
Abstract
IL-15 is a promising cytokine to expand NK and CD8+ T cells for cancer immunotherapy, but its application is limited by dose-limiting, on-target off-tumor toxicity. Here, we have developed a next-generation IL-15 that is activated inside the tumor microenvironment (TME). This pro-IL-15 has the extracellular domain of IL-15Rβ fused to the N-terminus of sIL-15-Fc through a tumor-enriched Matrix Metalloproteinase (MMP) cleavable peptide linker to block its activity. Unlike sIL-15-Fc, pro-IL-15 does not activate the peripheral expansion of NK cells and T cells, thus reducing systemic toxicity, but it still preserves efficient anti-tumor abilities. In various mouse tumors, the anti-tumor effect of pro-IL-15 depends on intratumoral CD8+ T cells and IFN-γ. Pro-IL-15 increases the stem-like TCF1+Tim-3−CD8+ T cells within tumor tissue and helps overcome immune checkpoint blockade (ICB) resistance. Moreover, pro-IL-15 synergizes with current tyrosine kinase inhibitor (TKI) targeted-therapy in a poorly inflamed TUBO tumor model, suggesting that pro-IL-15 helps overcome targeted-therapy resistance. Our results demonstrate a next-generation IL-15 cytokine that can stimulate potent anti-tumor activity without severe toxicity.
Similar content being viewed by others


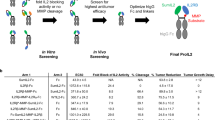
Introduction
Most tumor immunotherapies focus on blocking immune inhibitory signals or activating immune stimulatory pathways. However, several issues hinder clinical application, including severe treatment-associated organ toxicity and poor tumor control due to off-tumor distribution. IL-2, a pleiotropic cytokine which can expand T cells and NK cells, is one of the earliest FDA approved immunotherapy drugs for metastatic melanoma and renal cell cancer.1 However, IL-2 has not been widely used in the clinic due to its short half-life, the activation of immune inhibitory regulatory T cells (Tregs), and severe toxicity resulting from stimulation of vascular endothelium.2,3
IL-15 is another T cell growth factor whose receptor shares the same β and γ chain with IL-2. The IL-15Rα chain is typically expressed by myeloid cells, which binds to IL-15 first and trans-presents to the βγ-receptor-positive responding NK or CD8+ T cells.4 IL-15 is thought to be superior to IL-2 in the aspects of inducing lower vascular endothelium associated toxicity, much weaker Treg-stimulating activity, strong expanding NK and CD8+ T cells abilities, and avoiding activation-induced cell death (AICD).5 The administration of recombinant IL-15 is not effective in solid tumor therapy4,5 partly due to short half-life, toxicity and the lack of tumor activating property. Indeed, it can expand peripheral NK cells non-specifically and induce IFN-γ expression, which results in systemic toxicity.6 Recent studies showed that IL-15 within the TME is crucial for the optimal anti-tumor response. IL-15 is a component of the inflammatory milieu within tumor tissue, which is required for establishing a regular number of CD8+ T cells and NK cells.7 Loss of IL-15 within colorectal tumors is correlated with decreased T cell proliferation, higher tumor recurrence, and decreased patient survival.8 This dilemma highlights the need to develop new forms of IL-15 to activate NK cells or T cells within the tumor and avoid the expansion of peripheral NK cells.
Several forms of IL-15 have been developed to improve its activity and prolong its half-life for preclinical and clinical studies, including IL-15/IL-15Rα complex and several IL-15 muteins.5,9,10,11 However, this increased bioactivity without tumor targeting may also lead to severe toxicity.4,9 Here, we sought to develop a tumor-conditional pro-IL-15 to fulfill tumor-targeted delivery. The extracellular domain of IL-15Rβ is fused into the N-terminus of IL-15-IL-15Rα-Fc by an MMP-14 cleavable peptide linker. We observed that this targeted pro-IL-15 reduced the off-tumor expansion of NK cells, diminished systemic toxicity, and efficiently inhibited tumor growth. Importantly, pro-IL15 can overcome tumor resistance to ICB and tyrosine kinase inhibitor (TKI) therapies.
Results
Poor tumor control and substantial toxicity after systemic delivery of sIL-15-Fc
When we screened the correlation between CD8+ T cells and various cytokines in human skin melanoma cancer samples, we noticed that high CD8+ T cell infiltration was always correlated with high IL-15 levels (Supplementary Information, Fig. S1a). Moreover, both CD8+ T cell quantity and IL-15 expression level within tumors positively correlate with better survival in human cancer patients (Supplementary Information, Fig. S1b, c). These data suggest that IL-15 may contribute to CD8+ T cell infiltration for tumor inhibition.
We proposed that insufficient IL-15 secretion within the tumor may cause the failure of T-cell mediated tumor control. We then explored whether providing additional IL-15 to expand T cells could improve tumor control. In light of the mechanism of trans-presentation for IL-15, it is reported that IL-15 fused with the IL-15Rα sushi domain (super IL-15) has dramatically increased activity and stability compared to IL-15 alone.11 Fc fusion is reported to increase the half-life of cytokines and binding affinity through dimerization. Together, we designed and generated both IL-15-Fc and IL-15-IL-15Rα-Fc (super IL-15-Fc or sIL-15-Fc) proteins. Indeed, sIL-15-Fc had 1000-fold higher in vitro activity than IL-15-Fc (Supplementary Information, Fig. S2a). Consistently, sIL-15-Fc exhibited a much potent in vivo tumor-inhibiting effect than IL-15-Fc in the A20 lymphoma model (Supplementary Information, Fig. S2b). Though the in vitro activity of sIL-15-Fc was similar to sIL-15-his, sIL-15-Fc offered much better tumor control than sIL-15-his, suggesting that the Fc fusion to sIL-15 indeed increases its in vivo activity (Fig. 1a, b). In experimental animal tumor models and clinical patients, IL-15 treatment alone shows rather limited efficacy but significant toxicity in therapeutic doses.5 Consistently, we observed that 0.2 nmol (20 µg) sIL-15-Fc could only partially control tumor growth in the A20 tumor model (Fig. 1b). However, increasing the dose of sIL-15-Fc (0.8 nmol) treatment resulted in severe systemic toxicity manifesting in dramatic body-weight loss and decreased survival (Fig. 1c, d).

a The activity of the indicated proteins was detected by a CTLL-2 proliferation assay. b Balb/c mice (n = 5/group) were inoculated with 3 × 106 A20 cells. After the tumor was established (about 40 mm3), mice were treated with 0.2 nmol sIL-15-Fc (20 µg) or sIL-15-his (10 µg) by i.v. injection on days 8 and 11. The tumor size was measured. c, d C57BL/6 mice were inoculated with 5 × 105 MC38 cells. After the tumor was established (about 40 mm3), mice (n = 7–16/group) were treated with 0.8 nmol (80 µg) sIL-15-Fc by i.v. injection on days 8 and 11. For NK cell depletion, mice were injected with 20 µL of anti-Asialo GM1 antibody on days 8 and 11. Body-weight (c) and mouse survival (d) were monitored. e Tumor-bearing mice were treated as shown in c. On day 5 after the first anti-Asialo GM1 treatment, the numbers of the indicated cell subsets in peripheral blood were counted by FACS. Data are represented as means ± SEM and are representative of at least two independent experiments. c and d are the pool of two independent experiments. *P < 0.05; **P < 0.01.
After sIL-15-Fc treatment, CD8+ T cells, NK cells, and NKT cells impressively increased 10 to 20-fold in peripheral blood, while the quantity of CD4+ T cells was not affected (Supplementary Information, Fig. S3a). CD8+ T cells and NK cells were reported to contribute to tumor inhibition, but the systemic activation of broad non-tumor specific T cells and NK cells can result in off-tumor toxicities. To explore the subtypes of toxicity-inducing immune cells, we depleted CD8+ T cells, NK cells, NKT cells, or CD4+ T cells separately with the respective depleting antibodies (Supplementary Information, Fig. S3b–d). The depletion of CD8+ T cells or CD4+ T cells moderately increased the short-term survival of the sIL-15 treated mice, suggesting that CD8+ T cells or CD4+ T cells are only partially responsible for sIL-15-Fc mediated toxicity (Supplementary Information, Fig. S3b, c). However, anti-NK1.1 (clone PK136) antibody administration prevented most mice from dying from the sIL-15-Fc treatment (Supplementary Information, Fig. S3d). The anti-NK1.1 antibody could deplete both NK cells and NKT cells. To further determine whether NK cells or NKT cells are detrimental, we used an anti-Asialo GM1 antibody to deplete NK cells but not NKT cells (Fig. 1e). The depletion of only NK cells completely prevented mice from body weight loss and death, suggesting that NK cells are the main cellular component responsible for sIL-15-Fc induced toxicity (Fig. 1c, d). These data suggest that the systemic administration of sIL-15-Fc could mediate peripheral NK cell expansion and result in off-tumor toxicity that should be noted in the clinic.
Engineering a tumor-conditional pro-IL-15
IL-15 within the TME is required for establishing a regular number of CD8+ T cells and NK cells and is crucial for optimal anti-tumor response.7,8 We proposed that delivering IL-15 into tumors will improve solid tumor control. We compared the anti-tumor effect of sIL-15-Fc administrated by local intratumoral injection to that through systemic intravenous delivery. Intratumoral delivery of IL-15 was more effective in inhibiting tumor growth than systemic treatment in the MC38 model (Fig. 2a). Additionally, we treated other tumors, including A20 lymphoma, and observed similar results (Supplementary Information, Fig. S4a). These data suggest that sIL-15-Fc anti-tumor efficacy could be significantly improved if its delivery is targeted to the TME specifically. To create an IL-15 that can be preferentially activated inside the TME, we carefully designed a next-generation IL-15, pro-IL-15, by adding the extracellular domain of IL-15Rβ to the N-terminus of sIL-15-Fc to restrain IL-15’s activity. The MMP-14 cleavable peptide was used as a linker. In contrast to many MMPs that are soluble and easily leak out systemically, MMP-14 is a membrane protein highly expressed on tumor cells and tumor-associated macrophages.12,13 We choose a highly sensitive linker (amino acid sequence: SGFIANPVTA) that could be cleaved by MMP-14, acting as a switch to expose IL-15 in the MMP enriched tumor microenvironment (Fig. 2b). To optimize the release of active IL-15 after cleavage of the linker, we also designed different lengths of IL-15Rβ: whole IL-15Rβ extracellular domain versus a truncated extracellular domain of IL-15Rβ (RβD1) to reduce its binding affinity to pro-IL-15 (Fig. 2b).

a MC38 tumor-bearing mice (n = 4–5/group) were treated with 0.2 nmol (20 µg) sIL-15-Fc by i.t. or i.v. injection on days 11 and 14. The tumor size was measured twice weekly. b Schematic representation of the pro-IL-15 protein. c Pro-IL-15(RβD1) was incubated with pre-activated MMP-14 at 37 °C in vitro for 12 h. The released activity of proteins was detected by CTLL-2 proliferation assay. d MC38 tumor-bearing mice (n = 7 or 8/group) were i.v. treated with 0.2 nmol (26 µg) pro-IL-15(RβD1, MMP) or pro-IL-15(RβD1, G4S) on days 7, 10 and 13. Tumor growth was measured. Data are represented as means ± SEM and are representative of at least two independent experiments. ***P < 0.001; ****P < 0.0001.
Another advantage of using Fc is that it makes purification of pro-IL-15 or sIL-15-Fc easy. After a single step of Protein-A purification, reducing SDS-PAGE results showed a single band for sIL-15 or pro-IL-15. The MW of monomeric sIL-15-Fc, pro-IL-15(Rβ), and pro-IL-15(RβD1) is approximately 70 Kd, 120 Kd, and 100 Kd, respectively, which are larger than expected and possibly caused by glycosylation (Supplementary Information, Fig. S4b). After incubation with MMP-14, pro-IL-15 was cleaved into sIL-15-Fc and Rβ (or RβD1) (Supplementary Information, Fig. S4b). Then we compared the bioactivity of pro-IL-15 before and after MMP-14 digestion through a CTLL-2 proliferation assay. sIL-15-Fc stimulated CTLL-2 cells to proliferate in a dose-dependent manner (Fig. 2c). Pro-IL-15(Rβ) has about 1000-fold decreased activity compared to that of sIL-15-Fc (Supplementary Information, Fig. S4c), while pro-IL-15(RβD1) reduced activity approximately 100-fold (Fig. 2c). After incubation with MMP-14, the activities of both forms of pro-IL-15 recovered significantly and were comparable to sIL-15-Fc (Fig. 2c and Supplementary Information, Fig. S4c). In the syngeneic MC38 tumor model, pro-IL-15(RβD1) inhibited tumor growth much more effectively than that with Rβ (Supplementary Information, Fig. S4d), suggesting pro-IL-15(RβD1) has better in vivo activity and thus was used in the subsequent studies. Moreover, pro-IL-15 (RβD1, G4S) protein containing non-cleavable G4S linker showed much reduced anti-tumor effect in vivo (Fig. 2d and Supplementary Information, Fig. S4b), demonstrating that in vivo cleavage is required to release the pro-IL-15’s bioactivity. Together, these data suggest that the bioactivity of pro-IL-15 can be unleashed by MMP-14 in vivo and effectively inhibits tumor growth.
pro-IL-15 avoids peripheral NK expansion mediated toxicity
Because membrane protein MMP-14 is preferentially and highly expressed on tumor cells and tumor-associated macrophages, we expected that pro-IL-15 might avoid the expansion of peripheral lymphocytes and induce less toxicity. To test this assumption, we administrated a lethal dose of sIL-15-Fc or a comparable molecular amount of pro-IL-15 into tumor-bearing mice. Indeed, sIL-15-Fc (0.8 nmol/80 µg) induced significant body-weight loss, and eventually, 70% of mice died within only five days after treatment (Fig. 3a, b). However, high dose pro-IL-15 treated mice rapidly recovered after a transient body-weight loss, and no mice died even with a dose of 1.5 nmol treatment (Fig. 3a, b and Supplementary information, Fig. S5a, b). To further characterize the toxicity induced by cytokine treatment, we performed blood tests for tumor-bearing mice 24 h and 96 h after treatment, particularly alanine aminotransferase (ALT) as a liver damage marker and aspartate aminotransferase (AST) as a tissue damage marker. The serum levels of both ALT and AST were increased 96 hours after sIL-15 treatment, but not pro-IL-15 (Fig. 3c, d). Furthermore, sIL-15-Fc increased serum inflammatory cytokines such as IFN-γ, MCP-1, and IL-6, but pro-IL-15 did not induce inflammatory cytokines (Fig. 3e). Consistently, in contrast to sIL-15-Fc causing a sharp increase in peripheral blood NK cells, pro-IL-15 induced much less NK expansion (Fig. 3f). The expansion of peripheral lymphocytes may result in severe inflammation or injury to solid organs such as the liver, kidney, and lung. Indeed, sIL-15-Fc induced significant immune cell infiltration into organ tissues (Supplementary Information, Fig. S5c). In contrast, pro-IL-15 induced no organ inflammation in the treated mice (Supplementary Information, Fig. S5c). The serum half-life of pro-IL-15 and sIL-15-Fc is similar, suggesting that the decreased toxicity of peripheral pro-IL-15 is not caused by a shorter half-life (Supplementary Information, Fig. S5d). To test which cells are targeted by sIL-15-Fc but not pro-IL-15, we compared their binding ability to lymphocytes. sIL-15-Fc efficiently bound to splenic NK cells, NKT cells, and CD8+ T cells, with little binding to CD4+ T cells (Supplementary Information, Fig. S6). However, pro-IL-15 showed much weaker binding to splenic lymphocytes than sIL-15-Fc (Supplementary Information, Fig. S6). Altogether, pro-IL-15 was sheltered efficiently and successfully avoided inducing peripheral lymphocyte expansion and toxicity, compared to sIL-15-Fc.

a, b MC38 tumor-bearing mice were treated with PBS (n = 6), 0.8 nmol sIL-15-Fc (n = 12) or pro-IL-15 (n = 6) by i.v. injection on days 8 and 11. The body weight change of mice (a) and survival (b) were monitored. c, d MC38 tumor-bearing mice (n = 6/group) were treated the same as in a. The levels of ALT (c) and AST (d) in the peripheral serum were quantified on days 1 and 4 after the first treatment. e, f MC38 tumor-bearing mice (n = 4/group) were treated with 0.5 nmol sIL-15-Fc (50 µg) or pro-IL-15 (65 µg) on days 8 and 11 after tumor inoculation. Blood was collected at 24 h after the first treatment. Cytokine levels in the serum were measured by cytometric bead array (e). The lymphocyte expansion in the peripheral blood was tested by FACS on day 5 after the first treatment (f). Data are represented as means ± SEM. a, b are the pool of two independent experiments. Other data are representative of at least two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
pro-IL-15 re-activates intratumoral CD8+ T cells
We hypothesized that pro-IL-15 preferentially accumulates and is activated inside the TME to activate lymphocytes for tumor control. To trace where pro-IL-15 travels, we conjugated pro-IL-15 with a Cy5.5-maleimide tracer and injected it into tumor-bearing mice for whole-body imaging. The bioluminescence data showed that pro-IL-15 could accumulate within tumor tissue specifically (Fig. 4a). To further trace pro-IL15 distribution, we injected mice with fusion protein and collected various tissues three days after injection. The in vivo biodistribution of pro-IL-15 displayed considerable specificity; high concentrations of pro-IL-15 were retained within the tumor, and much less was found in normal tissues (Fig. 4b). Next, we compared the anti-tumor effect. Mice bearing MC38 or A20 tumor were treated with sIL-15-Fc or pro-IL-15 protein through intravenous injection. As shown, pro-IL-15 efficiently inhibited tumor growth as well as sIL-15-Fc (Fig. 4c, d and Supplementary Information, Fig. S7a, b). These data demonstrate that pro-IL-15 could accumulate and be activated within tumor tissues to inhibit tumor growth.

a 25 μg of Cy5.5-labeled pro-IL-15 protein was i.v. injected into MC38 tumor-bearing mice. One day after injection, pro-IL-15 accumulation in the tumor was tracked by the IVIS Spectrum in vivo imaging system. b MC38 tumor-bearing mice (n = 4/group) were i.v. injected with 25 μg of pro-IL-15 protein. Tissues were collected on day 3 after injection. The concentrations of the fusion protein were measured by ELISA. c MC38 tumor-bearing mice (n = 5/group) were treated with 0.2 nmol sIL-15-Fc (20 µg) or pro-IL-15 (26 µg) by i.v. injection on days 8 and 11. The tumor growth was monitored. d A20 tumor-bearing mice (n = 6/group) were treated with 0.2 nmol sIL-15-Fc or pro-IL-15 by i.v. injection on days 8 and 11. Tumor growth was monitored. Data are represented as means ± SEM and are representative of at least two independent experiments.
To test which cells contribute to pro-IL-15-mediated solid tumor control, we separately depleted individual types of immune cells using antibodies. When NK cells and NKT cells were depleted, the anti-tumor effect of pro-IL-15 was not impaired (Fig. 5a). In contrast, the depletion of both CD4+ T cells and CD8+ T cells completely abrogated the anti-tumor effect of pro-IL-15, suggesting T lymphocytes are required for the anti-tumor effect (Supplementary Information, Fig. S8a). Further study showed that CD8+ T cells alone are essential to mediate IL-15 mediated tumor killing (Fig. 5b). We further checked the quantity change of T cells within tumor tissue after pro-IL-15 treatment. Pro-IL-15 treatment significantly increased the numbers of effector CD8+ T cells but not Treg cells in the tumor tissues (Fig. 5c and Supplementary Information, Fig. S8b).

a, b MC38 tumor-bearing mice were treated with 0.2 nmol pro-IL-15 (26 µg) by i.p. injection on days 8, 11, and 14. For cell depletion, mice were injected with 400 μg of anti-NK1.1 Ab (n = 6/group) (a) or 200 µg anti-CD8 Ab (n = 5/group) (b) on days 8, 12, and 16. The tumor growth curve was monitored twice weekly. c MC38 tumor-bearing mice (n = 5/group) were treated with pro-IL-15 (26 µg) the same as in a. Two days after the last treatment, tumor tissues were collected. The quantity of CD8+ T cells in the tumor was evaluated by flow cytometry. d MC38 tumor-bearing mice (n = 5/group) were treated with pro-IL-15 (26 µg) by i.p. injection on days 13, 16, and 19. To block T cell migrating from LN into the tumor, mice were treated with FTY720 from day 13. The tumor volume was measured twice weekly. e MC38 tumor-bearing mice (n = 6/group) were treated with pro-IL-15 (26 µg) by i.p. injection on days 10, 14, and 18. For IFN-γ blocking, mice were injected with 500 μg of anti-IFN-γ Ab (clone R4-6A2) on days 10, 14, 18 and 22. The tumor volume was measured twice weekly. f MC38 tumor-bearing mice (n = 4/group) were treated with pro-IL-15(26 µg) by i.p. injection on days 8, 11 and 14. Two days after the last treatment, tumor tissues were harvested and TCF-1+Tim3− cells in CD8+ T cells were determined by flow cytometry. Data are represented as means ± SEM and are representative of at least two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
Next, we wanted to determine whether the pre-existing CD8+ T cells within the tumor or migrating T cells are essential for tumor control after pro-IL-15 treatment. We used FTY720, which can block lymphocytes efferent from lymph nodes into tumor tissues. Pro-IL-15 still preserved anti-tumor ability similar to the non-FTY720 treated group (Fig. 5d). This result and Fig. 5c suggest that pro-IL-15 expanded tumor-specific CD8+ T cells within the TME, which are essential and sufficient to inhibit tumor growth. IFN-γ acts as one of the effector cytokines of CD8+ T cells. To test whether IFN-γ contributes to pro-IL-15-mediated solid tumor control, mice were treated with anti-IFN-γ blocking antibody during pro-IL-15 treatment. The completely abrogated anti-tumor effects suggested that IFN-γ played an essential role in pro-IL-15-mediated tumor inhibition. It has been found that intratumoral TCF1+TIM3–CD8+ T cells exhibited a stem-like phenotype and expanded more efficiently after ICB therapy.14,15,16 After pro-IL-15 treatment, both the percentage and quantity of stem-like CD8+ T cells increased within tumor tissue, suggesting that it may help improve ICB therapy (Fig. 5f). Collectively, these results demonstrate that pro-IL-15 preferentially expands pre-existing CD8+ T cells within tumor tissues, which is critical for tumor control through IFN-γ.
pro-IL-15 overcomes checkpoint blockade resistance to control advanced tumors
PD-1 or PD-L1 blockade has been widely used to treat many types of cancer, but monotherapy of either one has shown limited anti-tumor effects in both animal models and clinic patients. Primary or secondary resistance has become a significant clinical problem. We speculated that blocking inhibitory PD-L1 could improve tumor-specific T cell responses but not enough to expand TILs and pro-IL-15 could expand those TILs and synergize with anti-PD-L1 antibody to amplify the anti-tumor effect. In fact, we did observe that PD-L1 was most highly expressed in myeloid cells within tumor tissues, and both myeloid cells and PD-L1 were increased after sIL-15 treatment (Supplementary Information, Fig. S8c, d). To test the hypothesis that PD-1/PD-L1 blockade could synergize with pro-IL-15 treatment, MC38 tumor-bearing mice were treated with pro-IL-15 or anti-PD-L1 when the tumor was well established (Fig. 6a, b). Either anti-PD-L1 or pro-IL-15 alone partially delayed tumor growth. Tumors rapidly relapsed in mice treated with anti-PD-L1, but the combination with pro-IL-15 greatly enhanced the tumor clearance for all tumor-bearing mice (Fig. 6a). In the combination-treatment group, 60% of mice were completely tumor-free, but only 10% of mice in the single anti-PD-L1 treatment group were tumor-free (Fig. 6b). We next explored whether the combination treatment could help to establish prolonged protective immunity and prevent tumor relapse, which are valuable in the clinic. Mice with complete tumor regression after combination therapy were re-challenged with a lethal dose of tumor cells (Fig. 6c). All of the previously tumor-cleared mice successfully rejected the re-challenge, indicating a robust anti-tumor memory response (Fig. 6c). Overall, pro-IL-15 can overcome anti-PD-L1 resistance and help establish protective memory immunity.

a, b MC38 tumor-bearing mice (n = 5–6/group) were i.p. treated with 0.2 nmol of pro-IL-15, 150 µg anti-PD-L1 Ab, or the combination of pro-IL-15 and anti-PD-L1 on days 12 and 16. The tumor volume was measured twice weekly and recorded as in a. Mice survival was recorded as in b. c Naïve mice or mice with complete tumor regression after combination therapy in a were re-challenged with 1.5 × 106 MC38 cells. The tumor growth was monitored twice weekly. Data are represented as means ± SEM and are representative of at least two independent experiments. *P < 0.05; **P < 0.01; ****P < 0.0001.
pro-IL-15 therapy can overcome TKI therapy resistance
The second and third generation EGFR-TKI therapies can induce a high response rate in patients with EGFR− or Her2− dependent cancers, but a high relapse rate is a major clinical problem.17 The TUBO model, a HER2/neu-dependent mammary carcinoma, fails to respond to immunotherapy due to insufficient TILs in established tumors, similar to clinical EGFR/HER2-driven tumors.18 We proposed that TKI therapy may expand the time window for immunotherapy by reducing the tumor burden. Furthermore, TKI will induce an inflammatory TME and increase T lymphocyte infiltration. Pro-IL-15 expands those TILs in the TME and may synergize with targeted therapy to overcome resistance. In our study, afatinib (a second-generation TKI agent) could only partially delay tumor growth, but later, all the treated tumors relapsed (Fig. 7a). However, the addition of pro-IL-15 could effectively control tumor growth and prevent tumor relapse (Fig. 7a, b). Altogether, these data suggest that proper use of pro-IL-15 could combine with TKI to treat poor inflamed tumors.

a, b Balb/c mice (n = 5–9/group) were subcutaneously inoculated with 5 × 105 of TUBO cells and then were i.p. treated with 0.2 nmol of pro-IL-15 on days 12, 15 and 19. For TKI therapy, mice were treated orally with 0.5 mg of afatinib (a second-generation TKI) on days 12 and 19. The tumor growth was measured and shown in a. The mice survival curve was shown in b. Data are represented as means ± SEM and are representative of at least two independent experiments. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Finally, to explore the possible application in the clinic, we constructed a human pro-IL-15. In a PBMC humanized tumor model, human pro-IL-15 treatment exhibited a similar anti-tumor effect to sIL-15-Fc (Supplementary Information, Fig. S9a, b). sIL-15-Fc treatment induced significant body weight loss, and all mice died quickly after the third treatment (Supplementary Information, Fig. S8c, d). Impressively, pro-IL-15-treated mice exhibited slight body weight loss, and most mice survived after treatment. These data suggest that the design of pro-IL-15 could be applied to human cytokines, which preserves the anti-tumor ability but reduces the adverse effects.
Discussion
CD8+ T cells infiltration and activation within the tumor are strongly correlated with better prognosis in the clinic. IL-15 is an attractive cytokine for activating CD8+ T cells and NK cells potently. However, the application of IL-15 in the clinic for cancer immunotherapy is limited by its short half-life, low efficacy, and dose-limiting toxicity due to the off-tumor binding with peripheral lymphocytes. Here, we have developed a pro-IL-15 to fulfill tumor-targeted delivery in the following ways. 1) To block IL-15’s bioactivity in peripheral tissues and specifically release it within the tumor, the extracellular domain of IL-15Rβ, as a natural blocking receptor, is fused to the N-terminus of IL-15-IL-15Rα-Fc through an MMP-14 cleavable peptide linker. 2) After IL-15Rβ is released by tumor-enriched MMP-14 digestion, the bioactivity of pro-IL-15 can be recovered. We observed that this pro-IL-15 protein prevents the peripheral expansion of NK cells and T cells, the release of inflammatory cytokine in the blood, and organ toxicity, but can still efficiently inhibit tumor growth. 3) Moreover, this next-generation IL-15 specifically expands CD8+ T cells within the tumor and overcomes ICB therapy resistance and TKI targeted therapy resistance, suggesting that pro-IL-15 treatment may benefit those patients resistant to current therapies.
Cytokines are potent targets for cancer immunotherapies, but they are limited by low efficacy and dose-limiting toxicity resulting from binding to peripheral receptors. IL-15 is usually intravenously injected, or subcutaneously injected frequently to achieve slow-release into the blood and reduce peripheral toxicity, but significant lymphoid organ retention and peripheral blood lymphocyte increases could still be observed.9,19 Complexes of IL-15 with IL-15Rα (sIL-15) increased the stability and activity, but may incur stronger peripheral lymphocyte activation.9,10,11 Consistent with previous research, we found that NK cells within peripheral blood were expanded significantly and mediated deleterious toxicity to the host after systemic injection of sIL-15-Fc.20 Moreover, it has been reported that IL-15 induced lymphocyte expansion within TME is crucial for the optimal anti-tumor response.7,8 We observed that intratumoral injection of sIL-15-Fc achieved better tumor treatment effects than systemic administration. These data highlight the need to develop a new generation IL-15 to target tumor tissues for a more potent anti-tumor efficacy. Cytokine fusion with tumor-targeting antibodies (such as anti-EGFR) has been exploited to target several cytokines into tumor tissues.21,22,23 However, the high affinity of cytokines to their receptors may override the antibody-mediated targeting. It is found that antibody-IL-2 fusion protein does not selectively home to tumors, but instead, the localization of this fusion protein in vivo is dominated by the IL-2 moiety.24 In our study, we constructed a pro-IL-15, whose binding with receptor-expressing cells can be spatially regulated. In contrast to sIL-15-Fc, pro-IL-15 prevented toxicity mediated by peripheral NK proliferation but preserved the efficient anti-tumor effect.
The truncated extracellular part of the IL-15Rβ subunit (CD122) was used to block the activity of IL-15 reversibly. Unlike other probody designs, this selection of the blocking subunit is a natural receptor, thus may have much less immunogenicity.25,26,27 The release of pro-IL-15’s activity was controlled by an MMP-14 (MT1-MMP) cleavable linker. MMPs are mainly involved in the remodeling of extracellular matrix,28 and the increased expression and activity of MMPs are associated with multiple cancer progression.12,29,30,31 In contrast to many other MMPs soluble and easily leaking out systemically, MMP-14 is a membrane protein that is highly expressed in tumor cells and tumor-associated macrophages but rather limited to leak into peripheral tissues.12,13 After in vitro incubation with MMP-14, pro-IL-15 was cleaved into sIL-15-Fc, and the activity of pro-IL-15 recovered significantly. Compared to sIL-15-Fc, pro-IL-15 exhibits lower toxicity in vivo. There is almost no expansion of lymphocytes in the peripheral tissues of mice, no peripheral inflammatory factors, or no ALT/AST induced after pro-IL-15 treatment; there is only a transient, slight weight loss in mice after treatment. However, pro-IL-15 was found to accumulate within tumor tissues preferentially, resulting in notably increased intratumoral CD8+ T cells and inhibited tumor growth efficiently. Tumor-derived MMPs release sIL-15 activity from pro-IL-15 within TME. In the future, the addition of tumor-targeting moieties to pro-IL-15 might further increase its tumor-targeting.
By depleting different immune cells in the MC38 mouse colorectal cancer model, we found that the therapeutic effect of pro-IL-15 did not depend on NK cells but depended on CD8+ T cells. Additionally, FTY720 was used to prevent peripheral CD8+ T cells from migrating into the tumor tissue. Pro-IL-15 can still inhibit tumor growth in this situation, which suggests that pre-existing CD8+ T cells are essential and sufficient for tumor control. CD8+ T cells within tumor tissues exhibited an exhausted status, marked by upregulation of immune inhibitory molecules (PD-1, TIM-3, Lag-3, TIGIT), reduced effector function, and insufficient proliferative capacity.32 Two distinct populations of exhausted CD8+ T cells were identified recently: stem-like T cells and terminally exhausted T cells. Stem-like CD8+ T cells were found to proliferate and differentiate into effector-biased terminally exhausted CD8+ T cells for tumor control after ICB therapy.14,15,16 Interestingly, we found that pro-IL-15 expands stem-like TCF1+ Tim-3− CD8+ T cells within tumor tissues, suggesting that it may help to expand more TILs and improve ICB therapy.
Immune checkpoint blocking drugs have achieved encouraging clinical effects for tumor treatment, but most patients with advanced cancer do not respond to the drug or relapse in the late stage. How to break the tumor’s tolerance to immune checkpoint blocking drugs is still an urgent challenge. We proposed that the insufficient expansion of CD8+ T cells by ICB therapy alone may be overcome by combing IL-15 treatment. We have also observed that IL-15 treatment increased the infiltration of myeloid cells in the tumor microenvironment and the expression of PD-L1 on myeloid cells, which may dampen the therapeutic effect of IL-15. These exciting results drove us to explore the anti-tumor effect of pro-IL-15 and PD-1/PD-L1 blockade combination therapy. Indeed, we observed that the combination of pro-IL-15 and anti-PD-L1 antibodies demonstrated a synergistic therapeutic effect, and the combination therapy can produce a strong protective memory immune response and prevent tumor recurrence.
Tumor recurrence is broadly found in the clinic, especially in poorly inflamed cancer patients, and has become a major challenge for cancer treatment. TKI is the first-line therapy for EGFR mutant positive cancer patients, and most of these patients have fewer TILs and do not respond to immunotherapy.33 After standard TKI therapy, patients usually have impressive initial complete responses but undergo relapse in a few months.17 Our study showed that pro-IL-15 induced more TILs in the TME and synergized with TKI targeted-therapy to overcome resistance to either single treatment to better control poorly inflamed tumors.
Overall, we have developed a next-generation IL-15 cytokine, which presents restrained activity in the peripheral tissues but recovers its bioactivity within tumors through site-specific digestion by the tumor-enriched MMP-14. Thus, pro-IL-15 may maintain anti-tumor activity while avoiding peripheral toxicity. The ability to proliferate CD8+ T cells makes pro-IL-15 a potential treatment to overcome tumor resistance to ICB therapy and conventional TKI targeted therapy.
Materials and methods
Mice
Female (6–8 weeks old) BALB/c and C57BL/6 mice were purchased from Vital River Laboratories (Beijing, China). All mice were maintained under specific pathogen-free (SPF) conditions in the animal facility of the Institute of Biophysics, Chinese Academy of Sciences. Animal care and experiments were performed under the guidelines of the Institute of Biophysics, Chinese Academy of Sciences. Protocols were approved by the Institutional Laboratory Animal Care and Use Committee.
Cell lines and reagents
A20 and MC38 cell lines were purchased from ATCC (Manassas, VA). Invitrogen™ FreeStyle™ 293-F Cells (R79007) were grown in SMM 293-TI medium (M293TI, Sino Biological). TUBO was cloned from a spontaneous mammary tumor in a BALB/c Neu-transgenic mouse.34 All cells were cultured at 37 °C in 5% CO2. MC38 cells were maintained in Dulbecco’s modified Eagle’s medium, supplemented with 10% heat-inactivated fetal bovine serum, 2 mmol/L L-glutamine, 0.1 mmol/L Minimum Essential Medium nonessential amino acids, 100 U/mL penicillin, and 100 mg/mL streptomycin. A20 cells and CTLL-2 cells were maintained in RPMI 1640 medium. All cell lines were routinely tested using mycoplasma contamination kit (R&D). Anti-PD-L1 Ab (10 F.9G2) and anti-IFNγ Ab (R4-6A2) were purchased from BioXCell (West Lebanon, NH). Anti-CD8 Ab (TIB210), anti-CD4 Ab (GK1.5), FcγRII/III blocking Ab (2.4G2), and anti-NK1.1 Ab (PK136) were produced in house. Anti-Asialo-GM1 Ab (Poly21460) was purchased from Biolegend. FTY720 was purchased from Sigma.
Production of IL-15 and pro-IL-15 fusion protein
IL-15-Rα-Fc: the cDNA encoding mouse IL-15 and IL-15Rα-sushi domain (amino acids 1–78) was fused with a 26-amino acid linker (SGGGSGGGGSGGGGSGGGGSGGGSLQ). The signal peptide of the human IgGκ leading sequence and IgG1 Fc was used. The entire sequence was then cloned into the pEE12.4 vector (Lonza). Pro-IL-15: the cDNA encoding the extracellular domain of mouse IL-15Rβ or extracellular domain 1 of IL-15Rβ was fused with IL-15-Ra-Fc by a 20-amino acid linker (GGGGSSGFIANPVTAGGGGS). The entire sequence was then cloned into the pEE12.4 vector (Lonza). The plasmid was transiently transfected into 293-F cells. Supernatant was collected on day 7 after transfection. The fusion protein was purified using a Protein A-Sepharose column according to the manual (Repligen Corporation).
In vitro digestion conditions for pro-IL15
Recombinant hMMP-14 (R&D Systems) was activated by rhFurin in an Activation Buffer (50 mM Tris, 1 mM CaCl2, 0.5% (w/v) Brij-35, pH 9.0) at 37 °C for 2 h. Pro-IL-15 was co-cultured with activated rhMMP14 in an Assay Buffer (50 mM Tris, 3 mM CaCl2, 1 µM ZnCl2, pH 7.5) at 37 °C for 12 h. Protein cleavage was confirmed by reduced SDS-PAGE analysis. The CTLL-2 proliferation assay was used to detect the released activity of pro-IL-15.
CTLL-2 proliferation assay
Functional IL-15 was measured using CTLL-2 cells. 100 µL purified proteins or MMP cleaved products with series dilution were added to a 96-well plate. 3 × 103 of CTLL-2 cells in 100 µL medium were added in each well and incubated for 72 h at 37 °C in 5% CO2. 20 µL CCK8 reagent (Cell Counting Kit-8) was added, and the plate was incubated for 3–4 h at 37 °C in 5% CO2. Absorbance was read at 450 nm.
Tumor growth and treatments
Approximately 2–5 × 105 of MC38 cells were injected subcutaneously into the right flank of C57BL/6 mice. A20 cells (3 × 106) or TUBO cells (5 × 105) were subcutaneously injected into the right flank of Balb/c mice. Tumor volumes were measured and calculated (length × width × height/2). After the tumor was established, mice were treated with sIL-15-Fc or pro-IL-15. For depletion of different types of cells, anti-CD8 Ab (clone TIB210) or anti-CD4 Ab (clone GK1.5) was i.p. injected at a dose of 200 μg. Anti-NK1.1 Ab (clone PK136) was i.p. injected at a dose of 400 μg. Anti-Asialo GM1 Ab (20 μl) was i.p. injected. For blocking IFN-γ, anti-IFN-γ Ab (clone R4-6A2) was i.p. injected at a dose of 500 μg. To block lymphocyte trafficking, mice were i.p. injected with 25 μg FTY720 at the first injection, and 20 μg of FTY720 was administered every other day to maintain the blockade. For TKI treatment, TUBO tumor-bearing mice were treated orally with 0.5 mg of afatinib (TKI) every 7 days for a total of two doses.
Toxicity
MC38 tumor-bearing C57BL/6 mice were treated with a lethal dose of sIL-15-Fc (80 µg or 0.8 nmol) or a comparable molar of pro-IL-15 by i.v. injection on days 8 and 11 after tumor inoculation. The body weight change of mice was monitored. The ALT and AST levels in the peripheral serum were quantified on days 1 and 4 after the first treatment. Four days after the first treatment, mice were sacrificed, and liver, kidney, lung tissues were collected for HE staining.
Fluorescence imaging
Pro-IL-15 was labeled with Cy5.5 and purified by washing away the unbound Cy5.5. 25 µg fluorescently-labeled pro-IL-15 was i.v. injected into C57BL/6 mice bearing subcutaneous MC38 tumors. Fluorescence was measured with IVIS Spectrum at different time points.
Flow cytometry
Tumor tissues were collected, cut into small pieces, and re-suspended in digestion buffer (RPMI-1640 medium with 1 mg/mL type IV collagenase and 100 µg/mL DNase I). Tumors were digested for 45 min at 37 °C, then passed through a 70 μm cell strainer to make single-cell suspensions. Cells suspended in FACS buffer (1% bovine serum albumin and 0.05% NaN3) were blocked with anti-CD16/32 Ab (anti-FcγIII/II receptor, clone 2.4G2) for 30 min and then stained with specific antibodies for 30 min on ice. For intracellular TCF-1 staining, samples were fixed, permeabilized, and stained with anti-mouse TCF-1. All fluorescent-labeling mAbs were purchased from BioLegend or eBioscience. DAPI or LIVE/DEAD™ fixable yellow dye (ThermoFisher) was used to exclude dead cells. Samples were analyzed on a FACS Calibur or Fortessa flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (TreeStar).
Statistical analysis
Data are shown as the means ± SEM. Statistical analyses of survival curve were compared using a log-rank test. Other statistical analyses were compared using an unpaired Student’s two-tailed t-test. Analyses were performed using GraphPad Prism version 5.0 (GraphPad Software). Statistically significant differences of P < 0.05, P < 0.01, P < 0.001, and P < 0.0001 are noted with *, **, *** and ****.
References
Rosenberg, S. A. IL-2: the first effective immunotherapy for human cancer. J. Immunol. 192, 5451–5458 (2014).
Kolitz, J. E. et al. Recombinant interleukin-2 in patients aged younger than 60 years with acute myeloid leukemia in first complete remission: results from Cancer and Leukemia Group B 19808. Cancer 120, 1010–1017 (2014).
Sim, G. C. et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Invest. 124, 99–110 (2014).
Fiore, P. F. et al. Interleukin-15 and cancer: some solved and many unsolved questions. J. Immunother. Cancer 8, e001428 (2020).
Waldmann, T. A., Dubois, S., Miljkovic, M. D. & Conlon, K. C. IL-15 in the combination immunotherapy of cancer. Front Immunol. 11, 868 (2020).
Guo, Y. et al. IL-15 superagonist-mediated immunotoxicity: role of NK cells and IFN-gamma. J. Immunol. 195, 2353–2364 (2015).
Santana Carrero, R. M. et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc. Natl. Acad. Sci. USA 116, 599–608 (2019).
Mlecnik, B. et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci. Transl. Med. 6, 228ra237 (2014).
Rhode, P. R. et al. Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol. Res. 4, 49–60 (2016).
Chertova, E. et al. Characterization and favorablein vivoproperties of heterodimeric soluble IL-15·IL-15Rα cytokine compared to IL-15 monomer. J. Biol. Chem. 288, 18093–18103 (2013).
Mortier, E. et al. Soluble interleukin-15 receptor α (IL-15Rα)-sushi as a selective and potent agonist of IL-15 action through IL-15Rβ/γ. J. Biol. Chem. 281, 1612–1619 (2006).
McGowan, P. M. & Duffy, M. J. Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Ann. Oncol. 19, 1566–1572 (2008).
Turunen, S. P., Tatti-Bugaeva, O. & Lehti, K. Membrane-type matrix metalloproteases as diverse effectors of cancer progression. Bioch. Biophys. Acta Mol. Cell Res. 1864, 1974–1988 (2017).
Utzschneider, D. T. et al. T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016).
Im, S. J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016).
He, R. et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–416 (2016).
Ohashi, K., Maruvka, Y. E., Michor, F. & Pao, W. Epidermal growth factor receptor tyrosine kinase inhibitor–resistant disease. J. Clin. Oncol. 31, 1070–1080 (2013).
Sun, Z. et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat. Commun. 10, 3874 (2019).
Liu, B. et al. Evaluation of the biological activities of the IL-15 superagonist complex, ALT-803, following intravenous versus subcutaneous administration in murine models. Cytokine 107, 105–112 (2018).
Guo, Y. et al. IL-15 superagonist–mediated immunotoxicity: role of NK cells and IFN-γ. J. Immunol. 195, 2353–2364 (2015).
Yang, X. et al. Targeting the tumor microenvironment with interferon-β bridges innate and adaptive immune responses. Cancer Cell 25, 37–48 (2014).
Liang, Y. et al. Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat. Commun. 9, 4586 (2018).
Weiss, T. et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci. Transl. Med. 12, eabb2311 (2020).
Tzeng, A., Kwan, B. H., Opel, C. F., Navaratna, T. & Wittrup, K. D. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc. Natl. Acad. Sci. USA 112, 3320–3325 (2015).
Pai, C. C. S. et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J. Clin. Invest. 129, 349–363 (2019).
Desnoyers, L. R. et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 5, 207ra144 (2013).
Puskas, J. et al. Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology 133, 206–220 (2011).
Winkler, J., Abisoye-Ogunniyan, A., Metcalf, K. J. & Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120 (2020).
Jiang, W. et al. Expression of membrane type-1 matrix metalloproteinase, MT1-MMP in human breast cancer and its impact on invasiveness of breast cancer cells. Int. J. Mol. Med. 17, 583–590 (2006).
Wu, K.-P. et al. MT1-MMP is not a good prognosticator of cancer survival: evidence from 11 studies. Tumor Biol. 35, 12489–12495 (2014).
Isaacson, K. J., Jensen, M. M., Subrahmanyam, N. B. & Ghandehari, H. Matrix-metalloproteinases as targets for controlled delivery in cancer: an analysis of upregulation and expression. J. Control Release 259, 62–75 (2017).
Hashimoto, M. et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med. 69, 301–318 (2018).
Zou, W., Wolchok, J. D. & Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv4 (2016).
Rovero, S. et al. DNA vaccination against rat Her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J. Immunol. 165, 5133–5142 (2000).
Acknowledgements
We thank Yang Wang for her technical assistance and providing reagents. We thank Benjamin Moon for the excellent editing. We thank Hui Su and Sai Yang for technical assistance in IBP. This work was partly supported by the funding from the National Science Foundation of China (No. 81803086) to Y. Liang.
Author information
Authors and Affiliations
Contributions
J.G., Y.L., H.P., and Y.-X.F. conceptualized the project and designed experiments. J.G. and Y.L. performed experiments. J.G. and Y.L. analyzed data. J.G., Y.L., H.P., and Y.-X.F. contributed to manuscript preparation. D.X., J.S., Y.C., and J.Z. provided technical or material supports. Y.L., H.P., and Y.-X.F. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Guo, J., Liang, Y., Xue, D. et al. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res 31, 1190–1198 (2021). https://doi.org/10.1038/s41422-021-00543-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-021-00543-4
This article is cited by
-
Interleukins in Epilepsy: Friend or Foe
Neuroscience Bulletin (2024)
-
New insights into the stemness of adoptively transferred T cells by γc family cytokines
Cell Communication and Signaling (2023)
-
The screening, identification, design and clinical application of tumor-specific neoantigens for TCR-T cells
Molecular Cancer (2023)
-
Small extracellular vesicle TGF-β in cancer progression and immune evasion
Cancer Gene Therapy (2023)
-
Biomimetic nanovaccine-mediated multivalent IL-15 self-transpresentation (MIST) for potent and safe cancer immunotherapy
Nature Communications (2023)
