
Abstract
Background
Despite a modest association between tobacco smoking and breast cancer risk reported by recent epidemiological studies, it is still equivocal whether smoking is causally related to breast cancer risk.
Methods
We applied Mendelian randomisation (MR) to evaluate a potential causal effect of cigarette smoking on breast cancer risk. Both individual-level data as well as summary statistics for 164 single-nucleotide polymorphisms (SNPs) reported in genome-wide association studies of lifetime smoking index (LSI) or cigarette per day (CPD) were used to obtain MR effect estimates. Data from 108,420 invasive breast cancer cases and 87,681 controls were used for the LSI analysis and for the CPD analysis conducted among ever-smokers from 26,147 cancer cases and 26,072 controls. Sensitivity analyses were conducted to address pleiotropy.
Results
Genetically predicted LSI was associated with increased breast cancer risk (OR 1.18 per SD, 95% CI: 1.07–1.30, P = 0.11 × 10–2), but there was no evidence of association for genetically predicted CPD (OR 1.02, 95% CI: 0.78–1.19, P = 0.85). The sensitivity analyses yielded similar results and showed no strong evidence of pleiotropic effect.
Conclusion
Our MR study provides supportive evidence for a potential causal association with breast cancer risk for lifetime smoking exposure but not cigarettes per day among smokers.
Similar content being viewed by others

Background
Breast cancer is the most common cancer in women, representing approximately one-quarter of all cancers diagnosed in women worldwide.1 Besides well-established risk factors for breast cancer, tobacco smoking has been widely studied as a potential risk factor for breast cancer since it is also a leading modifiable risk factor for cancers at sites not directly reached by tobacco smoke.2,3 Carcinogens associated with tobacco smoke include polycyclic aromatic hydrocarbons, aromatic amines and N-nitrosamines.4 It is biologically plausible that tobacco smoking may affect risk of breast cancer since metabolites of lipophilic tobacco-associated carcinogens have been detected in breast adipose tissue,5,6 and specific DNA adducts as well as p53 gene mutations are found in the breast cancer tissue of smokers.7,8,9
Based on the experimental and epidemiologic findings, evidence is insufficient to establish a causal relationship between tobacco smoking and breast cancer risk.2,10 Some of the inconsistencies in findings could be attributed to a potential dual effect of smoking on breast cancer.11 The anti-oestrogenic effect of smoking may attenuate or mask the carcinogenic effects.12 In a recent pooled analysis of 14 prospective cohort studies, adjusting for potential confounding by alcohol intake as well as body mass index (BMI), education, reproductive factors and other risk factors, a modest association was found between smoking and breast cancer risk.13 There was also an increased risk with longer duration of smoking prior to first birth, particularly for oestrogen receptor-positive tumours. It is however difficult to establish causation based on these observational studies.
Mendelian randomisation (MR) has been increasingly used to strengthen causal inference in observational studies and under certain assumptions is less vulnerable to residual confounding, reverse causation and selection bias.14 MR uses genetic variants such as single-nucleotide polymorphisms (SNPs) associated with an exposure of interest as an instrumental variable (IV), which is robust if three assumptions are met: (1) the genetic variants are casually associated with the exposure, (2) the variants are not associated with known or potential confounders for the exposure–outcome relationships and (3) the variants are associated with outcome only via the exposure of interest and not through other pathways.15,16 Given the unclear causal nature of the findings in observational studies, we conducted a MR analysis to investigate the association between smoking traits and breast cancer risk.
Methods
Study population
We used data from 81 studies participating in the Breast Cancer Association Consortium (BCAC), including 108,420 cases and 87,681 controls of European ancestry. Genotyping was performed using two custom-made genotyping arrays: OncoArray in 68,242 invasive breast cancer cases and 52,367 controls (https://epi.grants.cancer.gov/oncoarray/)17 and iCOGS arrays in 40,178 cases and 35,314 controls (http://ccge.medschl.cam.ac.uk/research/consortia/icogs/).18 Genotype data were imputed based on the 1000 Genomes project Phase 3 as the reference panel using the programme IMPUTE2.19 SNPs with high imputation quality (imputation r2 > 0.5) were included. Overlapping participants between datasets were excluded from the iCOGS dataset as the OncoArray provides a better genomic coverage than the iCOGS array. Demographic and epidemiologic data were harmonised across BCAC sites based on a standardised protocol and derived with respect to a reference date, which was date at diagnosis for cases and date at interview for controls. For controls and cases from the nested case-control studies, data from the baseline interview were considered, or if available, follow-up information. Chosen characteristics of the two datasets, including self-reported smoking behaviours (e.g., smoking status, smoking heaviness and smoking duration, including age at smoking initiation and lifetime smoking exposure) are shown in Supplementary Table 1. Ethics approval was obtained from the relevant institutional review boards for all BCAC studies, and all participants provided written informed consent.
Selection of SNPs associated with smoking exposure
Exposure variables were selected based on the availability of associated genetic variants to reflect smoking exposure. We included two quantitative smoking behaviour-related traits, cigarettes per day (CPD; average number of cigarettes smoked per day by ever-smokers) and lifetime smoking index (LSI; composite score that captures lifetime smoking exposure by taking into account smoking status as well as smoking duration, heaviness and cessation in ever-smokers).20 The recent GWAS of cigarettes per day from GSCAN consortium identified 55 conditionally independent genome-wide significant SNPs, explaining 1.09% of the variance in a sample of 337,334 ever-smokers of European ancestry.21 For LSI, 126 significantly associated SNPs, 0.36% explained variance in LSI, were identified based on a sample of 426,690 individuals (never-smokers and ever-smokers) of European ancestry from the UK biobank.20 The genetic scores of the two smoking behaviour-related traits were reported to be associated with significantly higher risks of lung cancer.20,22
For our analysis, SNPs were selected if they were reported to be associated at genome-wide significance level (P ≤ 5 × 10−8) and had a minor allele frequency (MAF) above or equal 1%. For each behaviour phenotype, we filtered the list of behaviour-associated SNPs so that the remaining SNPs were not in linkage disequilibrium (LD) (r2 > 0.1). For CPD, the SNP with lowest P value (rs10519203) of the correlated SNPs (rs12438181, rs28438420, rs72740955, rs146009840, rs28681284, rs8040868, r2 > 0.01) was retained. One SNP (rs4886550) was not available in our data and without any proxy SNPs (LD, r2 > 0.8). Given that alcohol consumption is a recognised confounder of the association between smoking and breast cancer risk,23,24,25 we excluded nine SNPs (three of CPD, six of LSI, respectively) correlated with any alcohol consumption-associated SNPs (r2 > 0.1) from recent large-scale GWAS (drinks per week, P ≤ 5 × 10−8).21 After exclusions, we included a total of 164 variants associated with one of the smoking traits; 44 and 120 variants, respectively, for CPD and LSI (Supplementary Table 2). No variants overlapped between the two smoking traits.
Statistical methods
Statistical power
Power calculations were conducted to estimate the magnitude of effects detectable with our study size assuming 5% α level and an R2 of 0.0109 for CPD and R2 of 0.0036 for LSI, which correspond to the variance in each smoking behaviour explained by the SNPs used for this analysis. Power calculations were performed using an online tool available at http://cnsgenomics.com/shiny/mRnd/.26 Detailed power calculations for all outcomes (invasive breast cancer, ER-positive and ER-negative) to detect different odd ratios are shown in Supplementary Table 3.
wPGS-based analyses
For our primary analysis, we generated weighted polygenic scores (wPGSs) using individual-level data of BCAC participants as follow: wPGS =\(\mathop {\sum }\nolimits_{i = 1}^n \beta _{gx} \ast \alpha _i\), which is the sum of the effect allele dosage (αi) (ranging from 0 to 2) for each SNP weighted by the β-coefficient βgx for the effect of the genetic variant (g) related to the smoking quality or/and duration (CPD, and LSI) (x) (Supplementary Table 2).20,21 The wPGSs of LSI was weakly correlated with wPGSs of CPD (0.17 in iCOGS and 0.03 in OncoArray) in ever-smokers. Analysis of LSI was conducted using all participants (including ever- and never-smokers), whereas that of CPD was performed solely in ever-smokers, and based on 26,147 cases (7342 for iCOGS, 18,805 for OncoArray) and 26,072 controls (8489 for iCOGS, 17,583 for OncoArray). These inclusion criteria correspond to those used in the GWAS studies that identified the SNPs for the smoking traits.20,21
Association analysis of the wPGSs with breast cancer risk using logistic regression was performed using fixed effect meta-analyses combining iCOGS and OncoArray results based on heterogeneity evaluated by Cochran’s Q statistics. The basic model (Model 1) was adjusted for age (continuous), principal components (PCs) of genetic ancestry (first ten PCs for iCOGS and OncoArray, separately) and study site, as previously described.27 In order to assess if the genetic instrument is independent of established risk factors for breast cancer, associations of wPGSs with selected breast cancer risk factors were assessed by linear regression for continuous variables and logistic regression for categorical variables (Supplementary Table 4). We adjusted for the risk factors that were associated with wPGSs of at least one of the smoking traits in Model 2. We additionally adjusted for alcohol assumption, a well-known confounder of the association between breast cancer risk and smoking, separately in Model 3 because of the large amount of missing data. Participants with missing covariables were excluded from all of the analyses.
Stratified analysis was performed to assess potential differences in associations with breast cancer risk by menopausal status (pre-, postmenopausal women) adjusting for age, study site and top ten PCs. Heterogeneity was tested employing the likelihood ratio test (LRT) for evaluating the multiplicative interaction terms in nested models. Polytomous regression was used to estimate the association according to oestrogen receptor (ER) status.
Scaling was applied to convert the wPGSs for CPD into meaningful units through dividing them by linear regression coefficients of self-reported CPD (0.35 per pack of cigarettes per day). The regression coefficient of CPD was derived from a meta-analysis of iCOGS and OncoArray data on smoking behaviours among 26,072 among ever-smoker controls (8490 and 17,703 controls of iCOGS and OncoArray, respectively).
Two-sample MR analyses
Five different two-sample MR methods using summary association data were applied: inverse-variance weighted (IVW),28 MR Egger,29 weighted median,30 weighted mode,31 and robust adjusted profile score (RAPS).32 Each of these methods makes slightly different assumptions about the nature of pleiotropy and therefore a roughly consistent point estimate across the multiple methods provides the strongest evidence of causal inference.28 The IVW method was implemented since the instruments consisted of multiple SNPs.33 Multivariable MR methods34 were conducted also using summary association data from GWASs of alcohol consumption (drinks per week),21 body mass index (BMI) among females35 and education attainment.36 To produce valid results, the IVW method requires that all instruments are associated with the exposure of interest (relevance assumption), but neither directly with the outcome of interest (only via the exposure; exclusion restriction) nor any confounders of the relationship between the exposure and the outcome (independence assumption).37 The intercept from MR-Egger regression is a statistical test for horizontal pleiotropy, whereas the slope can be interpreted as the smoking behaviour effect on breast cancer adjusted for horizontal pleiotropy.29 This method assumes however that the pleiotropic effects are independent of the instrument strength (InSIDE assumption). The weighted median estimator provides a valid causal estimate when at least half of the instruments are valid.30 The estimate from the weighted-mode analysis is valid when the largest group of instruments with consistent MR estimates is valid.31 MR-RAPS test extends the basic IVW random-effects approach by making the weight each variant receives in the analysis a function of the causal effect and the precision of the SNP-exposure association.32 The MR pleiotropy residual sum and outlier test (MR-PRESSO) was also implemented to identify outlying genetic variants and analyses were re-run after excluding these variants.38
All two-sample MR analyses using summary association data were performed with respect to three cancer susceptibility phenotypes: overall breast cancer (108,067 cases/88,386 controls) as well as oestrogen receptor (ER)-positive (70,435 cases) and ER-negative tumours (17,365 cases). Due to the nature of summary-level data, the analyses of both LSI and CPD were conducted using all samples regardless of smoking status.
R version 3.4.3 was used to conduct analyses. R package “Mendelian randomisation”, “mr_raps” and “MR-PRESSO” were used for two-sample MR analysis. All tests were considered at the 0.05 level of significance.
Results
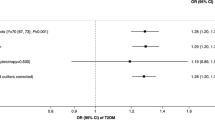
There was an association of wPGS for LSI with increased invasive breast cancer risk (OR per SD 1.18, 95% CI: 1.07–1.30, P = 0.11 × 10–2) whereas little evidence was found for an association between wPGSs for CPD and invasive breast cancer (OR 1.02 per pack of cigarettes per day, 95% CI: 0.78–1.19, P = 0.85) after adjustment for age and study (Model 1) (Table 1). Several breast cancer risk factors were associated with wPGSs of one of the smoking traits (CPD or/and LSI), including ever breastfeeding, menopausal status, age at menopause, BMI, age at first live birth, parity and education level (Supplementary Table 4). Adjustment for all of the identified risk factors did not change the association substantially (Model 2) (wPGSs for LSI, OR per SD 1.24, 95% CI: 1.06–1.45, P = 0.60 × 10−2) (Table 1). The point estimate of association between wPGS for LSI and invasive breast cancer risk remained unchanged after additional adjustment for alcohol consumption although imprecisely estimated (i.e. wide confidence intervals) (Model 3) (OR per SD 1.13, 95% CI: 0.86–1.49, P = 0.39). The association between wPGSs for CPD and invasive breast cancer did not change after adjustments.
There was no evidence for effect heterogeneity of the associations of LSI and CPD with breast cancer risk according to ER status or menopausal status (Supplementary Fig. 1).
Using IVW random-effects analysis, positive associations of genetically predicted LSI were found for overall breast cancer risk (OR per SD 1.14, 95% CI: 1.02–1.28, P = 0.02) and breast cancers according to ER status (OR per SD 1.14, 95% CI: 1.00–1.30, P = 0.04, for ER-positive and OR per SD 1.14, 95% CI: 0.95–1.37, P = 0.17, for ER-negative tumours) (Table 2, Supplementary Table 5 and Supplementary Figs. 2–4). There was no indication of horizontal pleiotropy based on the MR-Egger intercept test for any outcomes. The point estimates of associations were consistent across the different methods although the results based on MR-Egger regression, and weighted mode method were imprecisely estimated (i.e. wide confidence intervals). They remained substantially unchanged after multivariable adjustment for alcohol consumption, BMI and education. The MR-PRESSO analysis revealed three outliers for LSI. Removal of an outlier (rs2867112) with respect to risk for overall breast cancer and ER-negative cancer did not change the associations. No outlier was observed with ER-positive tumour (Table 2, Supplementary Table 5 and Supplementary Figs. 2–4).
For genetically predicted CPD, we found little evidence for an association with overall breast cancer and ER subtypes using IVW random-effects method (Table 2, Supplementary Table 5 and Supplementary Figs. 2–4). There was no indication of horizontal pleiotropy based on the MR-Egger intercept test. Results across two-sample MR analyses were consistent in overall breast cancer and ER subtypes without any indication of horizontal pleiotropy from the MR-Egger intercept. The MR-PRESSO analysis detected three outlying SNPs (rs11940255, rs1737894 and rs73229090) although the outcome of IVW analysis after outlier removed remained unchanged in overall breast cancer and ER-positive tumour risk. No outlier was observed with ER-negative disease (Table 2, Supplementary Table 5 and Supplementary Figs. 2–4).
Discussion
This MR study supports an association between genetically predicted lifetime smoking exposure and increasing invasive breast cancer risk but no clear association with cigarettes per day among smokers. The estimates based on the wPGS for the two smoking traits and several two-sample MR methods were consistent.
The LSI has not been assessed in studies of breast cancer risk but the modest association found in this analysis is in line with the modest associations of current and former smoking with invasive breast cancer risk reported in the recent large pooled analysis.13 We did not find support for an association between cigarettes per day in ever-smokers and invasive breast cancer risk, whereas modest associations were reported for cigarettes per day in current smokers compared with never-smokers in the pooled analysis. With the restriction to ever-smokers, the MR analysis of CPD had low statistical power for the very modest dose–response association estimated by our data (Table 1) as well as that reported in the pooled analysis.13
In addition to smoking exposure traits, we addressed two dichotomous smoking status traits, namely smoking initiation and smoking cessation, which account for the lifetime smoking exposure. Despite a pooled analysis of epidemiological studies reporting an increased risk of breast cancer associated with current smokers compared to non-smokers (OR of 1.02),13 a recent MR study found inconclusive evidence of association between genetically predicted smoking initiation and breast cancer risk using summary-level data (OR: 1.05, 95% CI: 0.99–1.12, P = 0.12 in BCAC; OR: 0.97, 95% CI: 0.90–1.06, P = 0.51 in UK Biobank).39 We conducted association analyses of wPGS of smoking initiation and smoking cessation with breast cancer risk (see Supplementary Note) and found no statically significant association for either smoking initiation (OR 1.05, 95% CI 0.99–1.11, P = 0.08) or smoking cessation (OR 1.06, 95% CI: 0.88–1.27, P = 0.52) (Supplementary Table 6). The results remained unchanged after adjusting for breast cancer risk factors (Supplementary Table 6). We cannot rule out that the MR analysis of the smoking status had low statistical power to detect the very modest association estimated by our data (Table 1) along with that reported in the pooled analysis.13 It is also possible that the MR analysis of smoking status especially smoking initiation alone does not capture the association between cigarette smoking and breast cancer risk comparing to the LSI which accounts for other smoking traits.
There are several not entirely resolved issues concerning the association between smoking and breast cancer risks, such as potential effect modification by timing of smoking exposure, menopausal status and oestrogen receptor (ER) status, potential confounding by alcohol consumption. We conducted an association analysis between wPGS of age at smoking initiation and invasive breast cancer (see Supplementary Note). The result showed an inverse association but statistically nonsignificant with low precision (OR 0.88, 95% CI: 0.25–3.07, P = 0.84) (Supplementary Table 6). This is in line with the result of a pooled analysis of epidemiological studies showing that women who started smoking later than 24 years old were at lower breast cancer risk than those who started smoking earlier when compared to non-smokers.13 Smoking initiation in relation to first birth has been considered an essential factor in the association with breast cancer risk since the undifferentiated breast epithelium is particularly susceptible to carcinogens before the first birth.40 Indeed this appears to be supported by findings of a stronger association with smoking with breast cancer risk if initiated before first birth and a stronger influence of smoking on breast cancer among women who started smoking more than 10 years before the first full-term pregnancy on breast cancer.13,41,42,43,44,45,46 Since the relevant information was only available for a subset of study participants, we did not have sufficient power to address potential differential associations according to timing of smoking exposure in relation to first birth.
Stronger associations between smoking and breast cancer among premenopausal women have been hypothesised since the morphology of the breast and the endogenous hormone levels change substantially during the menopausal transition, and menopausal status alter other breast cancer risk factors.10 The MR results confirmed the lack of effect modification by menopausal status also reported by previous epidemiological studies.13,47,48 Despite early evidence against a differential association by ER status,2,3 recent epidemiologic studies reported a stronger association for risk of ER-positive breast cancer.10,13,41 We did not find clear evidence for heterogeneity by ER status although power to detect effect heterogeneity was limited particularly due to the small sample size for ER-negative disease.
Previous epidemiologic studies have addressed the confounding effect by alcohol consumption and found an association of smoking with breast cancer risk after stratifying on alcohol consumption.13,41,42,43 We addressed this issue by excluding SNPs associated with alcohol intake from the wPGS. However, the results did not change significantly in sensitivity analyses with wPGS, including the overlapping alcohol consumption-associated SNPs (Supplementary Table 7). Also, our multivariable analyses adjusting for alcohol consumption yielded association estimates that remained unchanged. The lower precision can be attributed to the reduced dataset that required information on alcohol intake with the ensuing diminished power.
Despite epidemiological evidence indicating alcohol consumption as an established risk factor for breast cancer risk,49 a recent MR study reported no significant association of breast cancer risk with genetically predicted alcohol intake using summary association data.39 We conducted an analysis between wPGS of alcohol consumption and invasive breast cancer using individual-level data (see Supplementary Note). We found no clear evidence of association between genetically predicted alcohol consumption and invasive breast cancer (OR: 0.98, 95% CI: 0.86–1.11, P = 0.74). The association remained unchanged after adjusting for LSI (OR: 0.88, 95% CI: 0.72–1.09, P = 0.25).
Strengths and limitations
Despite the relatively large sample size used for our analyses, weak statistical power to detect modest associations should be considered when interpreting the results. The power calculation shows that our study had 68% power to detect an OR of 1.20 per SD change in LSI but only around 9% power for an OR of 1.05 in combined dataset (iCOGS and OncoArray) (Supplementary Table 3). Since the reported order of magnitude for associations with CPD is around 1.05 per ten cigarettes,13 we cannot exclude that the lack of association observed in the MR analysis particularly regarding CPD may be due to limited power. The unit for LSI is not scaled due to the nature of the phenotype. The reported order of magnitude for associations with LSI can therefore not be directly compared with the relative risk estimates for smoking from observational studies.
MR estimates have a causal interpretation only if the assumptions of the instrumental variable approach hold. Even though we performed extensive sensitivity analyses to detect potential violations, it is difficult to prove the validity of the assumptions. The LSI captures multiple aspects of smoking behaviours, which could have introduced more potential for horizontal pleiotropy. The more diffuse the definition of smoking, the more lifestyle factors might be correlated, making it especially important to test for horizontal pleiotropy. No evidence of pleiotropic effects was found by conducting various sensitivity analyses; however, residual pleiotropy is difficult to exclude and should be considered.
The genetic instrument for LSI allows for the use of the large entire sample to conduct MR analysis without stratifying on smoking status. The analyses for CPD were restricted to smokers by reason that the CPD-associated SNPs were identified among ever-smokers in the GWAS study,21 which reduce statistical power to detect an association. Moreover, we should note that restricting to ever-smokers may induce a sampling bias and invalidate the MR assumptions. By restricting to smokers, smoking initiation can open up the path from exposure (wPGS for CPD) to outcome (breast cancer risk). It can make the association between smoking and breast cancer risk appear weaker by removing a part of the association that is attributable to smoking initiation.
Another limitation is that our analysis was restricted to participants of European ancestry; therefore, our results may not apply to populations of other ethnicities. However, it reduces the potential bias caused by population stratification.
Conclusion
In conclusion, this Mendelian randomisation analysis using both individual-level data and summary statistics supports a causal association between lifetime smoking exposure and breast cancer risk. Larger studies for MR analysis are warranted to address additional aspects of smoking behaviour.
References
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A. & Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Tobacco smoke and involuntary smoking, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 83, 1–1438 (2004).
Centers for Disease C, Prevention, National Center for Chronic Disease P, Health P, Office on S, Health. Publications and reports of the surgeon general. in How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General Centers for Disease Control and Prevention (US): Atlanta (GA) (2010).
Hecht, S. S. Tobacco smoke carcinogens and breast cancer. Environ. Mol. Mutagen. 39, 119–126 (2002).
Petrakis, N. L., Gruenke, L. D., Beelen, T. C., Castagnoli, N. Jr. & Craig, J. C. Nicotine in breast fluid of nonlactating women. Science 199, 303–305 (1978).
Petrakis, N. L., Maack, C. A., Lee, R. E. & Lyon, M. Mutagenic activity in nipple aspirates of human breast fluid. Cancer Res. 40, 188–189 (1980).
Conway, K., Edmiston, S. N., Cui, L., Drouin, S. S., Pang, J., He, M. et al. Prevalence and spectrum of p53 mutations associated with smoking in breast cancer. Cancer Res. 62, 1987–1995 (2002).
Li, D., Zhang, W., Sahin, A. A. & Hittelman, W. N. DNA adducts in normal tissue adjacent to breast cancer: a review. Cancer Detect. Prev. 23, 454–462 (1999).
Rundle, A., Tang, D., Hibshoosh, H., Estabrook, A., Schnabel, F., Cao, W. et al. The relationship between genetic damage from polycyclic aromatic hydrocarbons in breast tissue and breast cancer. Carcinogenesis 21, 1281–1289 (2000).
Warren, G. W., Alberg, A. J., Kraft, A. S. & Cummings, K. M. The 2014 Surgeon General’s report: “the health consequences of smoking–50 years of progress”: a paradigm shift in cancer care. Cancer 120, 1914–1916 (2014).
Band, P. R., Le, N. D., Fang, R. & Deschamps, M. Carcinogenic and endocrine disrupting effects of cigarette smoke and risk of breast cancer. Lancet 360, 1044–1049 (2002).
Clemons, M. & Goss, P. Estrogen and the risk of breast cancer. New Engl. J. Med. 344, 276–285 (2001).
Gaudet, M. M., Carter, B. D., Brinton, L. A., Falk, R. T., Gram, I. T., Luo, J. et al. Pooled analysis of active cigarette smoking and invasive breast cancer risk in 14 cohort studies. Int. J. Epidemiol. 46, 881–893 (2017).
Smith, G. D. & Ebrahim, S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 32, 1–22 (2003).
Evans, D. M. & Davey Smith, G. Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu. Rev. Genomics Hum. Genet. 16, 327–350 (2015).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology 28, 30–42 (2017).
Michailidou, K., Lindstrom, S., Dennis, J., Beesley, J., Hui, S., Kar, S. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Michailidou, K., Hall, P., Gonzalez-Neira, A., Ghoussaini, M., Dennis, J., Milne, R. L. et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 45, 353–361 (2013).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Wootton, R. E., Richmond, R. C., Stuijfzand, B. G., Lawn, R. B., Sallis, H. M., Taylor, G. M. J. et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med, 50, 2435–2443 (2020).
Liu, M., Jiang, Y., Wedow, R., Li, Y., Brazel, D. M., Chen, F. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
Zhou, W., Liu, G., Hung, R. J., Haycock, P. C., Aldrich, M. C., Andrew, A. S. et al. Causal relationships between body mass index, smoking and lung cancer: univariable and multivariable Mendelian randomization. Int. J. Cancer https://doi.org/10.1002/ijc.33292 (2020).
Dumitrescu, R. G. & Shields, P. G. The etiology of alcohol-induced breast cancer. Alcohol 35, 213–225 (2005).
Hamajima, N., Hirose, K., Tajima, K., Rohan, T., Calle, E. E., Heath, C. W. Jr. et al. Alcohol, tobacco and breast cancer–collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br. J. Cancer 87, 1234–1245 (2002).
Moore, A. A., Gould, R., Reuben, D. B., Greendale, G. A., Carter, M. K., Zhou, K. et al. Longitudinal patterns and predictors of alcohol consumption in the United States. Am. J. Public Health 95, 458–465 (2005).
Brion, M. J., Shakhbazov, K. & Visscher, P. M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 42, 1497–1501 (2013).
Mavaddat, N., Michailidou, K., Dennis, J., Lush, M., Fachal, L., Lee, A. et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 104, 21–34 (2019).
Lawlor, D. A., Tilling, K. & Davey Smith, G. Triangulation in aetiological epidemiology. Int. J. Epidemiol. 45, 1866–1886 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genetic Epidemiol. 40, 304–314 (2016).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998 (2017).
Zhao, Q., Wang, J., Hemani, G., Bowden, J. & Small, D. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. Annals of Statistics, 48, 1742–1769 (2020).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genetic Epidemiol. 37, 658–665 (2013).
Sanderson, E., Davey Smith, G., Windmeijer, F. & Bowden, J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol. 48, 713–727 (2019).
Pulit, S. L., Stoneman, C., Morris, A. P., Wood, A. R., Glastonbury, C. A., Tyrrell, J. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174 (2019).
Lee, J. J., Wedow, R., Okbay, A., Kong, E., Maghzian, O., Zacher, M. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Haycock, P. C., Burgess, S., Wade, K. H., Bowden, J., Relton, C. & Davey Smith, G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am. J. Clin. Nutr. 103, 965–978 (2016).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Larsson, S. C., Carter, P., Kar, S., Vithayathil, M., Mason, A. M., Michaëlsson, K. et al. Smoking, alcohol consumption, and cancer: a Mendelian randomisation study in UK Biobank and international genetic consortia participants. PLoS Med. 17, e1003178–e1003178 (2020).
Russo, J., Hu, Y. F., Yang, X. & Russo, I. H. Developmental, cellular, and molecular basis of human breast cancer. J. Natl Cancer Inst. Monographs. 27, 17–37 (2000).
Dossus, L., Boutron-Ruault, M., Kaaks, R., Gram, I., Vilier, A., Fervers, B. et al. Active and passive cigarette smoking and breast cancer risk: results from the EPIC cohort. Int. J. Cancer 134, 1871–1888 (2014).
Gram, I. T., Little, M. A., Lund, E. & Braaten, T. The fraction of breast cancer attributable to smoking: the Norwegian women and cancer study 1991–2012. Br. J. Cancer 115, 616–623 (2016).
Gram, I. T., Park, S. Y., Kolonel, L. N., Maskarinec, G., Wilkens, L. R., Henderson, B. E. et al. Smoking and risk of breast cancer in a racially/ethnically diverse population of mainly women who do not drink alcohol: the MEC study. Am. J. Epidemiol. 182, 917–925 (2015).
Gaudet, M. M., Gapstur, S. M., Sun, J., Diver, W. R., Hannan, L. M. & Thun, M. J. Active smoking and breast cancer risk: original cohort data and meta-analysis. J. Natl Cancer Inst. 105, 515–525 (2013).
Catsburg, C., Miller, A. B. & Rohan, T. E. Active cigarette smoking and risk of breast cancer. Int. J. Cancer 136, 2204–2209 (2015).
Rosenberg, L., Boggs, D. A., Bethea, T. N., Wise, L. A., Adams-Campbell, L. L. & Palmer, J. R. A prospective study of smoking and breast cancer risk among African-American women. Cancer Causes Control 24, 2207–2215 (2013).
Egan, K. M., Stampfer, M. J., Hunter, D., Hankinson, S., Rosner, B. A., Holmes, M. et al. Active and passive smoking in breast cancer: prospective results from the Nurses’ Health Study. Epidemiology 13, 138–145 (2002).
Terry, P. D. & Rohan, T. E. Cigarette smoking and the risk of breast cancer in women: a review of the literature. Cancer Epidemiol. Biomark. Prev. 11, 953–971 (2002).
World Cancer Research Fund/American Institute for Cancer Research. Continuous Update Project Expert Report 2018. Diet, nutrition, physical activity and breast cacner. Avaiable at https://dietandcancerreport.org.
Acknowledgements
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out. The COGS study would not have been possible without the contributions of the following: Andrew Berchuck (OCAC), Rosalind A. Eeles, Ali Amin Al Olama, Zsofia Kote-Jarai, Sara Benlloch (PRACTICAL), Antonis Antoniou, and Lesley McGuffog (CIMBA), Andrew Lee, and Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Anna Gonzalez-Neira and the staff of the CNIO genotyping unit, and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Sune F. Nielsen and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility. ABCFS thank Maggie Angelakos, Judi Maskiell, Gillian Dite. ABCS thanks the Blood bank Sanquin, The Netherlands. ABCTB Investigators: Christine Clarke, Rosemary Balleine, Robert Baxter, Stephen Braye, Jane Carpenter, Jane Dahlstrom, John Forbes, Soon Lee, Debbie Marsh, Adrienne Morey, Nirmala Pathmanathan, Rodney Scott, Allan Spigelman, Nicholas Wilcken, Desmond Yip. Samples are made available to researchers on a non-exclusive basis. BBCS thanks Eileen Williams, Elaine Ryder-Mills, Kara Sargus. BCEES thanks Allyson Thomson, Christobel Saunders, Terry Slevin, BreastScreen Western Australia, Elizabeth Wylie, Rachel Lloyd. The BCINIS study would not have been possible without the contributions of Dr. K. Landsman, Dr. N. Gronich, Dr. A. Flugelman, Dr. W. Saliba, Dr. E. Liani, Dr. I. Cohen, Dr. S. Kalet, Dr. V. Friedman, Dr. O. Barnet of the NICCC in Haifa, and all the contributing family medicine, surgery, pathology and oncology teams in all medical institutes in Northern Israel. BIGGS thanks Niall McInerney, Gabrielle Colleran, Andrew Rowan, Angela Jones. The BREOGAN study would not have been possible without the contributions of the following: Manuela Gago-Dominguez, Jose Esteban Castelao, Angel Carracedo, Victor Muñoz Garzón, Alejandro Novo Domínguez, Maria Elena Martinez, Sara Miranda Ponte, Carmen Redondo Marey, Maite Peña Fernández, Manuel Enguix Castelo, Maria Torres, Manuel Calaza (BREOGAN), José Antúnez, Máximo Fraga and the staff of the Department of Pathology and Biobank of the University Hospital Complex of Santiago-CHUS, Instituto de Investigación Sanitaria de Santiago, IDIS, Xerencia de Xestion Integrada de Santiago-SERGAS; Joaquín González-Carreró and the staff of the Department of Pathology and Biobank of University Hospital Complex of Vigo, Instituto de Investigacion Biomedica Galicia Sur, SERGAS, Vigo, Spain. BSUCH thanks Peter Bugert, Medical Faculty Mannheim. CBCS thanks study participants, co-investigators, collaborators and staff of the Canadian Breast Cancer Study, and project coordinators Agnes Lai and Celine Morissette. CCGP thanks Styliani Apostolaki, Anna Margiolaki, Georgios Nintos, Maria Perraki, Georgia Saloustrou, Georgia Sevastaki, Konstantinos Pompodakis. CGPS thanks staff and participants of the Copenhagen General Population Study. For the excellent technical assistance: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, Dorthe Kjeldgård Hansen. The Danish Cancer Biobank is acknowledged for providing infrastructure for the collection of blood samples for the cases. CNIO-BCS thanks Guillermo Pita, Charo Alonso, Nuria Álvarez, Pilar Zamora, Primitiva Menendez, the Human Genotyping-CEGEN Unit (CNIO). Investigators from the CPS-II cohort thank the participants and Study Management Group for their invaluable contributions to this research. They also acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, as well as cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. The CTS Steering Committee includes Leslie Bernstein, Susan Neuhausen, Huiyan Ma, and Jessica Clague DeHart at the Beckman Research Institute of City of Hope, Dennis Deapen, Rich Pinder, and Eunjung Lee at the University of Southern California, Pam Horn-Ross, Peggy Reynolds, Christina Clarke Dur and David Nelson at the Cancer Prevention Institute of California, Hoda Anton-Culver, Argyrios Ziogas, and Hannah Park at the University of California Irvine, and Fred Schumacher at Case Western University. DIETCOMPLYF thanks the patients, nurses and clinical staff involved in the study. The DietCompLyf study was funded by the charity Against Breast Cancer (Registered Charity Number 1121258) and the NCRN. We thank the participants and the investigators of EPIC (European Prospective Investigation into Cancer and Nutrition). ESTHER thanks Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, Katja Butterbach. FHRISK thanks NIHR for funding. GC-HBOC thanks Stefanie Engert, Heide Hellebrand, Sandra Kröber and LIFE-Leipzig Research Centre for Civilization Diseases (Markus Loeffler, Joachim Thiery, Matthias Nüchter, Ronny Baber). The GENICA Network: Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany [HB, WYL], German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ) Partner Site Tübingen (HB), gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder—EXC 2180–390900677 (HB), Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany (Yon-Dschun Ko, Christian Baisch), Institute of Pathology, University of Bonn, Germany (Hans-Peter Fischer), Molecular Genetics of Breast Cancer, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany (Ute Hamann), Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany (Thomas Brüning, Beate Pesch, Sylvia Rabstein, Anne Lotz); and Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany (Volker Harth). GLACIER thanks Kelly Kohut, Patricia Gorman, Maria Troy. HABCS thanks Michael Bremer and Peter Hillemanns. HEBCS thanks Sofia Khan, Johanna Kiiski, Carl Blomqvist, Kristiina Aittomäki, Rainer Fagerholm, and Kirsimari Aaltonen. HMBCS thanks Hans Christiansen and Johann H. Karstens. HUBCS thanks Shamil Gantsev. ICICLE thanks Kelly Kohut, Michele Caneppele, Maria Troy. KARMA and SASBAC thank the Swedish Medical Research Counsel. KBCP thanks Eija Myöhänen, Helena Kemiläinen. kConFab/AOCS wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab. LMBC thanks Gilian Peuteman, Thomas Van Brussel, EvyVanderheyden and Kathleen Corthouts. MABCS thanks Milena Jakimovska (RCGEB “Georgi D. Efremov”), Snezhana Smichkoska, Emilija Lazarova (University Clinic of Radiotherapy and Oncology), Dzengis Jasar, Mitko Karadjozov (Adzibadem-Sistina Hospital), Andrej Arsovski and Liljana Stojanovska (Re-Medika Hospital) for their contributions and commitment to this study. MARIE thanks Petra Seibold, Dieter Flesch-Janys, Judith Heinz, Nadia Obi, Alina Vrieling, Sabine Behrens, Ursula Eilber, Muhabbet Celik, Til Olchers and Stefan Nickels. MBCSG (Milan Breast Cancer Study Group):, Siranoush Manoukian, Bernard Peissel, Jacopo Azzollini, Dario Zimbalatti, Daniela Zaffaroni, Bernardo Bonanni, Irene Feroce, Mariarosaria Calvello, Aliana Guerrieri Gonzaga, Monica Marabelli, Davide Bondavalli and the personnel of the Cogentech Cancer Genetic Test Laboratory The MCCS was made possible by the contribution of many people, including the original investigators, the teams that recruited the participants and continue working on follow-up, and the many thousands of Melbourne residents who continue to participate in the study. We thank the coordinators, the research staff and especially the MMHS participants for their continued collaboration on research studies in breast cancer. MSKCC thanks Marina Corines, Lauren Jacobs. MTLGEBCS would like to thank Martine Tranchant (CHU de Québec—Université Laval Research Center), Marie-France Valois, Annie Turgeon and Lea Heguy (McGill University Health Center, Royal Victoria Hospital; McGill University) for DNA extraction, sample management and skilful technical assistance. J.S. is Chair holder of the Canada Research Chair in Oncogenetics. The following are NBCS Collaborators: Kristine K. Sahlberg (PhD), Lars Ottestad (MD), Rolf Kåresen (Prof. Em.), Dr. Ellen Schlichting (MD), Marit Muri Holmen (MD), Toril Sauer (MD), Vilde Haakensen (MD), Olav Engebråten (MD), Bjørn Naume (MD), Alexander Fosså (MD), Cecile E. Kiserud (MD), Kristin V. Reinertsen (MD), Åslaug Helland (MD), Margit Riis (MD), Jürgen Geisler (MD), Anne-Lise Børresen-Dale (Prof. Em.), OSBREAC and Grethe I. Grenaker Alnæs (MSc). NBHS thank study participants and research staff for their contributions and commitment to the studies. For NHS and NHS2 the study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the NHS and NHS2 for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA and WY. The authors assume full responsibility for analyses and interpretation of these data. OBCS thanks Arja Jukkola-Vuorinen, Mervi Grip, Saila Kauppila, Meeri Otsukka, Leena Keskitalo and Kari Mononen for their contributions to this study. The OFBCR thanks Teresa Selander, Nayana Weerasooriya and Steve Gallinger. ORIGO thanks E. Krol-Warmerdam, and J. Blom for patient accrual, administering questionnaires, and managing clinical information. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner. The ethical approval for the POSH study is MREC /00/6/69, UKCRN ID: 1137. PREFACE thanks Sonja Oeser and Silke Landrith. PROCAS thanks NIHR for funding. The RBCS thanks Corine M. Beaufort, Jannet Blom, Renée Broeren-Foekens, Saskia Pelders, Wendy J.C. Prager-van der Smissen, Kirsten Ruigrok-Ritstier, Anita M.A.C. Trapman-Jansen, Michelle van der Vlugt-Daane, Vanja de Weerd, and the Erasmus MC Family Cancer Clinic. SBCS thanks Sue Higham, Helen Cramp, Dan Connley, Ian Brock, Sabapathy Balasubramanian and Malcolm W.R. Reed. We thank the SEARCH and EPIC teams. SKKDKFZS thanks all study participants, clinicians, family doctors, researchers and technicians for their contributions and commitment to this study. We thank the SUCCESS Study teams in Munich, Duessldorf, Erlangen and Ulm. SZBCS thanks Ewa Putresza. UCIBCS thanks Irene Masunaka. UKBGS thanks Breast Cancer Now and the Institute of Cancer Research for support and funding of the Breakthrough Generations Study, and the study participants, study staff, and the doctors, nurses and other health care providers and health information sources who have contributed to the study. We acknowledge NHS funding to the Royal Marsden/ICR NIHR Biomedical Research Centre. We thank the UKOPS participants who donated their samples and data and all the researchers, especially Andy Ryan, Simon Gayther and Ian Jacobs.
NBCS collaborators
Anne-Lise Børresen-Dale89,137,138,139,140,141,142,143,144,145,146,147, Grethe I. Grenaker Alnæs89,137,138,139,140,141,142,143,144,145,146,147, Kristine K. Sahlberg89,137,138,139,140,141,142,143,144,145,146,147, Lars Ottestad89,137,138,139,140,141,142,143,144,145,146,147, Rolf Kåresen89,137,138,139,140,141,142,143,144,145,146,147, Ellen Schlichting89,137,138,139,140,141,142,143,144,145,146,147, Marit Muri Holmen89,137,138,139,140,141,142,143,144,145,146,147, Toril Sauer89,137,138,139,140,141,142,143,144,145,146,147, Vilde Haakensen89,137,138,139,140,141,142,143,144,145,146,147, Olav Engebråten89,137,138,139,140,141,142,143,144,145,146,147, Bjørn Naume89,137,138,139,140,141,142,143,144,145,146,147, Alexander Fosså89,137,138,139,140,141,142,143,144,145,146,147, Cecile E. Kiserud89,137,138,139,140,141,142,143,144,145,146,147, Kristin V. Reinertsen89,137,138,139,140,141,142,143,144,145,146,147, Åslaug Helland89,137,138,139,140,141,142,143,144,145,146,147, Margit Riis89,137,138,139,140,141,142,143,144,145,146,147, Jürgen Geisler89,137,138,139,140,141,142,143,144,145,146,147
ABCTB investigators
Christine Clarke148, Deborah Marsh148, Rodney Scott148, Robert Baxter148, Desmond Yip148, Jane Carpenter148, Alison Davis148, Nirmala Pathmanathan148, Peter Simpson148, Dinny Graham148, Mythily Sachchithananthan148
kConFab investigators
David Amor149,150, Lesley Andrews149,150, Yoland Antill149,150, Rosemary Balleine149,150, Jonathan Beesley149,150, Ian Bennett149,150, Michael Bogwitz149,150, Leon Botes149,150, Meagan Brennan149,150, Melissa Brown149,150, Michael Buckley149,150, Jo Burke149,150, Phyllis Butow149,150, Liz Caldon149,150, Ian Campbell149,150, Deepa Chauhan149,150, Manisha Chauhan149,150, Georgia Chenevix-Trench149,150, Alice Christian149,150, Paul Cohen149,150, Alison Colley149,150, Ashley Crook149,150, James Cui149,150, Margaret Cummings149,150, Sarah-Jane Dawson149,150, Anna DeFazio149,150, Martin Delatycki149,150, Rebecca Dickson149,150, Joanne Dixon149,150, Ted Edkins149,150, Stacey Edwards149,150, Gelareh Farshid149,150, Andrew Fellows149,150, Georgina Fenton149,150, Michael Field149,150, James Flanagan149,150, Peter Fong149,150, Laura Forrest149,150, Stephen Fox149,150, Juliet French149,150, Michael Friedlander149,150, Clara Gaff149,150, Mike Gattas149,150, Peter George149,150, Sian Greening149,150, Marion Harris149,150, Stewart Hart149,150, Nick Hayward149,150, John Hopper149,150, Cass Hoskins149,150, Clare Hunt149,150, Paul James149,150, Mark Jenkins149,150, Alexa Kidd149,150, Judy Kirk149,150, Jessica Koehler149,150, James Kollias149,150, Sunil Lakhani149,150, Mitchell Lawrence149,150, Geoff Lindeman149,150, Lara Lipton149,150, Liz Lobb149,150, Graham Mann149,150, Deborah Marsh149,150, Sue Anne McLachlan149,150, Bettina Meiser149,150, Roger Milne149,150, Sophie Nightingale149,150, Shona O’Connell149,150, Sarah O’Sullivan149,150, David Gallego Ortega149,150, Nick Pachter149,150, Briony Patterson149,150, Amy Pearn149,150, Kelly Phillips149,150, Ellen Pieper149,150, Edwina Rickard149,150, Bridget Robinson149,150, Mona Saleh149,150, Elizabeth Salisbury149,150, Christobel Saunders149,150, Jodi Saunus149,150, Rodney Scott149,150, Clare Scott149,150, Adrienne Sexton149,150, Andrew Shelling149,150, Peter Simpson149,150, Melissa Southey149,150, Amanda Spurdle149,150, Jessica Taylor149,150, Renea Taylor149,150, Heather Thorne149,150, Alison Trainer149,150, Kathy Tucker149,150, Jane Visvader149,150, Logan Walker149,150, Rachael Williams149,150, Ingrid Winship149,150, Mary Ann Young149,150
Author information
Authors and Affiliations
Consortia
Contributions
J.C-C. conceived the project. H.A.P and J.C-C. conducted analyses, interpreted the results and wrote and edited the original and revised paper. S.N. contributed to the data analyses and the interpretation of the results. S.Be. and P.M.K. contributed to the data preparation. D.F.E., W.Z. and P.Kr. contributed to the results interpretation and the manuscript drafting and revision. D.F.E. coordinated the BCAC and led the iCOGS and OncoArray genotyping. P.H. led the COGS collaboration, J.S. led the OncoArray collaboration. M.K.B., Q.W. and J.Den. coordinated the BCAC database. K.Mi., T.U.A., I.L.A., H.A-C., N.N.A., V.A., K.J.A., A.Au., A.B., L.E.BF., H.Bec., M.W.B., S.Be., J.Ben., M.Berm., N.V.B., S.E.B., H.Bra., H.Bre., S.Y.B., B.Bur., D.Cam., F.C., J.E.C., S.J.C., G.C-T., C.L.C., N.C., D.M.C., F.J.C., A.Cox., S.S.C., K.Cz., M.B.D., P.D., T.D., I.D-S-S., M.Dw., D.M.E., AH.E., C.En., M.E., DG.E., P.A.F., H.F., L.F., M.G-C., J.A.G-S., M.M.G., G.G.G., G.G., M.S.G., D.E.G., A.G-N., M.G., P.G., E.H., C.A.H., N.H., P.H., U.H., S.H., E.F.H., S.N.H., W.H., B.A.M.H.-G., J.L.H., D.J.H., A.I., k.I., A.Jage., A.Jak., E.M.J., A.J., R.K., P.M.K., R.K., E.Kh., C.M.Ki., L.B.K., S.Kou., V.N.K., A.W.K., J.V.L., D.La., L.LM., W-L.L., J.Lu., A.Man., M.Man., S.Mar., ME.M., D.M., A.Me., U.Me., R.L.M., T.A.M., H.Ne., W.G.N., B.G.N., K.Of., A.F.O., H.O., T-W.P-S., P.Pe., J.Pet., D.P.K., N.Pre., P.Ra., G.R., H.S.R., A.Ro., E.S., E.J.S., M.K.S., R.K.S., M.J.S., L.Sch., C.Sc., M.Sh., X-O.S., J.Si., A.Sm., M.C.S., J.J.S., V.S., A.J.S., R.M.T., W.J.T., J.A.T., MB.T., I.T., M.A.T., T.T., C.M.V., E.M.VV., J.Vi., S.W., C.We., R.Wi., A.W., A.Z., P.D.P.P., W.Z., P.Kr. and J.C-C. led individual studies and contributed to the design of the study and data collection.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Collection of blood samples, urine samples and questionnaire information was undertaken with written informed consent and relevant ethical review board approval in accordance with the tenets of the Declaration of Helsinki (Supplementary Table 8).
Consent to publish
Not applicable.
Data availability
Availability of lifetime smoking index GWAS described by Wootton et al.20 Availability of self-reported cigarettes per day GWAS described by Liu et al.21 Individual genotyping data from BCAC will not be made publicly available due to restraints imposed by the ethics committees of individual studies; requests for data can be made to the Data Access Coordination Committee of BCAC (http://bcac.ccge.medschl.cam.ac.uk/). Summary results for all variants genotyped by BCAC are available at http://bcac.ccge.medschl.cam.ac.uk/.
Competing interests
U.Me. has reported a conflict of interest outside of the submitted work (owing stocks in Abcodia awarded to her by University College London). P.H., R.M.T. and A.Cox. are subject editors for the British Journal of Cancer. P.D.P. P., E.J.S. and K.Cz. are editorial board members for the British Journal of Cancer. The remaining authors declare no competing interests.
Funding information
BCAC is funded by Cancer Research UK [C1287/A16563, C1287/A10118], the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST respectively), and by the European Community´s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2–2009-223175) (COGS). The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation or writing of the report. Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (grant GPH-129344) and, the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSR-SIIRI-701 grant, and the Quebec Breast Cancer Foundation. Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112 - the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, and Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. The DRIVE Consortium was funded by U19 CA148065. The Australian Breast Cancer Family Study (ABCFS) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organisations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. M.C.S. is a NHMRC Senior Research Fellow. The ABCS study was supported by the Dutch Cancer Society [grants NKI 2007-3839; 2009 4363]. The Australian Breast Cancer Tissue Bank (ABCTB) was supported by the National Health and Medical Research Council of Australia, The Cancer Institute NSW and the National Breast Cancer Foundation. The AHS study is supported by the intramural research program of the National Institutes of Health, the National Cancer Institute (grant number Z01-CP010119), and the National Institute of Environmental Health Sciences (grant number Z01-ES049030). The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS is funded by Cancer Research UK and Breast Cancer Now and acknowledges NHS funding to the NIHR Biomedical Research Centre, and the National Cancer Research Network (NCRN). The BCEES was funded by the National Health and Medical Research Council, Australia. For the BCFR-NY, BCFR-PA, BCFR-UT, this work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organisations imply endorsement by the US Government or the BCFR. For BIGGS, ES is supported by NIHR Comprehensive Biomedical Research Centre, Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London, United Kingdom. IT is supported by the Oxford Biomedical Research Centre. The BREast Oncology GAlician Network (BREOGAN) is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigacion Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). CBCS is funded by the Canadian Cancer Society (grant # 313404) and the Canadian Institutes of Health Research. CCGP is supported by funding from the University of Crete. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l’Alimentation, de l’Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. The CNIO-BCS was supported by the Instituto de Salud Carlos III, the Red Temática de Investigación Cooperativa en Cáncer and grants from the Asociación Española Contra el Cáncer and the Fondo de Investigación Sanitario (PI11/00923 and PI12/00070). The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. The CTS was initially supported by the California Breast Cancer Act of 1993 and the California Breast Cancer Research Fund (contract 97-10500) and is currently funded through the National Institutes of Health (R01 CA77398, UM1 CA164917 and U01 CA199277). Collection of cancer incidence data was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885. The University of Westminster curates the DietCompLyf database funded by Against Breast Cancer Registered Charity No. 1121258 and the NCRN. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by: Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation, the Stavros Niarchos Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). FHRISK is funded from NIHR grant PGfAR 0707-10031. The GC-HBOC (German Consortium of Hereditary Breast and Ovarian Cancer) is supported by the German Cancer Aid (grant no 110837, coordinator: Rita K. Schmutzler, Cologne). This work was also funded by the European Regional Development Fund and Free State of Saxony, Germany (LIFE-Leipzig Research Centre for Civilization Diseases, project numbers 713-241202, 713-241202, 14505/2470, 14575/2470). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. Generation Scotland (GENSCOT) received core support from the Chief Scientist Office of the Scottish Government Health Directorates [CZD/16/6] and the Scottish Funding Council [HR03006]. Funding for identification of cases and contribution to BCAC funded in part by the Wellcome Trust Seed Award “Temporal trends in incidence and mortality of molecular subtypes of breast cancer to inform public health, policy and prevention” Reference 207800/Z/17/Z. The GEPARSIXTO study was conducted by the German Breast Group GmbH. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). GLACIER was supported by Breast Cancer Now, CRUK and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The HABCS study was supported by the Claudia von Schilling Foundation for Breast Cancer Research, by the Lower Saxonian Cancer Society, and by the Rudolf Bartling Foundation. The HEBCS was financially supported by the Helsinki University Hospital Research Fund, the Finnish Cancer Society, and the Sigrid Juselius Foundation. The HMBCS was supported by a grant from the Friends of Hannover Medical School and by the Rudolf Bartling Foundation. The HUBCS was supported by a grant from the German Federal Ministry of Research and Education (RUS08/017), M.B. was supported by grant 17-44-020498, 17-29-06014 of the Russian Foundation for Basic Research, E.K was supported by the program for support the bioresource collections №007-030164/2, and the study was performed as part of the assignment of the Ministry of Science and Higher Education of the Russian Federation (№АААА-А16-116020350032-1). Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (EVO) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command [DAMD17-01-1-0729], Cancer Council Victoria, Queensland Cancer Fund, Cancer Council New South Wales, Cancer Council South Australia, The Cancer Foundation of Western Australia, Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC; 400413, 400281, 199600). LMBC is supported by the ‘Stichting tegen Kanker’. D.L. is supported by the FWO. The MABCS study is funded by the Research Centre for Genetic Engineering and Biotechnology “Georgi D. Efremov”, MASA. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC) and by funds from the Italian citizens who allocated the 5/1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects “5×1000”). The MCBCS was supported by the NIH grants CA192393, CA116167, CA176785 an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [CA116201], and the Breast Cancer Research Foundation and a generous gift from the David F. and Margaret T. Grohne Family Foundation. The Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database. The MEC was supported by NIH grants CA63464, CA54281, CA098758, CA132839 and CA164973. The MISS study is supported by funding from ERC-2011-294576 Advanced grant, Swedish Cancer Society, Swedish Research Council, Local hospital funds, Berta Kamprad Foundation, Gunnar Nilsson. The MMHS study was supported by NIH grants CA97396, CA128931, CA116201, CA140286 and CA177150. MSKCC is supported by grants from the Breast Cancer Research Foundation and Robert and Kate Niehaus Clinical Cancer Genetics Initiative. The work of MTLGEBCS was supported by the Quebec Breast Cancer Foundation, the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program—grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade—grant # PSR-SIIRI-701. The NBCS has received funding from the K.G. Jebsen Centre for Breast Cancer Research; the Research Council of Norway grant 193387/V50 (to A.-L. Børresen-Dale and V.N. Kristensen) and grant 193387/H10 (to A.-L. Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (grant 39346 to A.-L. Børresen-Dale) and the Norwegian Cancer Society (to A.-L. Børresen-Dale and V.N. Kristensen). The NBHS was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The Northern California Breast Cancer Family Registry (NC-BCFR) and Ontario Familial Breast Cancer Registry (OFBCR) were supported by grants U01CA164920 and U01CA167551 from the USA National Cancer Institute of the National Institutes of Health. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the Breast Cancer Family Registry (BCFR) or the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organisations imply endorsement by the USA Government or the BCFR or CCFR. The Carolina Breast Cancer Study (NCBCS) was funded by Komen Foundation, the National Cancer Institute (P50 CA058223, U54 CA156733, U01 CA179715), and the North Carolina University Cancer Research Fund. The NHS was supported by NIH grants P01 CA87969, UM1 CA186107 and U19 CA148065. The NHS2 was supported by NIH grants UM1 CA176726 and U19 CA148065. The OBCS was supported by research grants from the Finnish Cancer Foundation, the Academy of Finland (grant number 250083, 122715 and Center of Excellence grant number 251314), the Finnish Cancer Foundation, the Sigrid Juselius Foundation, the University of Oulu, the University of Oulu Support Foundation and the special Governmental EVO funds for Oulu University Hospital-based research activities. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997-1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The POSH study is funded by Cancer Research UK (grants C1275/A11699, C1275/C22524, C1275/A19187, C1275/A15956 and Breast Cancer Campaign 2010PR62, 2013PR044. PROCAS is funded from NIHR grant PGfAR 0707-10031. The RBCS was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318). The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. The SBCS was supported by Sheffield Experimental Cancer Medicine Centre and Breast Cancer Now Tissue Bank. SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. The Two Sister Study (2SISTER) was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES102245), and, also by a grant from Susan G. Komen for the Cure, grant FAS0703856. SKKDKFZS is supported by the DKFZ. The SMC is funded by the Swedish Cancer Foundation and the Swedish Research Council (VR 2017-00644) grant for the Swedish Infrastructure for Medical Population-based Life-course Environmental Research (SIMPL+++ER). The SZBCS was supported by Grant PBZ_KBN_122/P05/2004 and the program of the Minister of Science and Higher Education under the name “Regional Initiative of Excellence” in 2019-2022 project number 002/RID/2018/19 amount of financing 12 000 000 PLN. The TNBCC was supported by: a Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation, a generous gift from the David F. and Margaret T. Grohne Family Foundation. The UCIBCS component of this research was supported by the NIH [CA58860, CA92044] and the Lon V Smith Foundation [LVS39420]. The UKBGS is funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. The UKOPS study was funded by The Eve Appeal (The Oak Foundation) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre and MRC core funding (MR_UU_12023). The USRT Study was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Open Access funding enabled and organized by Projekt DEAL.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, H.A., Neumeyer, S., Michailidou, K. et al. Mendelian randomisation study of smoking exposure in relation to breast cancer risk. Br J Cancer 125, 1135–1145 (2021). https://doi.org/10.1038/s41416-021-01432-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01432-8
This article is cited by
-
Global comparative transcriptomes uncover novel and population-specific gene expression in esophageal squamous cell carcinoma
Infectious Agents and Cancer (2023)
