
Bacterial Community Characteristics in the Gastrointestinal Tract of Yak (Bos grunniens) Fully Grazed on Pasture of the Qinghai-Tibetan Plateau of China

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Sampling and Measurements
2.3. Chemical Analysis
2.4. DNA Extraction and PCR Amplification
2.5. Bioinformatics Analysis
2.6. Statistical Analysis
3. Results
3.1. Chemical Determination Results
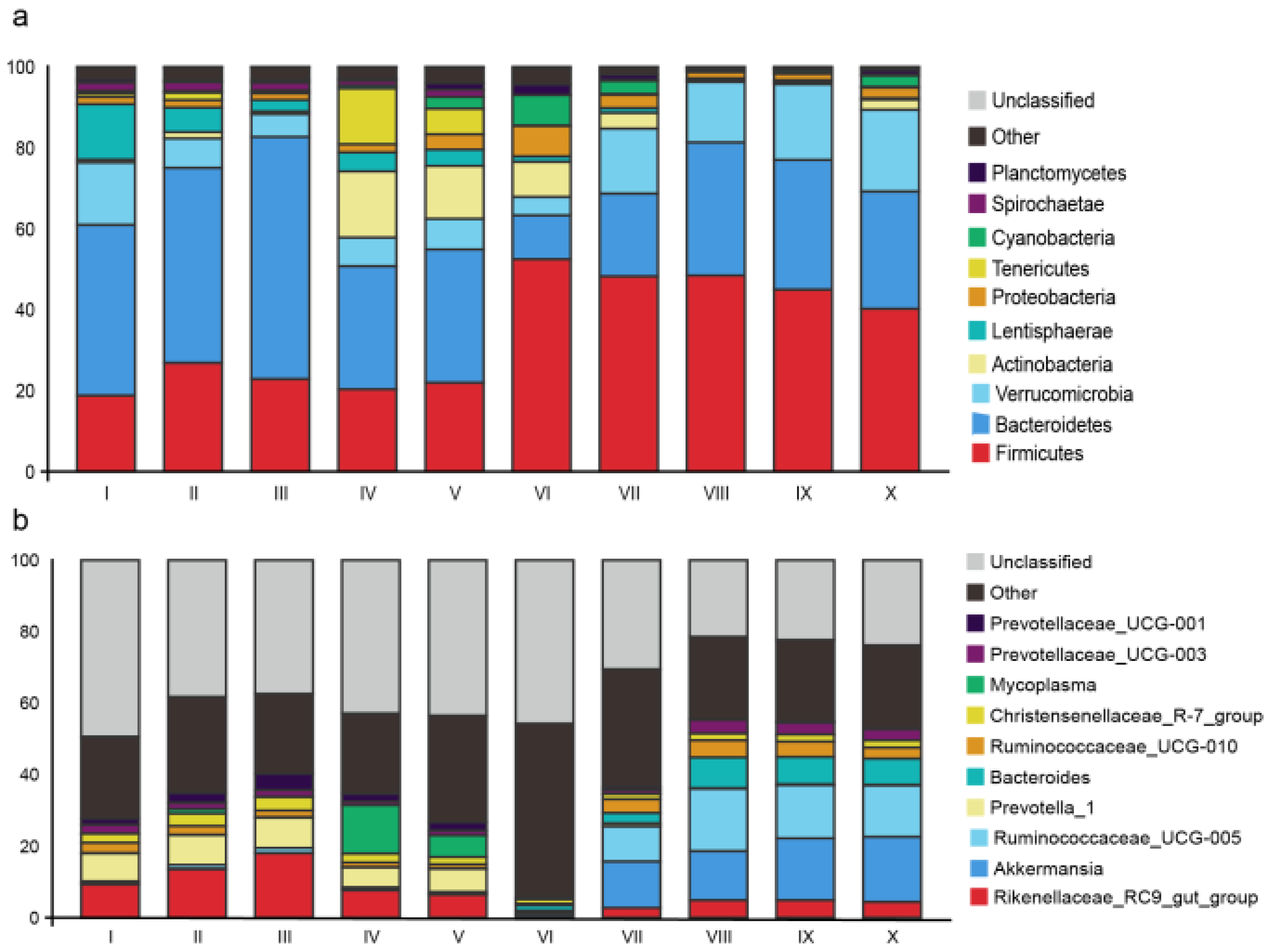
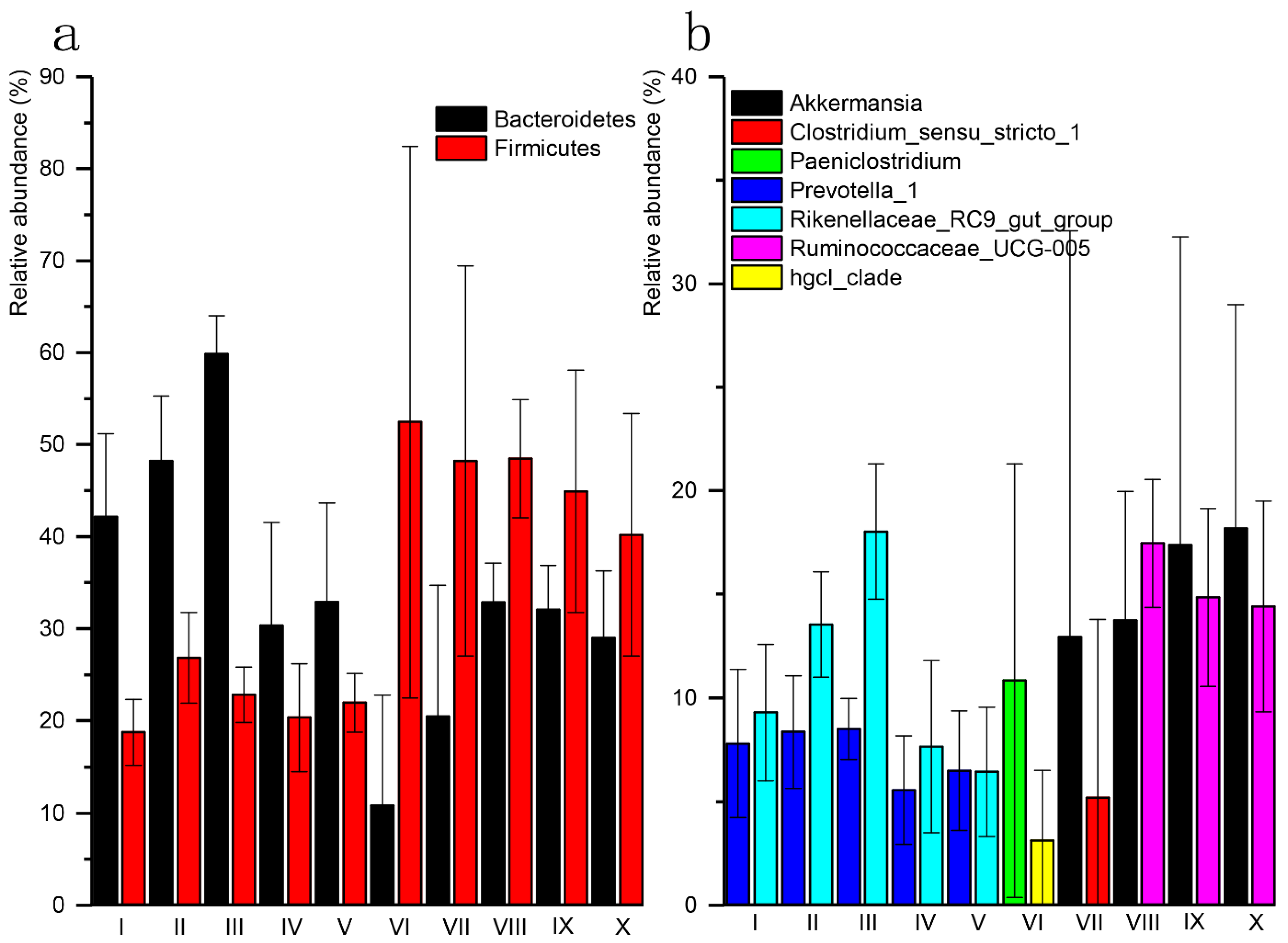
3.2. Sequencing and Taxonomic Composition of Bacterial Communities
3.3. Diversity and Composition Comparison
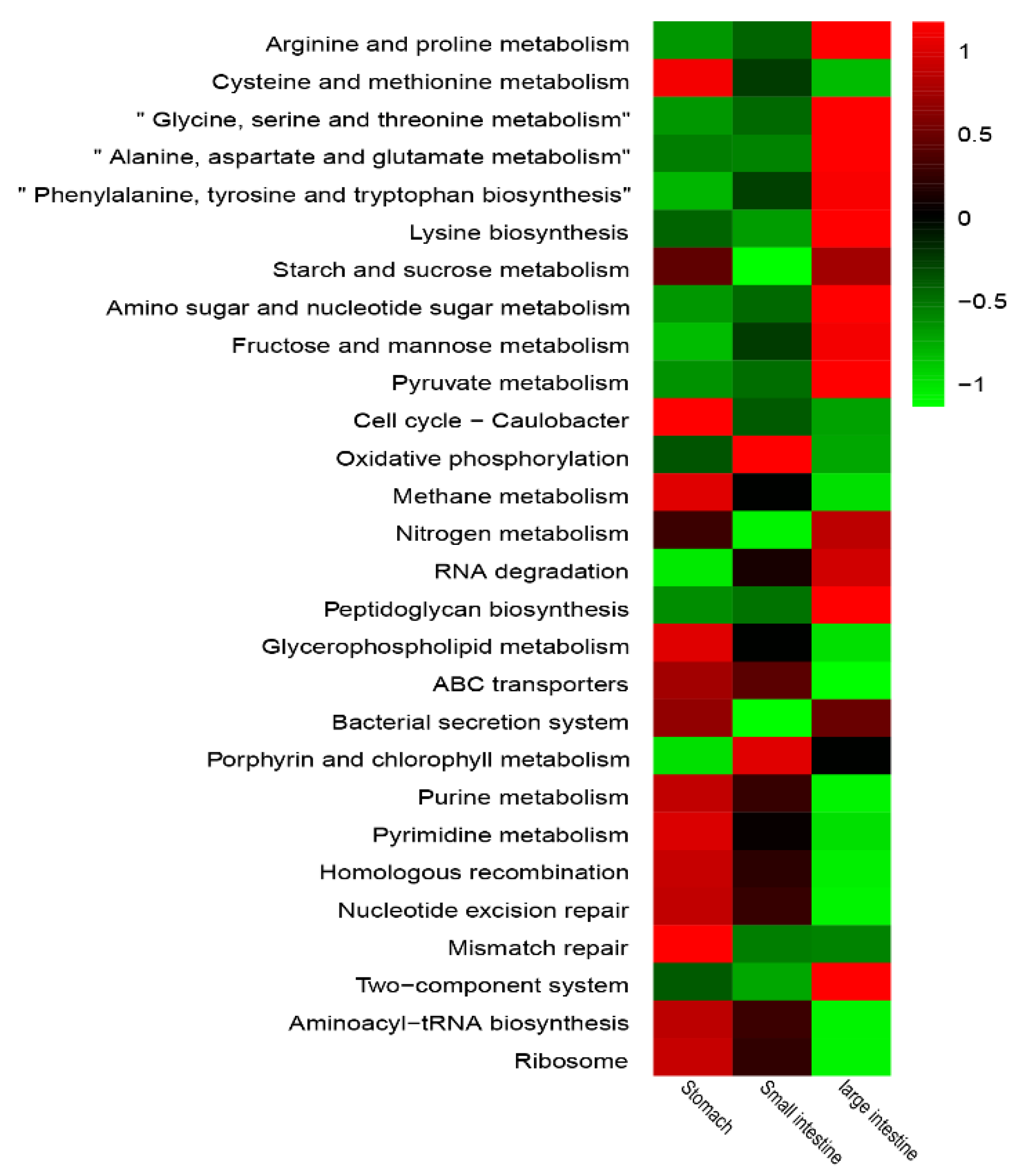
3.4. Predicted Functions in GIT Regions between the Sexes
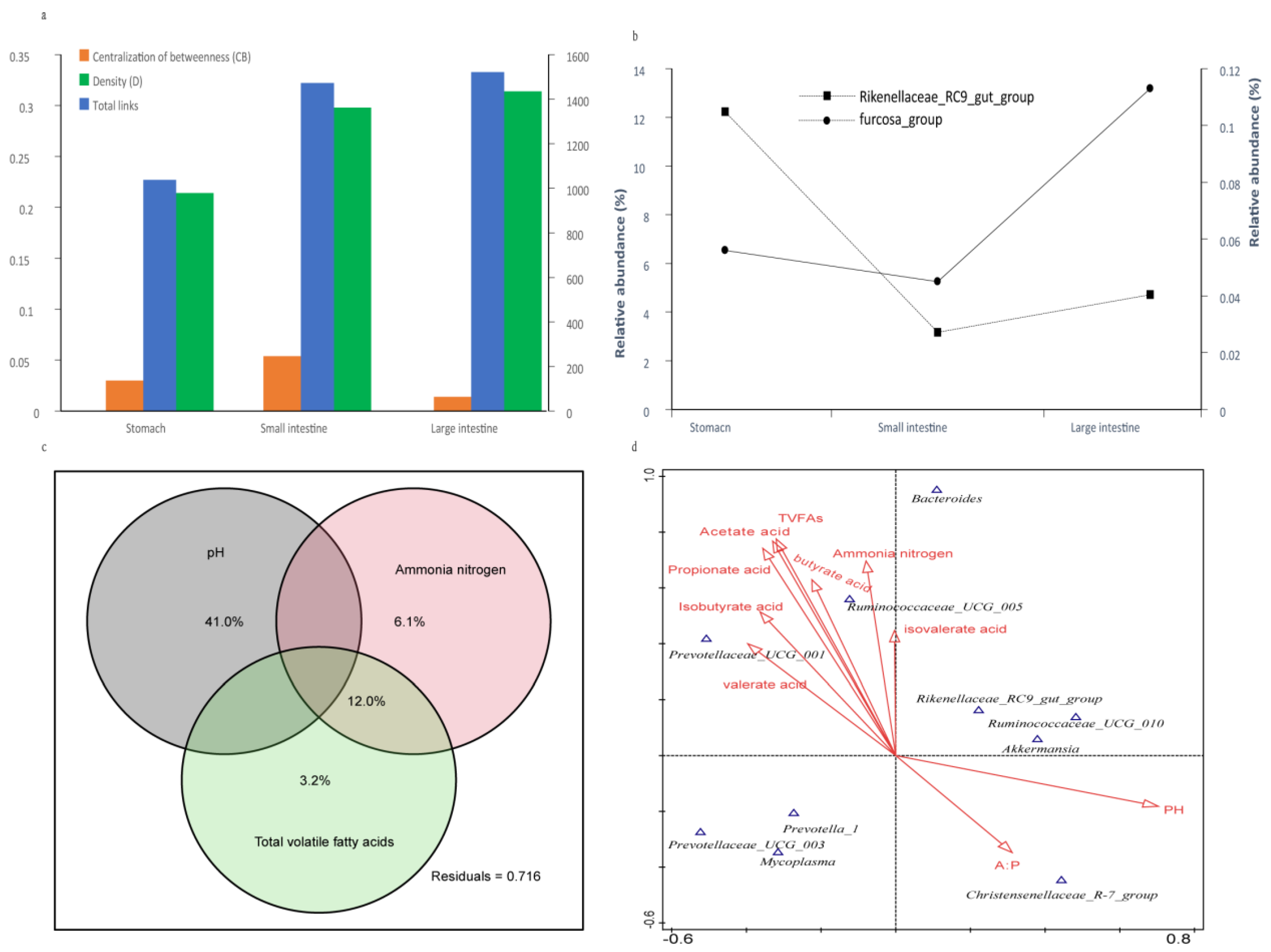
3.5. Correlation Analysis between Taxonomic Units and Fermentation Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Krause, D.O.; Nagaraja, T.G.; Wright, A.D.G.; Callaway, T.R. Board-invited review: Rumen microbiology: Leading the way in microbial ecology. J. Anim. Sci. 2013, 91, 331–341. [Google Scholar] [CrossRef]
- Dijkstra, J.; Tamminga, S. Simulation of the effects of diet on the contribution of rumen protozoa to degradation of fibre in the rumen. Br. J. Nutr. 1995, 74, 617–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Penner, G.B.; Hernandez-Sanabria, E.; Oba, M.; Guan, L.L. Effects of sampling location and time, and host animal on assessment of bacterial diversity and fermentation parameters sin the bovine rumen. J. Appl. Microbiol. 2010, 107, 1924–1934. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Morrison, M.; Yu, Z. Phylogenetic diversity of bacterial communities in bovine rumen as affected by diets and microenvironments. Folia Microbiol. 2011, 56, 453–458. [Google Scholar] [CrossRef]
- Han, X.; Yang, Y.; Yan, H.; Wang, X.; Qu, L.; Chen, Y. Rumen bacterial diversity of 80 to 110-day-old goats using 16S rRNA sequencing. PLoS ONE 2015, 10, e0117811. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wright, A.D.; Liu, H.; Bao, K.; Zhang, T.; Wang, K.; Cui, X.; Yang, F.; Zhang, Z.; Li, G. Bacterial community composition and fermentation patterns in the rumen of sika deer (Cervus nippon) fed three different diets. J. Microb. Ecol. 2015, 69, 307–318. [Google Scholar] [CrossRef]
- Chaucheyras-Durand, F.; Ossa, F. REVIEW: The rumen microbiome: Composition, abundance, diversity, and new investigative tools. Prof. Anim. Sci. 2014, 30, 1–12. [Google Scholar] [CrossRef]
- Sandra, K.; Henning, S.; Walters, W.A.; Clemente, J.C.; Rob, K.; Gordon, J.I.; Janssen, P.H. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE 2013, 8, e47879. [Google Scholar]
- Lee, J.E.; Lee, S.; Sung, J.; Ko, G.P. Analysis of human and animal fecal microbiota for microbial source tracking. ISME J. 2011, 5, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, G.T. Microbial community composition along the digestive tract in forage- and grain-fed bison. BMC Vet. Res. 2017, 13, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donia, M.S.; Cimermancic, P.; Schulze, C.J.; Wieland?Brown, L.C.; Martin, J.; Mitreva, M.; Clardy, J.; Linington, R.G.; Fischbach, M.A. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 2014, 158, 1402–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Ahrné, S.; Jeppsson, B.; Molin, G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16s rrna genes. FEMS Microbiol. Ecol. 2005, 54, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Kohl, K.D.; Miller, A.W.; Marvin, J.E.; Roderick, M.; M Denise, D. Herbivorous rodents (neotoma spp.) harbour abundant and active foregut microbiota. Environ. Microbiol. 2014, 16, 2869–2878. [Google Scholar] [CrossRef]
- Dougal, K.; Fuente, G.D.L.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Newbold, C.J. Identification of a core bacterial community within the large intestine of the horse. PLoS ONE 2013, 8, e77660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Zeng, D.; Ni, X.; Zhu, H.; Jian, P.; Zhou, Y.; Xu, S.; Lin, Y.; Li, Y.; Yin, Z. Microbial community compositions in the gastrointestinal tract of chinese mongolian sheep using illumina miseq sequencing revealed high microbial diversity. AMB Express 2017, 7, 75. [Google Scholar] [CrossRef]
- An, D.; Dong, X.; Dong, Z. Prokaryote diversity in the rumen of yak (bos grunniens) and jinnan cattle (bos taurus) estimated by 16s rdna homology analyses. Anaerobe 2005, 11, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.D.; Tan, H.Y.; Long, R.; Liang, J.B.; Wright, A.-D.G. Comparison of methanogen diversity of yak (bos grunniens) and cattle (bos taurus) from the qinghai-tibetan plateau, china. BMC Microbiol. 2012, 12, 237. [Google Scholar] [CrossRef] [Green Version]
- Lei, F.; Zhou, Z.; Ren, L.; Shi, F.; Can, M.; Chai, S.; Meng, Q. Ruminal bacterial diversity of yaks (bos grunniens) fed by grazing or indoor regime on the tibetan plateau by analysis of 16s rrna gene libraries. Ital. J. Anim. Sci. 2015, 14, 621–627. [Google Scholar]
- Ma, L.; Xu, S.; Liu, H.; Xu, T.; Hu, L.; Zhao, N.; Han, X.; Zhang, X. Yak rumen microbial diversity at different forage growth stages of an alpine meadow on the qinghai-tibet plateau. Peerj 2019, 7, e7645. [Google Scholar] [CrossRef]
- Guo, M.; Fahao, W.; Guangen, H.; Qin, Q.; Rong, L.; Ning, L.; Liangmeng, W.; Tongjie, C. Bacillus subtilis improves immunity and disease resistance in rabbits. Front. Immunol. 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 2012, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C. Uparse: Highly accurate otu sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rrna sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, Q.; Elmar, P.; Pelin, Y.; Jan, G.; Timmy, S.; Pablo, Y.; Jörg, P.; Oliver, G.F. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4fun: Predicting functional profiles from metagenomic 16s rrna data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Frey, J.C.; Rothman, J.M.; Pell, A.N.; Nizeyi, J.B.; Cranfield, M.R.; Angert, E.R. Fecal bacterial diversity in a wild gorilla. Appl. Environ. Microbiol. 2006, 72, 3788–3792. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Forster, R.J.; Yu, H.; Chambers, J.R.; Sabour, P.M.; Wheatcroft, R.; Chen, S. Diversity and phylogenetic analysis of bacteria in the mucosa of chicken ceca and comparison with bacteria in the cecal lumen. FEMS Microbiol. Lett. 2002, 208, 1–7. [Google Scholar] [CrossRef]
- Högberg, A.; Lindberg, J.E.; Leser, T.; Wallgren, P. Influence of cereal non-starch polysaccharides on ileo-caecal and rectal microbial populations in growing pigs. Acta Vet Scand 2004, 45, 87. [Google Scholar] [CrossRef] [PubMed]
- Kibe, R.; Sakamoto, M.; Hayashi, H.; Yokota, H.; Benno, Y. Maturation of the murine cecal microbiota as revealed by terminal restriction fragment length polymorphism and 16s rrna gene clone libraries. FEMS Microbiol. Lett. 2004, 235, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sait, L.; Galic, M.; Strugnell, R.A.; Janssen, P.H. Secretory antibodies do not affect the composition of the bacterial microbiota in the terminal ileum of 10-week-old mice. Appl. Environ. Microbiol. 2003, 69, 2100–2109. [Google Scholar] [CrossRef] [Green Version]
- Frey, J.C.; Pell, A.N.; Berthiaume, R.; Lapierre, H.; Lee, S.; Ha, J.K.; Mendell, J.E.; Angert, E.R. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. J. Appl. Microbiol. 2009, 108, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Chen, H.; Luo, X.; Guan, J.; He, Y.; Zhao, X. Microbial diversity in the rumen, reticulum, omasum, and abomasum of yak on a rapid fattening regime in an agro-pastoral transition zone. J. Microbiol. 2018, 56, 743. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.N.V.; Jewell, K.A.; Freitas, F.S.; Benjamin, L.A.; Tótola, M.R.; Borges, A.C.; Moraes, C.A.; Suen, G.J.V.M. Characterizing the microbiota across the gastrointestinal tract of a brazilian nelore steer. Vet. Microbiol. 2017, 164, 307–314. [Google Scholar] [CrossRef]
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.L.; Huse, S.M.; Sogin, M.L. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Thoetkiattikul, H.; Mhuantong…, W. Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16s rrna gene pyrosequencing. Curr. Microbiol. 2013, 67, 130–137. [Google Scholar] [CrossRef]
- Ishaq, S.L.; Wright, A.D.G. Insight into the bacterial gut microbiome of the north american moose (alces alces). BMC Microbiol. 2012, 12, 212. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Zhao, L. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar] [CrossRef] [Green Version]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, A.; Ben-Jacob, N.; Tayem, H.; Halperin, E.; Iraqi, F.A.; Gophna, U. Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb. Ecol. 2011, 61, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Elderman, M.; Hugenholtz, F.; Belzer, C.; Boekschoten, M.; van Beek, A.; de Haan, B.; Savelkoul, H.; de Vos, P.; Faas, M. Sex and strain dependent differences in mucosal immunology and microbiota composition in mice. Biol. Sex Differ. 2018, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Smidt, H.; Rijkers, G.T.; Vos, W.M.d. Intestinal microbiota in human health and disease: The impact of probiotics. Genes Nutr. 2011, 6, 209–240. [Google Scholar] [CrossRef] [Green Version]
- Booijink, C.C.; Zoetendal, E.G.; Kleerebezem, M.; De Vos, W.M. Microbial communities in the human small intestine: Coupling diversity to metagenomics. Future Microbiol. 2007, 3, 10. [Google Scholar] [CrossRef]
- Wheeler, W.; Noller, C. Gastrointestinal tract ph and starch in feces of ruminants. J. Anim. Sci. 1977, 44, 131–135. [Google Scholar] [CrossRef]
- Markle, J.; Frank, D.N.; Mortintoth, S.M.; Robertson, C.E.; Feazel, L.M.; Rollekampczyk, U.; Von Bergen, M.; Mccoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Kozik, A.; Nakatsu, C.H.; Chun, H.; Joneshall, Y.L. Age, sex, and tnf associated differences in the gut microbiota of mice and their impact on acute tnbs colitis. Exp. Mol. Pathol. 2017, 103, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Sinha, T.; Vila, A.V.; Garmaeva, S.; Jankipersadsing, S.A.; Imhann, F.; Collij, V.; Bonder, M.J.; Jiang, X.; Gurry, T.; Alm, E.J. Analysis of 1135 gut metagenomes identifies sex-specific resistome profiles. Gut Microbes 2019, 10, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K. Differences in gut microbiota associated with age, sex, and stool consistency in healthy japanese subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Cunha, I.S.; Barreto, C.C.; Costa, O.Y.; Bomfim, M.A.; Castro, A.P.; Kruger, R.H.; Quirino, B.F. Bacteria and archaea community structure in the rumen microbiome of goats (capra hircus) from the semiarid region of brazil. Anaerobe 2011, 17, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin, V.; Ransom, L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Gharechahi, J.; Zahiri, H.S.; Noghabi, K.A.; Salekdeh, G.H. In-depth diversity analysis of the bacterial community resident in the camel rumen. Syst. Appl. Microbiol. 2015, 38, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoetendal, E.G.; Plugge, C.M.; Akkermans, A.D.L.; Vos, W.M.D. Victivallis vadensis gen. Nov., sp. Nov., a sugar-fermenting anaerobe from human faeces. Int. J. Syst. Evol. Microbiol. 2003, 53, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Yin, J.; Liu, X.; Jia, B.; Chang, Z.; Lu, H.; Jiang, N.; Chen, Q. First insights into the microbial diversity in the omasum and reticulum of bovine using illumina sequencing. J. Appl. Genet. 2015, 56, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, C.; Wells, W.G.; Smith, C.J. Characterization of the primary starch utilization operon in the obligate anaerobe bacteroides fragilis: Regulation by carbon source and oxygen. J. Bacteriol. 2006, 188, 4663–4672. [Google Scholar] [CrossRef] [Green Version]
- Evans, N.J.; Brown, J.M.; Murray, R.D.; Getty, B.; Birtles, R.J.; Hart, C.A.; Carter, S.D.J.A.E.M. Characterization of novel bovine gastrointestinal tract treponema isolates and comparison with bovine digital dermatitis treponemes. Appl. Environ. Microbiol. 2011, 77, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Osborne, J.M.; Dehority, B. Synergism in degradation and utilization of intact forage cellulose, hemicellulose, and pectin by three pure cultures of ruminal bacteria. Appl. Environ. Microbiol. 1989, 55, 2247–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Bi, S.; Kang, J.; Zhang, Y.; Long, R.; Huang, X.; Shan, M.; Anderson, R.C. Bacterial communities related to 3-nitro-1-propionic acid degradation in the rumen of grazing ruminants in the qinghai-tibetan plateau. Anaerobe 2018, 54, 42–54. [Google Scholar] [CrossRef]
- Hamana, K.; Itoh, T.; Sakamoto, M.; Hayashi, H. Covalently linked polyamines in the cell wall peptidoglycan of the anaerobes belonging to the order selenomonadales. J. Gen. Appl. Microbiol. 2012, 58, 339–347. [Google Scholar] [CrossRef]
- Tian, W. Effects of different dietary concentrate to forage ratios on rumen fluid ph and vfa levels and blood vfa levels in dairy goats. Anim. Husb. Vet. Med. 2013, 4, 5–10. [Google Scholar]
- Lettat, A.; Noziere, P.; Silberberg, M.; Morgavi, D.P.; Berger, C.; Martin, C. Experimental feed induction of ruminal lactic, propionic, or butyric acidosis in sheep. J. Anim. Sci. 2010, 88, 3041–3046. [Google Scholar] [CrossRef]
- Minuti, A.; Ahmed, S.; Trevisi, E.; Picciolicappelli, F.; Bertoni, G.; Jahan, N.; Bani, P. Experimental acute rumen acidosis in sheep: Consequences on clinical, rumen, and gastrointestinal permeability conditions and blood chemistry. J. Anim. Sci. 2014, 92, 3966–3977. [Google Scholar] [CrossRef] [Green Version]
- Sadi, R.; Natsir, A.; Dagong, M.I.A. Evaluation of rumen fermentation performance and characteristic in local domestic goat fed with white teak leaf (gmelina arborea) or corncob-based complete feed. Int. J. Recent Innov. Acad. Res. 2019, 3, 7. [Google Scholar]
- Zhang, Z.; Xu, D.; Wang, L.; Hao, J.; Wang, J.; Zhou, X.; Wang, W.; Qiu, Q.; Huang, X.; Zhou, J. Convergent evolu-tion of rumen microbiomes in high-altitude mammals. Curr. Biol. 2016, 26, 1873–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhao, X.; Han, X.; Xu, S.; Chen, X. Comparative study of gut microbiota in Tibetan wild asses (Equus kiang) and domestic donkeys (Equus asinus) on the Qinghai-Tibet plateau. PeerJ 2020, 8, e9032. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Hua, Y.; Zeng, B.; Ning, R.; Li, Y.; Zhao, J. Gut microbiota signatures of longevity. Curr. Biol. 2016, 26, R832–R833. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Female Yaks | Male Yaks | p-Value |
|---|---|---|---|
| pH | 6.52 ± 0.06 | 6.44 ± 0.04 | 0.052 |
| Ammonia nitrogen(mg/L) | 104.60 ± 46.15 | 109.50 ± 20.92 | 0.834 |
| Acetate (mmol/L) | 14.00 ± 5.87 | 21.43 ± 12.21 | 0.268 |
| Propionate (mmol/L) | 2.87 ± 1.34 | 4.44 ± 2.54 | 0.268 |
| Isobutyrate (mmol/L) | 1.02 ± 0.46 | 1.30 ± 0.55 | 0.405 |
| butyrate (mmol/L) | 2.42 ± 2.02 | 2.87 ± 1.69 | 0.710 |
| isovalerate (mmol/L) | 1.30 ± 1.56 | 0.98 ± 0.61 | 0.682 |
| valerate (mmol/L) | 0.79 ± 0.34 | 1.02 ± 0.38 | 0.354 |
| Total volatile fatty acids (mmol/L) | 21.79 ± 9.59 | 32.03 ± 17.60 | 0.296 |
| Acetate to propionate ratio (A:P) | 5.05 ± 0.97 | 4.9 ± 0.75 | 0.789 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; Liu, H.; Hu, L.; Zhao, N.; Xu, S.; Lin, Z.; Chen, Y. Bacterial Community Characteristics in the Gastrointestinal Tract of Yak (Bos grunniens) Fully Grazed on Pasture of the Qinghai-Tibetan Plateau of China. Animals 2021, 11, 2243. https://doi.org/10.3390/ani11082243
Han X, Liu H, Hu L, Zhao N, Xu S, Lin Z, Chen Y. Bacterial Community Characteristics in the Gastrointestinal Tract of Yak (Bos grunniens) Fully Grazed on Pasture of the Qinghai-Tibetan Plateau of China. Animals. 2021; 11(8):2243. https://doi.org/10.3390/ani11082243
Chicago/Turabian StyleHan, Xueping, Hongjin Liu, Linyong Hu, Na Zhao, Shixiao Xu, Zhijia Lin, and Yongwei Chen. 2021. "Bacterial Community Characteristics in the Gastrointestinal Tract of Yak (Bos grunniens) Fully Grazed on Pasture of the Qinghai-Tibetan Plateau of China" Animals 11, no. 8: 2243. https://doi.org/10.3390/ani11082243