+ storage to achieve a clear understanding of the structure-performance relationship. Fig. S3 (Supporting Information) shows the first three CV curves of NGF-2 at a scan rate of 0.1 mV s−1. The overlap of the 2nd and 3rd cycles reveals the good reversibility of Li storage after the 1st cycle, suggesting that a stable solid electrolyte interface (SEI) film is formed during the initial cycle. In Fig. 5a, even at high scan rates, the peaks related to lithium intercalation/deintercalation maintain their original shape and become broader compared to peaks at low scan rates. Due to diffusion-related polarization, the cathodic peaks become steeper, and the anodic peaks shift to higher voltages with an increased scan rate.

a CV curves at different scan rates. b The relationship between logarithm of peak current and scan rates based on the CV analysis. c The separation of surface capacitive current (shaded area) from total current (red line) at the scan rate of 1 mV s−1. d The capacitive and diffusive capacity histogram at different scan rates. e GITT profiles of discharge and charge process (inset shows the corresponding diffusion coefficients as a function of depth of lithiation and delithiation). f Schematic illustration showing the initial stage of capacitive and bulk storage in NGF-2.
Generally, the relationship between the peak current and scan rate can be expressed by a power law56,57:
where i is the peak current, ν stands for the scan rate, and a and b are system-dependent coefficients. From the relationship of log(i) against log(ν), the value of b can be calculated. When the value of b is 0.5, it indicates an ideal faradaic intercalation process, while a value close to 1 represents a surficial charge storage procedure without a diffusion process. Figure 5b displays the relationship between the logarithm of the peak current and scan rate during the delithiation process. Regarding NGF-2, the well-defined linear relationship and the b value of 0.86 strongly imply fast surface-governed kinetics.
The capacitive charge in the NGF-2 electrode can be quantified according to Dunn’s approach. The current response at a specific potential is the combination of capacitive surface effects (k1 v) and diffusion-controlled Li+ intercalation (k2 v1/2)7,58:
in which i(V) is the current, v is the scan rate, and k1 and k2 are constants. In Fig. 5c, the percentage of the capacitive process (blue shade) in the total current response at a scan rate of 1 mV s−1 is calculated to be as high as 72%. Moreover, with increasing scan rates (Fig. 5d), the capacitive contribution gradually becomes 82%. This increase in the capacitive charge storage of NGF-2 strongly benefits its fast kinetics when cycling in LICs.
Galvanostatic intermittent titration (GITT) measurements were performed to understand the fast lithium diffusion kinetics in the bulk of NCF electrodes. Figure 5e shows the electrode potential response during GITT and the dependence of the diffusion coefficient (Dk) on the depth of discharge/charge, which are obtained based on Fick’s second law and Eq. (4):
in which τ is the current pulse duration, ΔES represents the change in the quasi-thermodynamic equilibrium potential before and after the current pulse, ΔEτ stands for the potential difference during the current pulse, and me and S are the electrode mass load and geometric area of the electrode, respectively. The material density Me/Ve (Ve is the molar volume of electrode materials and Me is the molar mass of carbon) can be inferred according to Eq. (5):
where Vt (cm3 g−1) corresponds to the total pore volume calculated from the N2 isotherm, and ρc is the true density of carbon (2 g cm−3). The results confirm a higher Dk for NGF-2 than NGF-0 during the lithiation and delithiation processes, revealing that reasonable N doping in graphene is beneficial for faster lithium diffusion.
The outstanding surface-governed electrochemical performance of NGF-2 is strongly attributed to its unique structural and chemical features, as illustrated in Fig. 5f. (i) The graphene framework in NGF-2 greatly reduces the contact interlayer resistance to promote electron transfer in the electrode matrix, whereas the large number of mesopores can serve as multiple channels for fast ion transport59. (ii) The large surface area leads to a sufficient electrode/electrolyte interface to absorb Li+ and promote a rapid charge-transfer reaction to reduce the ion transport length. (iii) High-level N doping enlarges the surface-dominant reaction of lithium ions, boosts fast ion shuttling across the graphene layers and increases the lithium storage capacity60. Moreover, these N-rich sites in NGF-2 also enhance the electrical conductivity to 10555 S m−1, which additionally contributes to the exceptional electrochemical reactivity and surface-dominated energy-storage performance.
To unravel this surface reaction mechanism and elucidate any phase change of the NCF-2 electrode during the Li+ insertion and extraction processes, in situ XRD patterns were collected between 0.03 and 3.0 V to monitor the structural evolution of the anode. The (002) peak located at 25.7° was carefully examined to probe the interaction between the Li+ and carbon lattices. In the 1D XRD spectra (Fig. 6a) of NCF-2, the similar shape and dimension of the XRD patterns in one complete charge and discharge cycle indicate the robust structural features of the NCF-2 anode, while the unchanged (002) peak position suggests that the Li storage reaction is mainly confined on the electrode surface because a large amount of Li insertion would inevitably cause a substantial peak shift toward lower angles61. The 2D XRD spectra help to disclose more information on the structural change of the electrode. In Fig. 6b, the contour maps of two lithiation/delithiation cycles in the selected 2θ angle ranges clearly show this progressive structural transition process in the carbon lattice. As discharging proceeds, the (002) peak gradually decreases in intensity and exhibits no obvious peak position change, indicating that Li+ is mainly stored by the adsorption mechanism on the surface of graphene to cause a less graphitic character. During the charging process, the structural evolution is basically inverted compared to the discharging process and finally returns to the delithiated state, revealing a reversible reaction. The structural evolution of NCF-2 during lithiation/delithiation in the second cycle is exactly the same as that in the first cycle.

a 1D XRD of the NGF-2 anode in one cycle and the magnified range showing the (002) peak of graphene. b 2D XRD versus galvanostatic discharge/charge time for two continuous cycles.
To gain further insight into the role of different N heteroatoms on Li adsorption behavior, we performed first-principles calculations based on density functional theory (DFT) using the Vienna Ab initio Simulation Package (VASP)39. All the geometric configurations of pristine graphene and graphene doped with graphitic (NQ), pyridinic (N6), and pyrrolic (N5) N heteroatoms are optimized (Fig. 7a–d), and the adsorption energy (ΔEads) of a Li atom is calculated as follows62:
where Etot is the total energy of the compound, ELi represents the energy per Li atom, Ecarbon stands for the energy of the carbon materials, and n denotes the number of Li atoms in the compound. Compared with pristine graphene (−1.18 eV) and NQ-doped (−0.74 eV) graphene, N5- and N6-doped graphene exhibit the lowest ΔEads of −3.62 and −2.07 eV, respectively, indicating that pyrrolic and pyridinic nitrogen heteroatoms have strongly enhanced the affinity to Li compared to graphitic nitrogen. According to the XPS results in this work, NGF-2 is endowed with higher pyrrolic and pyridinic N-doping levels than NGF-1 and NGF-3, which reasonably leads to a remarkably enhanced capacity and excellent rate capability. This phenomenon highlights the important role of selective N doping for improving the Li storage performance of graphene.

a–d Top view of the optimized molecular structure for a single lithium atom adsorbed on pristine graphene and graphene with graphitic pyridinic and pyrrolic N doping. e–h Ddifferential charge density of pristine and N-doped graphene adsorbed with lithium and the corresponding adsorption energy. Blue and yellow contours indicate the reduced and augmented charge density with an isosurface value of 0.001 e Bohr−3, respectively. i–l 2D cross-sectional differential charge density of lithium adsorbing on graphene without and with N doping. m–p The projected densities of states of pristine and N-doped graphene showing the s and p orbitals of carbon and nitrogen atoms and the s orbital of lithium atoms.
Isosurface plots of differential charge density were provided to understand the interaction of Li adsorbed on various N heteroatoms in graphene (Fig. 7e–h). The charge density tends to accumulate around pyridinic and pyrrolic N, which proves the effectiveness of our strategy. By subtracting the charge density of bare Li and carbon atoms from the compounds, a net gain of electronic charge emerges in the intermediate region between Li atoms and carbon layers, signifying an ionic bonding nature because of the charge transfer from the adsorbed Li to its nearest neighboring N atoms. The 2D cross-sectional differential charge density in Fig. 7i–l also clearly reveals this trend, in which the electron density around the pyrrolic N atom is the thickest among the different types of doped N. In fact, the electron-rich region around the N heteroatoms in graphene will cause the electron-deficient regions to gain electrons from Li atoms, which can be validated from the electron localization function (ELF) of pristine and N-doped graphene in Fig. S4a–h (Supporting Information). Bader charge analysis (Fig. S5, Table S4, Supporting Information) also reveals a substantial electron transfer between Li and the nearest carbon atoms, which quantitively confirms that the valence electron density of Li atoms has transferred almost totally to the bonding nitrogen due to the ionic nature of bonding.
The projected density of states (PDOS) of various N-doped graphene adsorbed with Li is computed to further interpret the electronic structure. The partially occupied Li 2s peak (Fig. 7m and Fig. S4i, Supporting Information) supports that the interaction between Li and graphene is dominated by charge transfer and an ionic bond is formed63. Regarding Li absorbed on graphene doped with graphitic N atoms, the increased DOS around the Fermi level (Fig. 7n and Fig. S4j, Supporting Information) points to a metallic bandgap and thus higher electronic conductivity. Particularly, the DOS near the Fermi level of pyridinic N- (Fig. 7o and Fig. S4k, Supporting Information) and pyrrolic N- (Fig. 7p and Fig. S4l, Supporting Information) doped systems diminishes to nearly zero due to the electron transfer from Li to NGFs, suggesting a stronger interaction between carbon and lithium atoms. The DFT analysis above reveals that proper pyrrolic and pyridinic nitrogen doping in graphene can efficiently enhance Li adsorption, theoretically validating the boosted lithium storage of NGFs. Therefore, it is highly expected that NGFs can earn a place as superior LIC anodes for fast lithium storage.
Fabrication of an all-graphene LIC using NGFs
Before the fabrication of LIC full cells, it is necessary to find a suitable cathode with both high capacity and rate capability. Typically, a high-performance cathode should exhibit a large surface area accessible to electrolyte ions and high electrical conductivity to boost charge transfer64. Unfortunately, commercial activated carbon (AC) cathodes usually provide undesirable capacity because the tortuous channels (typically less than 0.5 nm) in AC restrict the available surface area. The NGF-0 sample prepared in this work exhibits the best specific surface area (724 m2 g−1) among NGFs, and more importantly, it shows a highly porous structure and good electrical conductivity, which can simultaneously promote ion and electron transport. Therefore, we characterized the electrochemical behavior of the NGF-0 cathode in half-cells. Figure 8a displays the perfect rectangular shape of the CV curves at different scan rates within a wide potential window of 2-4 V vs. Li/Li+, indicating low electrode polarization as well as efficient capacitive energy storage at the electrode/electrolyte interface. The GCD results in Fig. 8b demonstrate highly linear profiles at various current densities due to the inherent nonfaradaic capacitive properties. At 0.5 A g−1, NGF-0 can deliver a high capacity of 82 mAh g−1, which represents a large improvement over commercial AC (<50 mAh g−1). Indeed, among the NGFs prepared in this work, NGF-0 also shows the highest specific capacity (Fig. S6, Supporting Information). The rate performance of NGF-0 in Fig. 8c also offers a good capacity of 61 mAh g−1 even at a higher current density of 8 A g−1. The Nyquist plot (Fig. 8d) in the low-frequency region gives a nearly vertical line perpendicular to the real axis, suggesting ideal capacitive characteristics.

a CV curves of the NGF-0 cathode at various scan rates from 2 to 4 V vs. Li/Li+. b Charge and discharge profiles. c Rate performance of NGF-0 from 0.5 to 8 A g−1. d Nyquist plot of NGF-0.
Based on the above considerations, we employed a high-rate NGF-0 cathode and high-capacity NGF-2 anode to fabricate an all-graphene NGF-0//NGF-2 LIC. The mass ratio of the cathode and anode (rc/a) was experimentally optimized from 1:1 to 4:1 to balance their different capacities and kinetics (Fig. S7, Supporting Information). Before testing the electrochemical performance, galvanostatic prelithiation of the NGF-2 anode was conducted at 0.5 A g−1 for five cycles in 1 M LiPF6 electrolyte from 0.03 to 3 V vs. Li/Li+ to eliminate the irreversible capacity and stabilize the anode potential. As displayed in Fig. 9a, NGF-0//NGF-2 presents quasi-rectangular CV curves at different scan rates from 5 to 20 mV s−1 within the voltage window of 2–4 V, indicating good electrochemical reversibility and high rate performance. The slight deviation of the CV curves from the ideal rectangular shape is ascribed to the synergistic energy-storage mechanisms of the capacitive cathode and faradaic anode. All the symmetric GCD profiles (Fig. 9b and Fig. S8, Supporting Information) suggest low electrode polarization at various current densities. The rate capability is shown in Fig. S9 (Supporting Information), which shows capacitances (normalized to the total mass of active materials) of 75 and 47 F g−1 at 0.5 and 20 A g−1, respectively.

a CV profiles of all-graphene NGF-0//NGF-2 at different scan rates with a wide voltage range of 1–4 V. b Charge and discharge profiles at various current densities. c Ragone plot of NGF-0//NGF-2 LIC compared with the reported representative hybrid capacitor. d Schematic of NGF-based cathode and anode enhancing the ionic diffusion and electron transport in NGF-0//NGF-2 LIC. e Electrochemical stability of NGF-0//NGF-2 at 4 A g−1 for 10,000 cycles. The inset shows the Nyquist plot (left) and Bode diagram (right).
From the GCD results, the energy density and power density based on the total mass of active materials on electrodes can be calculated. Specifically, this LIC can deliver a high energy density of 147 Wh kg−1 at a power density of 1.19 kW kg−1 and still preserve a good energy density of 86 Wh kg−1 even at an ultrahigh power output of 48.9 kW kg−1. A summary of the energy and power densities of state-of-the-art LICs is plotted in the Ragone plot (Fig. 9c), clearly illustrating the superior energy and power performance of NGF-0//NGF-2 to recently reported carbon-based LICs, including carbon sphere//Si/carbon65, graphene/AC//graphene51, N-doped AC//graphite66, AC//hard carbon67, and AC//rGO68. This energy-storage performance is also better than that of many LICs utilizing Li4Ti5O12 and transitional metal compound anodes, such as graphene//Li4Ti5O1269, AC//Li4Ti5O1270, activated polyaniline-derived carbon//VN/rGO7, pyridine-derived porous N-doped carbon//TiC58, AC//TiO2/graphene71, and porous N-doped AC//MnO/graphene72. More comparisons of the electrochemical performances of NGF-0//NGF-2 with various LIC systems are provided in Table S5 (Supporting Information), which validates the efficient energy-storage ability of NGF electrodes.
To better understand the fast ion transport properties, the energy-storage mechanism of NGF-0//NGF-2 is illustrated in Fig. 9d. In the charge process, the adsorption of PF6¯ ions onto the porous surface of the cathode elevates the voltage of this capacitor-type electrode; moreover, Li+ ions are intercalated and absorbed on the surface of the anode to cause the potential in this battery-type electrode to decrease. Similarly, the reverse movement of ions occurs in the discharge process to deliver the stored energy. As a result, a good capacity retention of 87% is achieved after 10,000 cycles at a high current density of 4 A g−1, and the coulombic efficiency remains at ~100% during the whole cycling stability test (Fig. 9e). Moreover, the electrochemical impedance characterization also points to fast Li storage in the LIC. In the Nyquist diagram (inset in Fig. 9e), the downsized semicircle in the high-frequency region and vertical line in the low-frequency part validate the mitigated charge-transfer resistance and ideal capacitive energy storage. The low internal resistance Rs (3.94 Ω) and equivalent series resistance (ESR, 8.64 Ω) can be ascribed to the high electrical conductivity and mesoporous feature of NGF electrodes, which not only provides a pathway for the fast transport of electrons but also enhances the diffusion capability of ions through the pore channels in the electrode bulk. The Bode plot clearly demonstrates a phase angle of nearly 90° in the low-frequency range, typical of capacitive energy-storage features. In addition, NGF-0//NGF-2 exhibits a high characteristic frequency f0 of 0.095 Hz at 45°, corresponding to a time constant τ0 of 10.52 s. This is the shortest time to release the energy stored in the LIC cell with an efficiency of >50%. The above study on the Li storage behavior of NGFs with proper N doping exemplifies their special advantage as high-performance electrode materials for LICs.
Conclusion
In summary, we developed a scalable, green and low-cost magnesiothermic conversion of CO2 to synthesize mesoporous NGFs with excellent electrical conductivity (10524 S m−1), a high doping of nitrogen (4.7%) and a controllable specific surface area (326–724 m2 g−1). Due to the synergistic electrode kinetics, these NGFs exhibit sufficient external active sites and rapid electrochemical kinetics for capacitive-dominated lithium-ion storage, demonstrating high capacity performance (1361 mA h g−1 at 0.1 A g−1) as well as outstanding cycling stability (capacity loss of 0.008% per cycle). Using these NGFs as both the cathode and anode, an all-graphene LIC was assembled to operate within a cell voltage of 2–4 V. The energy density of the NGF-based LIC is up to 151 Wh kg−1 at a power density of 1.2 kW kg−1 and still retains an energy density of 86 Wh kg−1 when the power output reaches an ultrahigh level of 49 kW kg−1. The present work opens a new path for the large-scale fabrication of heteroatom-doped graphene materials with enhanced electrochemical performance. Thus, these NGFs may find application as promising electrode materials in future high energy density and high-power output storage devices.
References
Winter, M., Barnett, B. & Xu, K. Before Li ion batteries. Chem. Rev. 118, 11433–11456 (2018).
Dubal, D. P., Ayyad, O., Ruiz, V. & Gomez-Romero, P. Hybrid energy storage: the merging of battery and supercapacitor chemistries. Chem. Soc. Rev. 44, 1777–1790 (2015).
Li, B. et al. Electrode materials, electrolytes, and challenges in nonaqueous lithium-ion capacitors. Adv. Mater. 30, 1705670 (2018).
Zhu, C. et al. Construction of SnO2-graphene composite with half-supported cluster structure as anode toward superior lithium storage properties. Sci. Rep. 7, 3276 (2017).
Halim, M. et al. Pseudocapacitive characteristics of low-carbon silicon oxycarbide for lithium-ion capacitors. ACS Appl. Mater. Interfaces 9, 20566–20576 (2017).
Wang, H. et al. Hybrid device employing three-dimensional arrays of mno in carbon nanosheets bridges battery-supercapacitor divide. Nano Lett. 14, 1987–1994 (2014).
Wang, R. T., Lang, J. W., Zhang, P., Lin, Z. Y. & Yan, X. B. Fast and large lithium storage in 3D porous VN nanowires-graphene composite as a superior anode toward high-performance hybrid supercapacitors. Adv. Funct. Mater. 25, 2270–2278 (2015).
Wang, R. et al. Engineering layer structure of MoS2-graphene composites with robust and fast lithium storage for high-performance Li-ion capacitors. Energy Storage Mater. 9, 195–205 (2017).
Zhang, F. et al. A high-performance supercapacitor-battery hybrid energy storage device based on graphene-enhanced electrode materials with ultrahigh energy density. Energy Environ. Sci. 6, 1623–1632 (2013).
Naoi, K., Ishimoto, S., Isobe, Y. & Aoyagi, S. High-rate nano-crystalline Li4Ti5O12 attached on carbon nano-fibers for hybrid supercapacitors. J. Power Sources 195, 6250–6254 (2010).
Chen, Z. et al. High-performance supercapacitors based on intertwined CNT/V2O5 nanowire nanocomposites. Adv. Mater. 23, 791–795 (2011).
Kim, H.-K. et al. TiO2-reduced graphene oxide nanocomposites by microwave-assisted forced hydrolysis as excellent insertion anode for Li-ion battery and capacitor. J. Power Sources 327, 171–177 (2016).
Deng, B. H., Lei, T. Y., Zhu, W. H., Xiao, L. & Liu, J. P. In-plane assembled orthorhombic Nb2O5 nanorod films with high-rate Li+ intercalation for high-performance flexible Li-ion capacitors. Adv. Funct. Mater. 28, 1704330 (2018).
Aravindan, V., Gnanaraj, J., Lee, Y. S. & Madhavi, S. Insertion-type electrodes for nonaqueous Li-ion capacitors. Chem. Rev. 114, 11619–11635 (2014).
Raccichini, R., Varzi, A., Passerini, S. & Scrosati, B. The role of graphene for electrochemical energy storage. Nat. Mater. 14, 271–279 (2015).
Wang, G., Shen, X., Yao, J. & Park, J. Graphene nanosheets for enhanced lithium storage in lithium ion batteries. Carbon 47, 2049–2053 (2009).
Chou, S.-L. et al. Enhanced reversible lithium storage in a nanosize silicon/graphene composite. Electrochem. Commun. 12, 303–306 (2010).
Yoo, E. et al. Large reversible Li storage of graphene nanosheet families for use in rechargeable lithium ion batteries. Nano Lett. 8, 2277–2282 (2008).
Yang, J. et al. Enhanced capacity and rate capability of nitrogen/oxygen dual-doped hard carbon in capacitive potassium-ion storage. Adv. Mater. 30, 1700104 (2018).
Wen, Y. et al. Expanded graphite as superior anode for sodium-ion batteries. Nat. Commun. 5, 4033 (2014).
Zhan, C. et al. High-performance sodium-ion hybrid capacitors based on an interlayer-expanded MoS2/RGO composite: surpassing the performance of lithium-ion capacitors in a uniform system. NPG Asia Mater. 10, 775–787 (2018).
Lee, S.-H., Kim, K.-Y. & Yoon, J.-R. Binder- and conductive additive-free laser-induced graphene/LiNi1/3Mn1/3Co1/3O2 for advanced hybrid supercapacitors. NPG Asia Mater. 12, 28 (2020).
Fang, Y. et al. Two-dimensional mesoporous carbon nanosheets and their derived graphene nanosheets: synthesis and efficient lithium ion storage. J. Am. Chem. Soc. 135, 1524–1530 (2013).
Ren, L. et al. 3D hierarchical porous graphene aerogel with tunable meso-pores on graphene nanosheets for high-performance energy storage. Sci. Rep. 5, 14229 (2015).
Zhang, W. et al. Direct pyrolysis of supermolecules: an ultrahigh edge-nitrogen doping strategy of carbon anodes for potassium-ion batteries. Adv. Mater. 32, 2000732 (2020).
Yun, Y. S. et al. Effects of sulfur doping on graphene-based nanosheets for use as anode materials in lithium-ion batteries. J. Power Sources 262, 79–85 (2014).
Zhang, C., Mahmood, N., Yin, H., Liu, F. & Hou, Y. Synthesis of phosphorus-doped graphene and its multifunctional applications for oxygen reduction reaction and lithium ion batteries. Adv. Mater. 25, 4932–4937 (2013).
Wu, Z. S., Ren, W. C., Xu, L., Li, F. & Cheng, H. M. Doped graphene sheets as anode materials with superhigh rate and large capacity for lithium ion batteries. ACS Nano 5, 5463–5471 (2011).
Wang, X. et al. Heteroatom-doped graphene materials: syntheses, properties and applications. Chem. Soc. Rev. 43, 7067–7098 (2014).
Rao, C. N. R., Gopalakrishnan, K. & Govindaraj, A. Synthesis, properties and applications of graphene doped with boron, nitrogen and other elements. Nano Today 9, 324–343 (2014).
Reddy, A. L. et al. Synthesis of nitrogen-doped graphene films for lithium battery application. ACS Nano 4, 6337–6342 (2010).
Wang, H. et al. Nitrogen-doped graphene nanosheets with excellent lithium storage properties. J. Mater. Chem. 21, 5430 (2011).
Li, X. et al. Superior cycle stability of nitrogen-doped graphene nanosheets as anodes for lithium ion batteries. Electrochem. Commun. 13, 822–825 (2011).
Wang, X. et al. High-density monolith of n-doped holey graphene for ultrahigh volumetric capacity of Li-ion batteries. Adv. Energy Mater. 6, 1502100 (2016).
Li, Y. et al. Enhanced thermoelectric properties of Cu2SnSe3 by (Ag,In)-co-doping. Adv. Funct. Mater. 26, 6025–6032 (2016).
Huczko, A. et al. Self-propagating high-temperature synthesis (SHS) of crystalline nanomaterials. J. Cryst. Growth 401, 469–473 (2014).
Merzhanov, A. G. The chemistry of self-propagating high-temperature synthesis. J. Mater. Chem. 14, 1779 (2004).
Su, X. et al. Self-propagating high-temperature synthesis for compound thermoelectrics and new criterion for combustion processing. Nat. Commun. 5, 4908 (2014).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Yuan, W., Chen, J. & Shi, G. Nanoporous graphene materials. Mater. Today 17, 77–85 (2014).
Li, C. et al. Scalable self-propagating high-temperature synthesis of graphene for supercapacitors with superior power density and cyclic stability. Adv. Mater. 29, 1604690 (2017).
Ferrari, A. C. et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 97, 187401 (2006).
Ruan, J. et al. Rational construction of nitrogen‐doped hierarchical dual‐carbon for advanced potassium‐ion hybrid capacitors. Adv. Energy Mater. 10, 1904045 (2020).
Kim, T., Jung, G., Yoo, S., Suh, K. S. & Ruoff, R. S. Activated graphene-based carbons as supercapacitor electrodes with macro- and mesopores. ACS Nano 7, 6899–6905 (2013).
Qie, L. et al. Synthesis of functionalized 3D hierarchical porous carbon for high-performance supercapacitors. Energy Environ. Sci. 6, 2497–2504 (2013).
Li, C. et al. Scalable combustion synthesis of graphene-welded activated carbon for high-performance supercapacitors. Chem. Eng. J. 414, 128781 (2021).
Hu, Y.-S. et al. Synthesis of hierarchically porous carbon monoliths with highly ordered microstructure and their application in rechargeable lithium batteries with high-rate capability. Adv. Funct. Mater. 17, 1873–1878 (2007).
Ren, J. J. et al. Pre-lithiated graphene nanosheets as negative electrode materials for Li-ion capacitors with high power and energy density. J. Power Sources 264, 108–113 (2014).
Zhang, T. F. et al. High energy density li-ion capacitor assembled with all graphene-based electrodes. Carbon 92, 106–118 (2015).
Sun, Y. G. et al. Hybrid lithium-ion capacitors with asymmetric graphene electrodes. J. Mater. Chem. A. 5, 13601–13609 (2017).
Song, H., Yang, G. & Wang, C. General scalable strategy toward heterogeneously doped hierarchical porous graphitic carbon bubbles for lithium-ion battery anodes. ACS Appl. Mater. Interfaces 6, 21661–21668 (2014).
Sun, J. et al. Carbon nanorings and their enhanced lithium storage properties. Adv. Mater. 25, 1125–1130 (2013). 1124.
Qie, L. et al. Nitrogen-doped porous carbon nanofiber webs as anodes for lithium ion batteries with a superhigh capacity and rate capability. Adv. Mater. 24, 2047–2050 (2012).
Brezesinski, T., Wang, J., Tolbert, S. H. & Dunn, B. Ordered mesoporous alpha-MoO3 with iso-oriented nanocrystalline walls for thin-film pseudocapacitors. Nat. Mater. 9, 146–151 (2010).
Augustyn, V. et al. High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance. Nat. Mater. 12, 518–522 (2013).
Wang, H. W. et al. A high-energy lithium-ion capacitor by integration of a 3D interconnected titanium carbide nanoparticle chain anode with a pyridine-derived porous nitrogen-doped carbon cathode. Adv. Func. Mater. 26, 3082–3093 (2016).
Kim, K.-W. et al. Extremely fast electrochromic supercapacitors based on mesoporous WO3 prepared by an evaporation-induced self-assembly. NPG Asia Mater. 12, 84 (2020).
Bokhari, S. W. et al. Nitrogen doping in the carbon matrix for Li-ion hybrid supercapacitors: state of the art, challenges and future prospective. RSC Adv. 7, 18926–18936 (2017).
Park, G. O. et al. In operando monitoring of the pore dynamics in ordered mesoporous electrode materials by small angle X-ray scattering. ACS Nano 9, 5470–5477 (2015).
Liu, B. & Zhou, K. Recent progress on graphene-analogous 2D nanomaterials: properties, modeling and applications. Prog. Mater. Sci. 100, 99–169 (2019).
Fan, X., Zheng, W. T. & Kuo, J. L. Adsorption and diffusion of Li on pristine and defective graphene. ACS Appl. Mater. Interfaces 4, 2432–2438 (2012).
Li, C., Zhang, X., Wang, K., Sun, X. & Ma, Y. High-power and long-life lithium-ion capacitors constructed from n-doped hierarchical carbon nanolayer cathode and mesoporous graphene anode. Carbon 140, 237–248 (2018).
Yi, R. et al. High-performance hybrid supercapacitor enabled by a high-rate Si-based anode. Adv. Funct. Mater. 24, 7433–7439 (2014).
Yu, X. et al. Ultrahigh-rate and high-density lithium-ion capacitors through hybriding nitrogen-enriched hierarchical porous carbon cathode with prelithiated microcrystalline graphite anode. Nano Energy 15, 43–53 (2015).
Zhang, J., Liu, X. F., Wang, J., Shi, J. L. & Shi, Z. Q. Different types of pre-lithiated hard carbon as negative electrode material for lithium-ion capacitors. Electrochim. Acta 187, 134–142 (2016).
Lee, J. H. et al. Functionalized graphene for high performance lithium ion capacitors. ChemSusChem 5, 2328–2333 (2012).
Ye, L. et al. A high performance Li-ion capacitor constructed with Li4Ti5O12/C hybrid and porous graphene macroform. J. Power Sources 282, 174–178 (2015).
Jain, A. et al. Activated carbons derived from coconut shells as high energy density cathode material for Li-ion capacitors. Sci. Rep. 3, 3002 (2013).
Kim, H. et al. A novel high-energy hybrid supercapacitor with an anatase TiO2-reduced graphene oxide anode and an activated carbon cathode. Adv. Energy Mater. 3, 1500–1506 (2013).
Yang, M. et al. Fabrication of high-power Li-ion hybrid supercapacitors by enhancing the exterior surface charge storage. Adv. Energy Mater. 5, 1500550 (2015).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 51907193 and 51677182), the Beijing Municipal Science and Technology Commission (No. Z181100000118006), the Dalian National Laboratory for Clean Energy (DNL) Cooperation Fund, CAS (Nos. DNL201915 and DNL201912), the Key Research Program of Frontier Sciences, CAS (No. ZDBS-LY-JSC047), the International Partnership Program of Chinese Academy of Sciences (No. 182111KYSB20160014), and the Youth Innovation Promotion Association, CAS (No. 2020145).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, C., Zhang, X., Wang, K. et al. Nitrogen-enriched graphene framework from a large-scale magnesiothermic conversion of CO2 with synergistic kinetics for high-power lithium-ion capacitors. NPG Asia Mater 13, 59 (2021). https://doi.org/10.1038/s41427-021-00327-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-021-00327-7
This article is cited by
-
Matched MnO@C anode and porous carbon cathode for Li-ion hybrid supercapacitors
Rare Metals (2023)
-
Advancing silicon-based Li-ion batteries: enhanced stability and performance through carbon-coated Si and rGO linkage
Journal of Materials Science (2023)
