
Abstract
The current classification of sporadic Creutzfeldt–Jakob disease (sCJD) includes six major clinicopathological subtypes defined by the physicochemical properties of the protease-resistant core of the pathologic prion protein (PrPSc), defining two major PrPSc types (i.e., 1 and 2), and the methionine (M)/valine (V) polymorphic codon 129 of the prion protein gene (PRNP). How these sCJD subtypes relate to the well-documented phenotypic heterogeneity of genetic CJD (gCJD) is not fully understood. We analyzed molecular and phenotypic features in 208 individuals affected by gCJD, carrying 17 different mutations, and compared them with those of a large series of sCJD cases. We identified six major groups of gCJD based on the combination PrPSc type and codon 129 genotype on PRNP mutated allele, each showing distinctive histopathological characteristics, irrespectively of the PRNP associated mutation. Five gCJD groups, named M1, M2C, M2T, V1, and V2, largely reproduced those previously described in sCJD subtypes. The sixth group shared phenotypic traits with the V2 group and was only detected in patients carrying the E200K-129M haplotype in association with a PrPSc type of intermediate size (“i”) between type 1 and type 2. Additional mutation-specific effects involved the pattern of PrP deposition (e.g., a “thickened” synaptic pattern in E200K carriers, cerebellar “stripe-like linear granular deposits” in those with insertion mutations, and intraneuronal globular dots in E200K-V2 or -M”i”). A few isolated cases linked to rare PRNP haplotypes (e.g., T183A-129M), showed atypical phenotypic features, which prevented their classification into the six major groups. The phenotypic variability of gCJD is mostly consistent with that previously found in sCJD. As in sCJD, the codon 129 genotype and physicochemical properties of PrPSc significantly correlated with the phenotypic variability of gCJD. The most common mutations linked to CJD appear to have a variable and overall less significant effect on the disease phenotype, but they significantly influence disease susceptibility often in a strain-specific manner. The criteria currently used for sCJD subtypes can be expanded and adapted to gCJD to provide an updated classification of the disease with a molecular basis.
Similar content being viewed by others


Introduction
Prion diseases are invariably fatal, largely transmissible, neurodegenerative disorders that affect humans and other mammals. The central pathogenic event in prion disease is the accumulation of a misfolded, prone-to-aggregate isoform of the cellular prion protein (PrPC), named scrapie isoform (PrPSc). The transition to PrPSc involves the refolding of PrPC into high‐β‐sheet conformers that assemble into amyloid fibrils. Once formed, PrPSc replicates itself by a seeded-conversion mechanism in which PrPSc multimers bind to PrPC and mediate its conversion into PrPSc [87].
Prion diseases typically manifest a wide range of phenotypic variability, depending on genetic factors, namely, the primary sequence of the prion protein gene (PRNP), and PrPSc intrinsic properties likely related to the conformational heterogeneity of PrPSc multimers. PrPSc structural diversity often generates biological variants, termed strains, that can be serially propagated upon experimental or natural transmission independently from the host genotype [7, 11, 78, 98]. Prion strains can be distinguished after transmission to syngeneic hosts by the regional lesion profile, morphology of PrPSc deposits, and biochemical properties of PrPSc reflecting the distinct quaternary structure of PrPSc aggregates [2, 5]. Following their initial discovery and characterization through the experimental transmission of scrapie isolates [11], prion strains have also been associated with human prion diseases' phenotypes [8, 46, 51, 71, 84, 92, 101]. The current classification of sporadic Creutzfeldt–Jakob disease (sCJD), the most common human prion disease, recognizes six major phenotypic subtypes with distinctive clinicopathological features, broadly correlating at the molecular level with the genotype at the polymorphic codon 129 (methionine, M; or valine, V) in the prion protein gene (PRNP) and the size of the protease-resistant PrPSc core (type 1 migrating at 21 kDa and type 2 at 19 kDa), which is a prion strain marker in individuals with the same codon 129 genotype [79, 92].
In contrast to animal prion diseases, the human prion disease includes genetic forms linked to mutations in the PRNP gene encoding for PrPC. The genetic prion disease accounts for up to 15% of all cases and manifests an autosomal dominant inheritance pattern with variable penetrance. PRNP mutations are associated with a wide range of clinicopathological entities, encompassing CJD, Gerstmann–Sträussler–Scheinker disease (GSS), and fatal familial insomnia (FFI) [26, 29, 56]. The existence of PRNP mutations that segregate with familial prion diseases [38, 39, 64, 75] raises the critical question of these mutations' pathogenic role, including their effect on disease phenotype and the generation of strain-specific properties. The issue is particularly relevant for the mutations linked to CJD and FFI, for which sporadic and genetic disease forms are firmly established. Notably, a significant number of patients with CJD carrying a mutation lack a positive family history, while only a subgroup of PRNP mutations show high or full penetrance [52, 66].
In genetic prion disorders, the PRNP primary sequence was initially thought to be the main determinant of phenotypical heterogeneity [10]. However, increasing evidence indicates that genetic phenotypes largely reproduce the spectrum of the sporadic ones. In line with this view, initial experimental studies demonstrated that the transmission of genetic CJD (gCJD) to mice, monkeys, and bank voles induces disease phenotypes indistinguishable from those determined by sCJD inocula [4, 51, 58, 61, 71, 84, 96]. Notably, preliminary data support the hypothesis that distinct phenotypes in genetic prion diseases also behave as specific strains and that the same strain can affect patients carrying different PRNP mutations [71, 84, 98]. Despite the above, a systematic analysis of molecular and clinicopathological features across the spectrum of PRNP variants and their associated disorders is lacking. Indeed, given the low disease prevalence, most prior studies have focused on phenotypes related to a single PRNP mutation and a limited number of participants. To contribute to the full understanding of these issues and reach a better classification of genetic prion diseases, we performed a comprehensive study of molecular and phenotypic features in an extensive patient cohort, and compared them with their sporadic counterparts. Since the GSS phenotype does not have an established sporadic phenotype, we focused on patients with CJD and fatal insomnia (FI).
Materials and methods
Selection of patients
We studied 208 PRNP mutation carriers with a neuropathologically confirmed diagnosis of either CJD or FFI.
One hundred and fifty-one patients died in Europe (Italy, 98; Germany, 36; UK, 10; France, 6, and Austria, 1) and 57 in the USA (for a detailed list of providing Centers see Supplementary methods, online resource). Comprehensive data for 20 subjects and partial information of clinical and/or pathological and/or molecular genetic features for additional 17 patients have been previously reported [9, 10, 12, 16, 19, 24, 27, 31,32,33, 35, 40, 55, 57, 60, 65, 68, 69, 76, 77, 89,90,91, 95, 99]. Inclusion criteria were limited to the availability of CNS frozen tissue for PrPSc typing and determination of the PRNP haplotype (i.e., mutation plus the genotype at codon 129 in cis). As the only exception, given the rarity of the molecular combination, two cases that lacked frozen tissue but belonged to well-characterized families carrying the D178N-129V haplotype were also included [12, 60].
Finally, to compare the biochemical and pathological heterogeneity between genetic and sporadic forms of CJD and FI, we used a previously published cohort of 225 individuals with sCJD and 6 with sporadic FI [1, 80, 82].
Histological examination
Semiquantitative evaluation of spongiform change and astrogliosis was carried out in 193 brains by comparing hematoxylin and eosin stained sections from the affected participants and the sporadic CJD and FI subjects. Spongiform change was scored on a 0–4 scale (not detectable, mild, moderate, severe, and status spongiosus), whereas astrogliosis was scored on a 0–3 scale (not detectable, mild, moderate, and severe). A lesion profile for each patient was obtained by averaging the two scores. Only in the FI group, we analyzed the extent of spongiform change and astrogliosis separately. At least nine anatomical regions were always analyzed, including frontal, temporal and occipital neocortices, CA1 sector of hippocampus, entorhinal cortex, striatum (caudate nucleus and putamen), midbrain (substantia nigra and periaqueductal gray), medial thalamus, and cerebellum (vermis and hemisphere).
Immunohistochemistry
Paraffin sections from formalin-fixed tissue blocks of frontal (n = 175) and occipital cortices (n = 176), thalamus (n = 146) and cerebellum (n = 186) obtained from 190 brains, were immunolabelled using the monoclonal antibodies 3F4, 12F10 or L42 (German cases), as previously described [82, 85, 100]. The main pattern of PrP deposition, reflecting the morphology of PrP aggregates was analyzed in each brain region, and compared with those previously described in sCJD (e.g., “synaptic”; coarse/perivacuolar; plaque-like) [79]. In a subgroup of cases showing the plaque-like pattern, we also performed an analysis of the aggregate size with the Aperio Imagescope software using 10 sections (363.5 × 241.8 μm, 5 from cerebellum and 5 from the neocortex) for each case.
Molecular genetic analysis
PRNP (RefSeq NM_0003111) open reading frame (ORF) was analyzed as previously described [16, 79]. Briefly, genomic DNA was used to amplify the PRNP coding region in the polymerase chain reaction with the primers Forward 5´-GCAGTCATTATGGCGAACCTTGGCTG-3´ and Reverse 5´-GTACTGAGGATCCTCCTCATCCCACTATCAGGAAGA-3´. Mutation and codon 129 genotype were determined by direct sequencing of the PRNP ORF using the BigDye Terminator 3.1 Cycle Sequencing Kit according to manufacturer’s instructions and the Applied Biosystems ABI3500 Dx Genetic Analyzer sequencer (Thermo Fisher Scientific, Waltham, MA). To ascertain the codon 129 genotype in the mutated allele in MV heterozygotes, the amplified ORF was digested with a mutation-specific restriction enzyme, and then re-amplified with the same primers. The codon 129 genotype of the allele bearing the mutation was then determined by direct sequencing. For insertion/deletion mutations, the genotype at codon 129 in cis with the mutation was determined as described [3].
Protein studies
PrPSc typing
Immunoblot analysis of PrPSc was carried out as previously described [83]. One or multiple samples from different brain regions, including the frontal cortex (n = 202), temporal cortex (n = 158), occipital cortex (n = 155), putamen (n = 149), medial thalamus (n = 152), and cerebellum (n = 165) were examined in all but two subjects with the D178N-129V PRNP haplotype (see also patient selection). To evaluate in depth the extent of co-occurrence of mixed PrPSc types, we performed immunoblots in samples from all six regions in 142 cases. Sample preparation and immunoblot analysis of PrPSc was carried out as previously described using 7 or 15 cm long separating gels [73, 83, 91].
PK titration curves
Serial PK digestion of total homogenates (THs) from the frontal cortex was performed as described [91]. Briefly, THs were adjusted to a total protein concentration of 6 mg/ml, and the applied PK activity ranged from 2 to 256 U/ml. ED50 expresses the PK concentration needed to digest 50% of PrPSc.
Thermo-solubilization assay (TSA)
The analysis of thermostability of PrPSc aggregates was performed as described [22]. Briefly, THs were digested with 8 U/ml PK for 1 h at 37 °C with mild shaking (300 rpm). After PK inactivation by PMSF (3.6 mM final concentration), aliquots were mixed with an equal volume of loading buffer (final concentration 1.5% SDS, 2% β-mercaptoethanol, 5% glycerol, 1 mM EDTA, 31.2 mM Tris, pH 6.8) and heated to temperatures ranging from 25 °C to 95 °C (∆T = 10 °C) for 6 min before immunoblot analysis. T50 expresses the temperature needed to solubilize 50% of PrPSc.
PK titration curves and TSA analyses were performed in 50 gCJD and 47 sCJD cases, which are described in detail in Supplementary Table 1, online resource.
Clinical data and diagnostic investigations
Clinical data including at least those obtained from one neurological examination were available in 192 (179 gCJD and 13 FFI) patients. Disease duration was calculated from the time of presentation of neurological signs to death (i.e., prodromal nonspecific symptoms were not considered). We also reviewed the clinical charts for the following: symptoms at onset and during disease evolution, results of electroencephalographic recordings (EEG), cerebral magnetic resonance imaging (MRI) studies, and cerebrospinal fluid analyses, including results of proteins 14-3-3, and total-tau (t-tau), and of the prion real-time quaking-induced conversion (RT-QuIC) assay (see also Supplementary Materials, online resource).
Results
Molecular genetic analysis
All participants carried a single heterozygous PRNP mutation, except for one who was homozygous for the R208H mutation. The distribution in the cohort of PRNP haplotypes determined by the mutation and the codon 129 polymorphism in both mutated and normal alleles is summarized in Table 1.
PrPSc typing
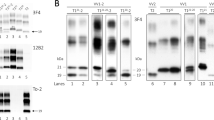
In most cases, the immunoblot profile of proteinase-K (PK)-resistant PrPSc reproduced the type 1 (~ 21 kDa) and type 2 (~ 19 kDa) profiles previously described in sCJD (Fig. 1) [78]. However, eight participants carrying the E200K-129M haplotype and V at codon 129 in the wild-type allele showed a type “i” (= intermediate) profile characterized by a 20 kDa unglycosylated PrPSc core (Fig. 1a). Moreover, as previously observed in sCJD MV2K [72, 82], three MV heterozygotes carrying an insertional mutation of 5 or 6 octapeptide repeats in cis with 129V (5/6-OPRI-129V) showed a doublet of unglycosylated fragments migrating at 19 kDa (type 2) and 20 kDa (type “i”) (Fig. 1b). Similarly, participants carrying the D178N-129V and T188R-129V haplotypes showed an unglycosylated PrPSc band comprising a doublet of fragments, in variable relative proportion, including the 21 kDa (type 1) peptide and a second slightly faster-migrating fragment (Fig. 1c). Finally, a subgroup of participants carrying 5/6-OPRI-129V also showed a PrPSc type 1 profile comprised of a doublet of fragments, including a second fragment migrating slightly slower than the typical 21 kDa (type 1) associated band (Supplementary Fig. 1, online resource).

Immunoblot profile of PrPSc in genetic CJD. a Genetic CJD haplotypes associated with PrPSc type 1 (lanes 3, 5) and “i” (lane 6). As in sCJD MM1, monoglycosylated PrPSc was predominant in the V210I mutation, while the diglycosylated was the most represented isoform in patients carrying the E200K variant. D178N-129V carriers showed an equal mix of mono- and diglycosylated isoforms, whereas the unglycosylated band was underrepresented. b Genetic CJD haplotypes associated with PrPSc type 2 (lanes 2, 3, 5 and 7) and 1 + 2 (lane 6). The unglycosylated isoform in a 5-OPRI-129V case with heterozygosity (MV) at codon 129 (lane 5) is characterized by a doublet migrating at 19 and 20 kDa (PrPSc type 2 + “i”). The T183A mutation showed a profile characterized by a marked under-representation of the diglycosylated isoform as compared with the monoglycosylated band. c Immunoblot profile of D178N-129V cases. The unglycosylated isoform is represented by doublet with variable dominance of either the band migrating at 21kDa (PrPSc type 1) or the one migrating slightly faster. A similar pattern of migration was observed in sCJD VV1 (lane 1). a2 and c3 lanes showed the same case. Samples were resolved in 7 (a, b) and 15 cm (c) long gels and probed with the primary antibody 3F4
PrPSc type 1 and type 2 were significantly associated with both codon 129 genotype and mutation type (Table 1). Similarly to sCJD (Supplementary Table 2, online resource), the individuals carrying the prevalent mutations, i.e., E200K, V210I, and OPRI, generally showed PrPSc type 1 when coupled with 129M, and type 2 (or type 2 + “i”) when associated with 129V. Exceptions included the eight cases carrying E200K-129M combined with PrPSc type “i”, and five individuals with E200K-129M or 5-OPRI-129M in association with PrPSc type 2. In contrast, a few rare mutations (T183A, T188R), and the prevalent D178N variant showed either an exclusive association with PrPSc type 2 when in cis with M (T183A) or an almost invariable association with PrPSc type 1 when in cis with V (T188R) or both associations (D178N) (Table 1).
Regional immunoblot analysis showed PrPSc types 1 + 2 co-occurrence in a minority (6.3% overall) of participants carrying the following haplotypes: V210I-129M, 5/6-OPRI-129M, V203I-129M, E196K-129M, E200K-129V, T188R-129V, and 5/6-OPRI-129V (Table 1). A sub-analysis limited to the cases with at least six brain regions available for immunoblotting demonstrated only a slight increase in the prevalence of PrPSc types co-occurrence (7.0%) (Supplementary Table 3, online resource).
As previously shown [21, 32, 37, 81], the relative proportion of the three bands that correspond to the di-, mono-, and unglycosylated PrPSc differed significantly between participants carrying different PRNP mutations (Supplementary Table 4, online resource). As in sCJD, monoglycosylated PrPSc was generally predominant. However, the diglycosylated form was the most represented isoform in those carrying E200K and D178N-129M. Moreover, participants carrying the D178N-129V haplotype showed an equal mix of mono- and diglycosylated isoforms, whereas the unglycosylated band was underrepresented. Finally, the T183A and the 5/6-OPRI-129V subgroup with PrPSc types 1 and 2 co-occurrence had peculiar immunoblot profiles characterized by a marked under-representation of the diglycosylated isoform (Fig. 1, Supplementary Figs. 1 and 2, online resource).
Histotype classification of gCJD
Given the strong molecular phenotypic correlation, we classified the study cohort into five major groups based on PrPSc type and codon 129 genotype of the PRNP mutated allele. Thereafter, we analyzed the effect of PRNP mutations within each of the five groups, namely 129M-type 1, 129V-type 2, 129M-type 2, 129V-type 1, and 129M-type “i.” Given the atypical PrPSc features associated with T183A-129M cases and a subgroup of participants carrying 5 or 6 OPRI and 129V, we characterized these cases separately.
Group 129M-type 1
The 129M-PrPSc type 1 combination was the most frequent (n = 141) and included several PRNP mutations (Table 1). The type of spongiform change, lesion profile, and PrP deposition pattern were highly consistent among cases. However, some mutation-specific effects were evident, particularly in the morphology of PrP deposits. As in sCJD MM(V)1 [85], the spongiform change, characterized by small, non-confluent vacuoles, mainly affected the cerebral cortices, neostriatum, thalamus, and cerebellum, whereas the hippocampus and midbrain were relatively spared (Figs. 2a and 3a). Moreover, like in sCJD MM(V)1, immunohistochemistry demonstrated a synaptic pattern of PrP deposition in the cerebellum and the cerebral cortex (Table 2, Fig. 3b, Supplementary Table 5, online resource). Mutation-specific phenotypic variations involved the E200K, E196K/A, and OPRI carriers (Fig. 4). Participants with E200K and E196K/A (47.8% and 66.7%, respectively) showed a modified synaptic pattern (hereafter designated as “thickened” synaptic), characterized by focal, patchy PrP aggregates in the molecular layer of the cerebellum and, to a lesser extent, in the cerebral cortices (Fig. 4a). Moreover, 10 out of 12 (83.3%) OPRI cases displayed small granular deposits with a striking linear distribution in the cerebellum molecular layer (Fig. 4b, c). These stripes extended perpendicularly from the molecular layer's surface to the Purkinje cell layer in 5/6-OPRI participants, while they were shorter and thicker in the carriers of 3 or 4 OPRI (3/4-OPRI). Of the two cases not displaying stripes, one had very severe, panencephalopathic changes and showed extensive synaptic PrP deposition in the cerebellar molecular layer and plaque-like deposits in the granular layer. The other one showed fine PrP deposits in the molecular layer, sometimes assuming a striking linear orientation perpendicular to the surface. Moreover, there were clusters ofgranular PrP deposits in round patches, often associated with non-confluent spongiform changes, in the deeper cortical layers layers (IV–V) of 8 out of 12 (66.7%) OPRI cases (Fig. 4d). Finally, one of the two participants (50%) carrying E211Q-129M showed numerous small, sometimes confluent PrP-amyloid plaques in the cerebellar molecular layer (Fig. 4e).

Comparison of lesion profiles between the different genetic CJD groups classified according to the PRNP haplotype/PrPSc type combination and the corresponding sCJD subtypes. The following anatomical regions were analyzed: frontal (FC), temporal (TC), and occipital (OC) neocortices, hippocampus (HI) (CA 1 region), entorhinal cortex (EC), neostriatum (ST) (nuclei caudatus and putamen), thalamus (TH) (mediodorsal nucleus), midbrain (MD) (substantia nigra and periaqueductal gray), and cerebellum (CE). Spongiform change was scored on a 0–4 scale (not detectable, mild, moderate, severe, and status spongiosus), and gliosis on a 0–3 scale (not detectable, mild, moderate, and severe). Lesion profiles were obtained by averaging the two scores for each brain region examined. Spongiform change and gliosis are shown separately in FFI and sCJD MM2T. Data are expressed as mean ± SD values

Main histopathological features of genetic CJD phenotypes. a, b Spongiform change characterized by small, non-confluent vacuoles (a, frontal cortex, E200K-129M), and synaptic type of PrP deposition (b, cerebellum, V210I-129M) in subjects of the 129M-type 1 group. c Prominent PrP immunoreactivity in the deep cortical layers, i.e., laminar distribution, in a subject carrying E200K-129V (frontal cortex; details at higher magnification in c1 and c2). d Spongiform change with intermediate size vacuoles in a D178N-129V carrier (temporal cortex). e–h Plaque-like PrP deposition in the cerebellar granular layer (e) and in the frontal cortex (f); kuru-type plaques (g, h) in the cerebellar granular layer of a codon 129 heterozygous patient with 5-OPRI-129V. i Mini plaque-like PrP deposits in the subiculum of a D178N-129V carrier. j, k Spongiform change with large and confluent vacuoles (j) and coarse PrP deposits with perivacuolar distribution (k) in the occipital cortex of an individual of the E200K-type 2C group. l Patchy PrP deposits in the molecular layer of a case of the E200K-type 2C group. Haematoxylin–eosin staining (a, d, g, j), and immunohistochemistry for PrP with mAb 3F4 (b, e, f, h, i, k, l), and 12F10 (c)

Mutation-specific histopathological variations across the spectrum of gCJD phenotypes. a Modified, “thickened” synaptic pattern of PrP deposition in the molecular layer of cerebellum in a patient carrying the E200K-129M haplotype. b, c Granular PrP deposits with long-thin (b) and short-thick (c) stripe-like appearance distributed perpendicularly to the surface in the molecular layer of cerebellum in 6- and 4-OPRI-129M carriers, respectively. d Round patches of fine, granular PrP deposits in the deep frontal cortex of a 4-OPRI-129M case. e Multiple, small PrP-amyloid plaques in the cerebellar molecular layer in a E211Q-129M carrier. f, g Multiple, intraneuronal globular PrP deposits distributed in the perikaryon in a E200K-129V individual with PrPSc type 2 (f, deep layers of frontal cortex) and E200K-129M with PrPSc type “i” (g, pons). h–k Spongiform change characterized by small and intermediate, non-confluent vacuoles in the caudate nucleus, and granular/mini plaque-like PrP deposits in the striatum and molecular layer of cerebellum of patients with T183A-129M (h, i) and atypical 6-OPRI-129V (j, k), respectively. Immunohistochemistry for PrP with mAb 3F4 (a–g, i, k), and haematoxylin–eosin staining (h, j)
Group 129V-type 2
In contrast to 129M-type 1, the lesion profile of 129V-type 2 cases showed a predominant involvement of the limbic cortices, deep brain nuclei, and midbrain. The cerebellum was variably affected, while pathological changes in the neocortices were less severe than those in the subcortical structures, particularly in the occipital lobe (Fig. 2b). Notably, as in sCJD VV2 and MV2K [85], the cortical spongiform change featured non-confluent vacuoles of intermediate size and often showed a laminar distribution with a predominant involvement of deeper layers.
We detected PrP-immunopositive amyloid plaques of the kuru type in the cerebellum of all the codon 129 MV patients carrying OPRI mutations (5-OPRI n = 2, 6-OPRI n = 1) and in the VV participant with R208H substitution (Fig. 3g, h). In contrast, kuru-type plaques were absent in codon 129 MV patients carrying the E200K mutation. Immunohistochemistry also revealed plaque-like PrP deposits in the cerebellar granular layer (Fig. 3e), thalamus, and, to a lesser extent, deep cortical layers close to the gray–white matter junction (Fig. 3f). The plaque-like pattern of PrP deposition was generally more pronounced in 5/6-OPRI than in E200K cases. Finally, there was a synaptic pattern of staining in 82.3% of cases (Table 2). Neocortical synaptic deposits often co-distributed with the laminar spongiform change in the deeper layers (Fig. 3c). Notably, all E200K-129V brains showed peculiar intraneuronal globular deposits, mainly involving the perikaryon, which was most evident in the brainstem nuclei, deeper layers of the cerebral cortex, and cerebellar dentate nucleus (Fig. 4f). Finally, similar to the OPRI mutations of the 129M-type 1 group, all the 5/6-OPRI showed linear stripes of PrP deposits in the cerebellar molecular layer.
Group 129V-type 1
Patients belonging to this group carried either the D178N or T188R variant. As in sCJD VV1 [85], 129V-type 1 participants showed a predominant cortico-striatal pathology and a relative sparing of the cerebellum and brainstem. Moreover, the hippocampus (CA1 and dentate gyrus) and, particularly, the entorhinal cortex were consistently affected (Fig. 2c). There were no significant differences between codon 129 VV and MV carriers (Supplementary Fig. 3, online resource). As in 129V-type 2 cases, spongiform change consisted of mostly non-confluent vacuoles of intermediate size (Fig. 3d). In all the cases, immunohistochemistry revealed small, round PrP aggregates in the molecular layer of the cerebellum and neocortices, where they often co-distributed with the most severe spongiform change in the deeper layers (Fig. 3i). PrP deposits in the neocortices were more abundant and significantly smaller than those detected in 129V-type 2 brains (Supplementary Fig. 4, online resource); hence, the eponym “mini plaque-like” was used to distinguish them from the plaque-like pattern seen in 129V-type 2 brains.
The D178N mutation carriers showed a homogeneous pattern of PrP deposition between the cortex and cerebellum. In contrast, T188R participants displayed larger and more pronounced PrP deposits in the cerebellum than in the cerebral cortex, and slightly more severe lesions in the cerebellum compared to the D178N carriers (Supplementary Figs. 4 and 5). Finally, PrP mini plaque-like aggregates were more abundant and showed a larger size than in sCJD VV1 (Supplementary Table 4, online resource).
Group 129M-type 2
This group included cases carrying D178N (n = 12), E200K (n = 4), and 5-OPRI (n = 1). As in sCJD [85], this group comprised two histotypes with highly distinctive features, primarily affecting the cerebral cortex or the thalamus. In the former, the spongiform change consisted of large and confluent vacuoles showing a uniform distribution among the neocortices, entorhinal cortex, striatum, and thalamus, with relative sparing of the hippocampus, midbrain, and cerebellum as in sCJD MM2C (Fig. 2e) [85]. Similarly, PrP immunopositivity was typically coarse and distributed around the large vacuoles (perivacuolar pattern) (Fig. 3j, k, and Table 2). As the main difference with sCJD MM2C, all E200K cases uniquely showed “thickened” synaptic PrP deposits in the cerebellar molecular layer, while the 5-OPRI case with the most prolonged disease duration uniquely showed large, patchy, coarse PrP aggregates, often surrounding vacuoles, in the cerebellar molecular layer (Fig. 3l) in addition to the perivacuolar and coarse deposits. These deposits were readily distinguishable from the plaque-like pattern seen in the cerebellar granular layer and white matter of 129V-type 2 cases and from the mini plaque-like aggregates of the 129V-type 1 cases, based on topographic distribution, shape and size. Given the striking analogies with sCJD MM2C, we named this group gCJD 129M-type 2C.
In the second group, which only included participants carrying the D178N-129M haplotype, the lesion profile was highly consistent with that of sCJD MM2T (Fig. 2d). However, given the established correlation between the lesion profile and disease duration in FFI [26], and the longer average disease course in the FFI-129MV group, only the latter showed striking analogies with sCJD MM2T. In contrast, participants of the FFI-129MM group demonstrated a shorter mean disease course (129MM 9.1 ± 1.9 vs. 129MV 26.2 ± 3.9 months, p ≤ 0.0001), a relative sparing of the cerebral neocortex, and less consistent spongiform change (Supplementary Fig. 3, online resource) than those in the FFI-129MV group. As previously reported [26], PrP immunohistochemistry revealed in some of the cases faint synaptic or granular deposits confined to the cerebral cortices and often co-localizing with the spongiform change (Table 2).
Group 129M-type “i”
This group included codon 129 heterozygotes carrying the E200K-129M haplotype and showing the PrPSc type “i” (n = 8). Both the morphology and distribution of the spongiform change were somehow reminiscent of gCJD 129V-type 2 and sCJD MV2K (Fig. 2f). The spongiform change featured non-confluent vacuoles of intermediate size and showed a laminar pattern in the cerebral cortex with a predominant involvement of the deeper layers. There was a synaptic pattern of deposition in the cerebellum in all cases. Moreover, as in E200K of the 129V-type 2 group, PrP immunostaining showed intraneuronal, cytoplasmic PrP-immunoreactive globular inclusions in the cerebral cortex's deeper layers, thalamus, cerebellar dentate nucleus, and brainstem nuclei (Fig. 4g).
Atypical and mixed phenotypes
Atypical phenotypes
We applied the term “atypical” when, despite the consistent haplotype and PrPSc type, the clinicopathological features of gCJD did not match those of the participants (both sporadic and genetic) displaying the same molecular signature. For example, two patients carrying the T183A-129M haplotype associated with PrPSc type 2 did not match the features of either FFI or gCJD 129M-type 2C. The spongiform change, comprising small, non-confluent vacuoles (Fig. 4h), mainly affected the neocortices, entorhinal cortex, striatum, and thalamus. The hippocampus, midbrain, and, to a lesser extent, the cerebellum were relatively spared by the spongiform change. Immunohistochemistry for PrP revealed granular/mini focal aggregates in the cerebellar molecular layer, striatum, and thalamus (Fig. 4i).
In three patients carrying 5/6-OPRI-129V and PrPSc types 1 + 2, both the distribution and severity of the neuropathological change were similar to those associated with the T183A mutation (Supplementary Fig. 6, online resource). The spongiform change consisted of non-confluent vacuoles of mixed sizes (small and intermediate) (Fig. 4j). Immunohistochemistry showed PrP aggregates in the form of mini plaque-like, focal deposits in the neocortices. Besides, PrP stripe-like aggregates were detected in the cerebellum molecular layer, along with granular/mini plaque-like deposits (Fig. 4k).
Mixed phenotypes
Immunohistochemistry for PrP revealed large, confluent vacuoles and coarse/perivacuolar PrP deposits, hallmarks of PrPSc type 2 deposition, in 39 out of 127 (30.7%) participants in the 129M-type 1 group. The latter histopathologic features were virtually indistinguishable from those featuring the gCJD 129M-type 2C group and, in most cases, were restricted to focal areas of the occipital cortex. The resulting mixed phenotype was significantly more frequent in participants carrying V210I (48.0%) than in those carrying E200K (23.9%, p = 0.025) or other rarer mutations (10.5%, p = 0.005), while the comparison with the participants carrying 3–6 OPRI (33.3%) was not statistically significant. The above-mentioned mixed phenotype is virtually indistinguishable from MM(V)1 + 2C, the most frequent subtype combination documented in sCJD [82].
Besides the typical 129M-type 2C features, one participant carrying E200K-129MV showed a moderate to severe spongiform change characterized by non-confluent, intermediate-sized vacuoles in the hippocampus, midbrain, and cerebellum. In these regions, immunohistochemistry revealed plaque-like and pericellular PrP deposits and a strong synaptic positivity, which was more intense in the cerebellar granular layer than in the molecular layer. Altogether, these neuropathological features suggest the co-occurrence of two phenotypes, both associated with PrPSc type 2 (i.e., 129M-type 2C + 129V-type 2).
Finally, in all the four participants carrying E200K-129M with PrPSc type 2, we observed “thickened” synaptic PrP deposits in the cerebellar molecular layer. Although in both sporadic and genetic CJD, synaptic PrP in the cerebellar molecular layer is typically associated with PrPSc type 1, we did not detect PrPSc type 1 in any of these cases. Therefore, whether these cases represent mixed phenotypes (E200K-129M type 2C + 1) remains to be seen.
PrPSc resistance to protease digestion and thermostability
PrPSc biochemical properties, such as resistance to PK digestion and thermostability, contributed a molecular signature besides typing to sCJD histotypes [22, 94]. Therefore, we explored whether these PrPSc features also allow for the reliable differentiation of the gCJD spectrum. PrPSc resistance to PK digestion was higher in 129V-type 2 cases (ED50 18.0 ± 9.5 U/mL) than in 129M-type 1 (ED50 7.5 ± 2.8 U/mL, p = 0.003) or 129V-type 1 (ED50 8.6 ± 1.1 U/mL p = 0.033) groups, whereas it showed intermediate values in 129M-type 2C and 129M-type “i” cases (ED50 17.2 ± 7.9 U/mL and 15.3 ± 10.8 U/mL, respectively) (Fig. 5a). There were no differences within each group across mutations, except for a slightly lower ED50 of PrPSc type 1 in participants carrying V210I than those carrying E200K (ED50 5.2 ± 0.4 vs. 9.1 ± 2.9 U/mL, p = 0.044). Finally, we did not find any significant difference between gCJD and sCJD groups defined by the PrPSc type/codon 129 genotype combination (Fig. 5a).

Comparison of PrPSc biochemical properties between genetic and sporadic CJD and FI. a, b PrPSc PK resistance (a) and thermostability (b) across the spectrum of genetic and sporadic CJD and FI subtypes. The color and distribution of the curves were adapted to highlight the differences between genetic groups and the similarities with the corresponding sporadic subtypes. The dot lines represent the ED50 (PK concentration needed to digest 50% of PrPSc) (a) and the T50 (temperature needed to solubilize 50% of PrPSc) (b)
After exposure to increasing temperature, PrPSc in gCJD 129M-type 1 (T50 77.5 ± 4.9 °C), 129V-type 2 (T50 82.9 ± 5.0 °C) and 129M-type “i” (T50 75.2 ± 3.4 °C) cases showed a higher thermostability than in 129V-type 1 (T50 64.8 ± 12.8 °C, vs. 129M-type 1, p = 0.002; vs. 129V-type 2, p < 0.001) and 129M-type 2C cases (T50 56.9 ± 7.3 °C, vs. 129M-type 1, p < 0.001; vs. 129V-type 2, p < 0.0001, vs. 129M-type “i”, p = 0.006) (Fig. 5b). We did not detect any significant difference between gCJD 129M-type 1, 129V-type 2, 129M-type 2 cases, and the corresponding sCJD subtypes (Fig. 5b). Genetic CJD 129V-type 1 cases showed a higher PrPSc thermostability than sCJD VV1 (T50 64.8 ± 12.8 vs. < 25.0 °C), which was, at least partially, due to the higher stability of PrPSc associated with the T188R mutation compared with the D178N variant (T50 77.5 ± 3.4 vs. 57.2 ± 9.3 °C, p = 0.036). There were no other significant differences between mutations within each group (Supplementary Fig. 7, online resource).
Finally, PrPSc associated with FFI cases was highly sensitive to both PK and temperature denaturation. PrPSc thermostability was similar in FFI and sCJD MM2T groups (T50 60.1 ± 6.5 vs. 57.6 ± 3.2 °C, p = 0.394), while PK resistance was slightly lower in the former group (ED50 9.0 ± 0.3.4 vs. 13.2 ± 3.7 U/mL, p = 0.093).
Clinical findings
Age at onset and duration of symptoms
The age at onset and disease duration in gCJD also significantly reflected those of their sporadic counterparts. In particular, like sCJD VV1 and MM2T patients, those from the 129V-type 1 and FFI groups were younger than gCJD patients of other groups, while, like sCJD MM1, 129M-type 1 patients had the shortest disease duration (Table 3). A significant exception was observed for participants carrying 5/6-OPRI mutations in the 129M-type 1 group, which were significantly younger at onset and showed a more protracted clinical course than those with other mutations. Similarly, the 5-OPRI case in the 129M-type 2 group showed the most prolonged survival. However, we did not find a similar effect on the age at onset and disease duration in patients of the 129V-type 2 group carrying 5/6-OPRI. The mean age at onset was also slightly less in E200K carriers than in patients carrying all other mutations (61.6 ± 8.9 vs. 65.7 ± 9.7 years, p = 0.008) after excluding those with 5/6-OPRI and those of 129V-type 1 and FFI groups. In the 129V-type 1 group, D178N carriers were significantly younger at disease onset than patients with the T188R variant (p = 0.001). Furthermore, in the D178N group, 129VV patients were younger at disease onset and showed a trend toward a shorter disease duration than 129VM carriers (Supplementary Table 6, online resource). In the FFI group, 129MV subjects showed a significantly longer disease duration (Supplementary Table 6, online resource). Finally, the two participants carrying the T183A mutation showed a strikingly young age at onset (40 and 41 years) and prolonged disease duration (108 and 48 months).
Clinical symptoms and signs at onset
Genetic CJD 129V-type 2 and 129M-type “i” groups presented with ataxia in most cases (70%). In contrast, 129V-type 1 and 129M-type 2C groups showed cognitive impairment as the most frequent presenting sign (82% and 75% of cases, respectively) (Table 4).
Both cognitive decline and ataxia characterized the clinical onset in approximately half of the patients with 129M-type 1, without significant differences among PRNP mutations (Supplementary Table 7, online resource). Moreover, in about 20% of 129M-type 1 cases, the disease presented with unilateral neurological signs and visual symptoms. We also found a higher prevalence of isolated memory loss in the gCJD groups characterized by remarkable hippocampal spongiform change and gliosis (129V-type 2, 129V-type 1, and 129M-type “i”) (data not shown).
Clinical symptoms and signs during evolution
A variable combination of cognitive, behavioral/psychiatric, cerebellar, pyramidal, and extrapyramidal signs characterized the clinical course of most gCJD cases (Supplementary Table 8, online resource). However, similar to sCJD, visual symptoms were more frequent in the 129M-type 1 group than in other groups, whereas all 129V-type 2 patients presented with cerebellar symptoms or signs. Myoclonus was frequent in gCJD groups with predominant cortical involvement (129M-type 1, 129V-type 1, and 129M-type 2C), while it occurred less frequently and, possibly, at a later stage in the other groups.
Diagnostic investigations
CSF prion RT-QuIC demonstrated positive PrPSc seeding activity in 92.1% of the tested gCJD patients (35/38) and had a higher sensitivity than CSF t-tau (41/46, 89.1%) and 14-3-3 proteins (95/113, 84.1%) (Supplementary Table 9, online resource). Fifty-five out of 67 cases (82.1%) presented with definite signal hyperintensities in FLAIR and/or DWI-MRI sequences. Increased signal more frequently involved the striatum than neocortices (83.6% vs. 41.8% of cases) (Supplementary Table 10, online resource). Finally, EEG recordings disclosed diffuse, periodic sharp-wave complexes, a feature supporting the diagnosis of CJD, in 89 out of 167 cases (53.3%). The latter finding was most frequent in 129M-type 1 than in other gCJD groups (Supplementary Table 11, online resource). Further details about the results of diagnostic investigations are provided in the Supplementary Materials, online resource.
Discussion
To reach a comprehensive characterization of the gCJD phenotypic spectrum, we carried out for the first time a systematic analysis of biochemical, molecular, clinical, and histopathological features in a large cohort, including 17 different PRNP mutations that represented the most frequent pathogenic human PRNP mutations linked to gCJD. The added value of the present analysis compared to previous studies include:
-
The deepest characterization of the disease subtypes linked to the most prevalent human PRNP mutation (E200K), including the description of two previously unreported histo-molecular phenotypes (i.e., M2C-E200K and M”i”-E200K).
-
The definition of the phenotypic spectrum of gCJD V210I in the largest patient population gathered to date.
-
The phenotypic characterization of 27 previously unreported individuals carrying five different mutations in association with PrPSc type 2, which is extremely rare in gCJD, especially in combination with the codon 129M genotype.
-
The systematic analysis of the co-occurrence and regional distribution of PrPSc types 1 and 2 in multiple brain regions in 142 individuals with gCJD.
-
The characterization of PrPSc resistance to protease digestion and temperature denaturation in gCJD.
-
A systematic comparison of data obtained with the same methodology in two large cohorts of gCJD and sCJD.
Overall, the results provide an accurate definition and classification of gCJD subtypes and insights into significant aspects of human prion biology, such as the effect of PRNP variants on phenotypic expression and the origin of prion strains.
Our findings demonstrate a striking parallelism between genetic and sporadic CJD phenotypes [20]. While overall the combination of PRNP genotype and PrPSc type accounts for the ultimate clinicopathological phenotype in both the forms, the genotypic variability determined by most PRNP mutations has only a moderate or even minimal effect on the disease phenotype. Our finding of divergent phenotypes in participants carrying the same PRNP mutation in cis with distinct codon 129 genotypes in patients carrying the E200K, 5/6-OPRI, D178N, R208H, and E196K variants underlines the central role of codon 129 (on the mutated allele). Moreover, the detection of distinct PrPSc types (1, 2 or “i”) in participants carrying the same PRNP haplotype (E200K-129M, 5-OPRI-129M) firmly demonstrates that PrPSc can assume distinct conformations in individuals carrying the same PRNP sequence, even in the presence of a mutation. Finally, the fact that in gCJD, most mutations are preferentially associated with PrPSc type 1 when in cis with 129M, and with type 2 when in cis with 129V, further proves the similarities with sCJD. These findings strengthen the notion that in gCJD, as in sporadic and acquired CJD forms, the codon 129 genotype plays a fundamental role in directing PrPSc abnormal conformation and the emergence of a specific prion strain [30, 34, 44, 50, 63, 68, 93].
Our data indicate that specific mutations have a variable effect on phenotypic expression. Specifically, four groups can be distinguished. The first would include several PRNP variants linked to PrPSc type 1 and 129M in the mutated allele with apparently incomplete penetrance and no effect on PrPSc glycotype (e.g., V210I and V203I). These mutations have virtually no effect on the clinicopathological disease phenotype. The second group would include some PRNP variants know to have a high penetrance (E200K and OPRI) with or without a specific effect on PrPSc glycotype. Examples of the effect of these mutations on the pathological phenotype include the “thickened” synaptic pattern of PrP deposition, which is best seen in the molecular layer of cerebellum, the stripe-like granular deposits perpendicular to the surface of the cerebellar molecular layer, which are hallmarks of the E200K and 5/6-OPRI, respectively, and the intraneuronal globular deposits found in the V2-E200K and M”i”-E200K groups [14, 16, 41, 53, 61, 62]. Similar effects are the young age at onset and the relatively long disease duration in 5/6-OPRI cases, at least when combined with 129M and PrPSc type 1. The third group would include mutations like T183A located near the PrP glycosylation sites, which, by blocking the addition of the first glycan chain to residue 181 [17], profoundly affect the PrPSc glycoform profile and the resulting disease phenotype. Indeed, the T183A-129M haplotype has been linked to PrPSc type 2 with an atypical glycoform ratio and a histotype lacking several pathological distinctive features of the sCJD MM2 phenotypes, with whom it shares the 129M-type 2 combination. The observation is consistent with the results of experimental transmission studies demonstrating that the glycosylation status of host PrPC affects prion strain characteristics [15]. Finally, although D178N and T188R mutations do not induce a novel phenotype, they share the peculiarity of an association with molecular types, which are extremely rare in sCJD. Notably, only the rare 129M-type 2 or 129V-type1 combinations have been detected to date in patients carrying the D178N mutation, making these combinations more common in genetic prion diseases than in the sporadic form.
In this study, we focused on PRNP mutations linked to CJD or FFI, representing only a subset of the inherited prion diseases, albeit the most common. Consequently, it would be misleading to extend the conclusions driven by the present findings to the inherited PrP-amyloidosis. Indeed, the absence of a definite sporadic counterpart strongly suggests that, in contrast to CJD and FFI, phenotypic diversity in GSS and related syndromes is primarily driven by the PRNP mutation. However, the recent characterization of variably protease-sensitive prionopathy, a novel phenotype of sporadic prion disease associated with distinctive PrPSc properties and clinico-pathological features, has clearly shown that the formation of C-terminally truncated, unglycosylated and anchorless fragments, thought to be GSS-specific until recently, may also occur in sporadic prion disease in the absence of any mutation [102], a finding which supports the view that most PrPSc conformational diversity may develop independent of the presence of PRNP mutations.
To provide an updated definition of the spectrum of gCJD variants accounting for all the major molecular/genetic determinants of the phenotype, we suggest a novel terminology that, in a hierarchical order, include: a) the PRNP genotype at codon 129 in cis with the mutation, b) the type of PrPSc, and c) the PRNP mutation to propose a classification of gCJD in six main variants or subtypes (see Table 5), that largely, but not entirely, reproduces those already defined in sCJD. Additionally, we propose to provisionally classify the few cases not fulfilling the six groups as “atypical”.
The classification recognizes the prion strains as the critical molecular determinants of the phenotypic spectrum of all forms of CJD. Transmission studies conducted to date with sCJD inocula currently support the existence of five distinct strains of CJD prions: the M1 related to the typical CJD phenotype or myoclonic variant [MM(V)1], V2 linked to the ataxic and kuru-plaque variants (VV2 and MV2K), M2T causing the thalamic variant (MM2T or sporadic FI), M2C linked to the cortical MM2C subtype, and V1 associated with the VV1 subtype [8, 13, 42, 74, 79]. Transmission data for gCJD and FFI are available for the E200K-129M [4, 51, 71, 84] and V210I-129M [59] haplotypes (expressing PrPSc type 1), a few cases carrying insertion mutations coupled with M at codon 129 in the mutated allele [61], a single case carrying the E200K-129V haplotype [4], and several FFI cases [25, 58, 97, 98]. Therefore, the limited data gathered to date demonstrating the M1 and M2T strains in gCJD and FFI are in line with the idea that the spectrum of strain variation in gCJD significantly parallels that identified in sCJD. The data we contributed with the present study further corroborates this notion by strongly supporting the occurrence of V2, V1, and M2C strains in gCJD, in addition to M1 and M2T. However, further transmission studies will be needed to fully confirm our conclusions on the parallelism between gCJD and sCJD regarding the spectrum of prion strains.
Of note, the V2 strain is the one showing the most significant differences between the genetic and sporadic forms. Transmission studies have to date linked the V2 strain to CJD subtypes VV2 and MV2K, occurring in both sporadic and acquired forms of the prion disease [46, 48, 84, 88], although some authors recently argued that the MV2K subtype should be considered a distinct prion “strain” based on the unique PrPSc and histopathologic features in the natural host [70]. Remarkably, an additional subtype, named MM “i”K, which only affected patients with iatrogenic CJD showed the same transmission properties of the VV2 and MV2K subtypes in humanized knock-in transgenic mice [46, 48]. The existence of the latter subtype makes V2 the only strain affecting all the three codon 129 genotypes, although it preferentially converts and replicates the PrPC-129V [8, 46, 70]. As an additional peculiarity, the PrPSc conformation associated with the V2 strain changes according to the presence of V or M at position 129 [49, 84], resulting in a PK-resistant core of 19 kDa (type 2) in the patients carrying VV, a 20 kDa fragment (type “i”) in those with MM, and a “doublet” including both PrPSc fragments in those carrying MV [45, 72]. However, only patients with 5/6-OPRI-129V(M) showed a genuine MV2K histotype, including a western blot profile with the 20 and 21 kDa doublet. In contrast, those carrying E200K-129V(M), despite the similar lesion profile lacked the kuru-type amyloid plaques. The relative contribution of the wild-type and mutated alleles to PrPSc formation might explain the two mutations' different behavior: in patients carrying the E200K mutation, only the mutated allele contributes to PrPSc formation, whereas both mutant and wild-type PrPC convert to PrPSc in OPRI [23, 28].
The identification of the novel M“i”-E200K gCJD phenotype requires further comments. Both the clinicopathological and molecular data strongly suggest that also this histotype is linked to the V2 strain. Firstly, the lesion profile and other histopathological features of this histotype strikingly resemble those of the V2-E200K group. Moreover, both PK resistance and thermo-solubility of M“i”-E200K PrPSc show more similarities to those of the V2-E200K PrPSc than to those of M1-E200K PrPSc. These findings, combined with the notion that PrPSc type “i” (129M) has been linked to V2 prions in iatrogenic CJD, strongly support the idea that the M“i”-E200K phenotype reflects the adaptation of V2 strain to the 129M-E200K host genotype.
To explain why the 129M/type “i” combination does not have a sporadic counterpart, we hypothesized that the E200K mutation might act as a permissive factor for the V2 adaptation through a change in conformation or by favoring the selection of highly glycosylated isoforms [6]. The lack of kuru plaques in M”i”-E200K might, again, depend on the mono-allelic origin of PrPSc (from the mutant PrP allele), somehow preventing the interaction between PrP isoforms generated by different alleles, or, again on a mutation-specific effect, possibly related to the glycan chains inhibiting PrPSc-amyloid plaque formation. Experimental transmission studies are required to explore these issues further. Finally, given that all M“i”-E200K affected participants carried 129V in trans with mutation, we cannot rule out a possible role of the wild-type allele on the emergence of this phenotype.
Another interesting finding was the presence of intraneuronal cytoplasmic PrP inclusions in a subgroup of patients carrying the E200K mutation. Intraneuronal PrP immunoreactivity has been consistently observed in animals but not in human prion diseases [43]. Interestingly, Kovacs et al. [54] reported that a particular type of intraneuronal PrP immunoreactivity, they named type III, resembling the "globular" pattern we observed, distinguishes a subset of gCJD E200K individuals. In contrast, the presence of smaller dot-like intraneuronal PrP deposits characterized both genetic and sporadic CJD brains. Although, we could not replicate the whole spectrum of intraneuronal deposits described by Kovacs et al. [54], likely because of differences in the PrP immunostaining protocol, our findings are consistent with the intraneuronal globular inclusions being a specific feature of the molecular subtypes V2-E200K and M"i"-E200K linked to the V2 strain. The reason why the E200K mutation favors the formation of intracellular PrP deposits that are nor readily seen in sCJD might depend on the abnormal intracellular processing of the mutant PrP showing impaired transport to the cell surface [18].
By examining this large cohort of gCJD cases, we also detected rare “atypical” phenotypes, difficult to reconcile with one of the major histotypes included in the current sCJD classification. Notably, atypical phenotypes shared a PrPSc isoform characterized by a marked under-representation of the diglycosylated isoform. Furthermore, they showed an exclusive or predominant PrPSc type 2 migration profile. It is worth noting that a similar immunoblot profile also characterizes some sCJD with atypical features that remain unclassified to date. The lesion profiles and the PrP patterns of deposition were comparable between haplotypes suggesting that specific PrPSc properties, such as the glycotype, contributed to the phenotype more than the haplotype. The transmission of these cases would determine whether the atypical phenotype is driven by a novel prion strain or by the host genotype.
Another interesting phenomenon determining phenotypic diversity in prion disease is the coexistence of more than one prion strain in the natural host. We have previously shown [82] that the mixed phenotypes resulting from the co-occurrence of PrPSc types 1 and 2 affect about 35% of sporadic CJD cases, although other studies reported a wide range of estimates of the phenomenon likely due to differences in detection methodologies [36, 47, 86]. All 3 codon 129 genotypes may be affected although MM cases are more frequently involved, given that coexistence of the MM(V)1 and MM2C subtypes is the most prevalent association, involving almost 40% of MM cases [82]. The present results provide evidence that the mixed phenotypes also characterize the gCJD spectrum. They essentially reproduce the most common mixed variants previously described in sCJD [82]. However, when compared using the same protocol and methodology, the overall frequency appears slightly lower than in sCJD (21% vs. 35%), an effect that is, once again, mutation dependent. Specifically, mixed phenotypes were extremely rare in the M1-E200K group despite the large number of patients analyzed. In contrast, patients with M1-V210I showed a slightly higher prevalence compared to the corresponding sCJD group [82]. Altogether, these findings suggest that specific PRNP mutations, particularly those significantly affecting the disease phenotype like the E200K, may modulate disease susceptibility in a strain-specific manner. Accordingly, the E200K mutation would determine a reduced relative M2 vs M1 susceptibility. In contrast, PRNP mutations with incomplete penetrance such as the V210I, would be more permissive to reproduce the same prevalence and type of multiple stains/phenotype coexistence we observed in the sporadic form.
The age at onset in gCJD is another issue requiring specific comments. On an average, it is generally believed that the disease starts earlier in patients with gCJD compared to sCJD owing to the specific predisposing effect of the mutations [67]. However, our data depict a more complex picture, which underlines the role of the prion strain in both sCJD and gCJD, irrespective of the presence of a PRNP mutation. We have previously shown that the disease onset in sCJD occurs significantly earlier in sCJD VV1 and MM2T than in the other subtypes [1, 82]. Notably, we observed the same difference in the age at onset between the 129V-type 1, and FFI groups and most of the other gCJD groups. Exceptions concerned the PRNP mutations influencing PrPSc properties and disease phenotype, including E200K, T183A, and 5/6-OPRI, albeit to a different extent. Compared to sCJD, T183A and 5/6-OPRI-129M carriers showed a remarkably younger age at onset, whereas it was only slightly earlier, independent of the haplotype/PrPSc type combination, in those carrying E200K. Of note, 5/6-OPRI in combination with 129V cases did not show the same effect on age at onset, suggesting a strain-specific (earlier onset in M1 but not V2 affected participants) effect.
Strengths of our study include the comprehensive analysis of the molecular, histopathological, and, to a lesser extent, clinical data of the largest cohort of gCJD cases examined to date, and the direct comparison with a series of sCJD cases representative of currently identified disease subtypes. Limitations of this study include the absence of few rare mutations (e.g., G114V, E200G, and V189I) that have been linked to a CJD phenotype, and the small/imbalanced sample sizes of patient subgroups that are virtually inevitable, given the highly heterogeneous prevalence of the PRNP variants and phenotypes, which is also limiting the power of the statistical comparison within each of the gCJD groups, especially for demographic data and clinical features.
In conclusion, the results of the present study confirm and significantly extend previous data on gCJD collected in small case series or case reports. The results support an updated classification of gCJD in six groups or subtypes that largely, but not entirely, reproduce those already defined in sCJD. The most significant divergence between the two forms includes the 129M-type “i” combination in patients carrying the E200K variant, likely representing the adaptation of the V2 CJD strain to the 129M-E200K allele, which has no counterpart in the sporadic form. Additionally, some mutations significantly affect the morphology and cellular distribution of PrP deposits (E200K, OPRI, E211Q-129M, and R208H-129V) or, rarely, are linked to atypical phenotypes (T183A-129M). The latter features introduce further complexity to the histopathologic classification of CJD subtypes but may also help neuropathologists to suspect the genetic etiology of disease through the identification of mutation-specific patterns of PrP deposition.
Overall, our data show that most PRNP mutations have limited or even no effect on phenotypic expression compared to the PrPSc type/codon 129 (mutated allele) combination; however, they may act in conjunction with codon 129 genotype in modulating disease susceptibility to a specific PrPSc type/prion strain. They apparently do not generate novel, mutation-specific conformations, a conclusion that must be confirmed through experimental transmission studies.
References
Abu-Rumeileh S, Redaelli V, Baiardi S, Mackenzie G, Windl O, Ritchie DL et al (2018) Sporadic fatal insomnia in europe: phenotypic features and diagnostic challenges. Ann Neurol 84:347–360. https://doi.org/10.1002/ana.25300
Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. https://doi.org/10.1038/nrm2204
Areškevičiūtė A, Høgh P, Bartoletti-Stella A, Melchior LC, Nielsen PR, Parchi P et al (2019) A novel eight octapeptide repeat insertion in PRNP causing prion disease in a Danish family. J Neuropathol Exp Neurol 78:595–604. https://doi.org/10.1093/jnen/nlz037
Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R et al (2009) Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol 90:546–558. https://doi.org/10.1099/vir.0.007930-0
Bartz JC (2021) Environmental and host factors that contribute to prion strain evolution. Acta Neuropathol. https://doi.org/10.1007/s00401-021-02310-6
Baskakov IV, Katorcha E, Makarava N (2018) Prion strain-specific structure and pathology: a view from the perspective of glycobiology. Viruses 10:723. https://doi.org/10.3390/v10120723
Bessen RA, Marsh RF (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. https://doi.org/10.1128/JVI.68.12.7859-7868.1994
Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010. https://doi.org/10.1073/pnas.1004688107
Brown P, Goldfarb LG, Gibbs CJ Jr, Gajdusek DC (1991) The phenotypic expression of different mutations in transmissible familial Creutzfeldt-Jakob disease. Eur J Epidemiol 7:469–476. https://doi.org/10.1007/BF00143124
Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A et al (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35:513–529. https://doi.org/10.1002/ana.410350504
Bruce ME (2003) TSE strain variation. Br Med Bull 66:99–108. https://doi.org/10.1093/bmb/66.1.99
Buge A, Escourolle R, Brion S, Rancurel G, Hauw JJ, Mehaut M et al (1978) Maladie de Creutzfeldt-Jakob familiale. Etude clinique et anatomique de trois cas sur huit répartis sur trois générations. Transmission au singe écureuil [Familial Creutzfeldt-Jakob disease. A clinical and pathological study of three cases in a family with eight affected members in three generations (author’s transl)]. Rev Neurol (Paris) 134:165–181
Cali I, Puoti G, Smucny J, Curtiss PM, Cracco L, Kitamoto T et al (2020) Co-existence of PrPDtypes 1 and 2 in sporadic Creutzfeldt-Jakob disease of the VV subgroup: phenotypic and prion protein characteristics. Sci Rep 10:1503. https://doi.org/10.1038/s41598-020-58446-0
Campbell TA, Palmer MS, Will RG, Gibb WR, Luthert PJ, Collinge J (1996) A prion disease with a novel 96-base pair insertional mutation in the prion protein gene. Neurology 46:761–766. https://doi.org/10.1212/wnl.46.3.761
Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P et al (2013) Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 32:756–769. https://doi.org/10.1038/emboj.2013.6
Capellari S, Vital C, Parchi P, Petersen RB, Ferrer X, Jarnier D et al (1997) Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology 49:133–141. https://doi.org/10.1212/wnl.49.1.133
Capellari S, Zaidi SI, Long AC, Kwon EE, Petersen RB (2000) The Thr183Ala mutation, not the loss of the first glycosylation site, alters the physical properties of the prion protein. J Alzheimers Dis 2:27–35. https://doi.org/10.3233/jad-2000-2104
Capellari S, Parchi P, Russo CM, Sanford J, Sy MS, Gambetti P et al (2000) Effect of the E200K mutation on prion protein metabolism. Comparative study of a cell model and human brain. Am J Pathol 157(2):613–622. https://doi.org/10.1016/S0002-9440(10)64572-5
Capellari S, Cardone F, Notari S, Schininà ME, Maras B, Sità D et al (2005) Creutzfeldt-Jakob disease associated with the R208H mutation in the prion protein gene. Neurology 64:905–907. https://doi.org/10.1212/01.WNL.0000152837.82388.DE
Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P (2011) Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 121:21–37. https://doi.org/10.1007/s00401-010-0760-4
Cardone F, Liu QG, Petraroli R, Ladogana A, D’Alessandro M, Arpino C et al (1999) Prion protein glycotype analysis in familial and sporadic Creutzfeldt-Jakob disease patients. Brain Res Bull 49:429–433. https://doi.org/10.1016/s0361-9230(99)00077-5
Cescatti M, Saverioni D, Capellari S, Tagliavini F, Kitamoto T, Ironside J et al (2016) Analysis of Conformational Stability of Abnormal Prion Protein Aggregates across the Spectrum of Creutzfeldt-Jakob Disease Prions. J Virol 90:6244–6254. https://doi.org/10.1128/JVI.00144-16
Chen SG, Parchi P, Brown P, Capellari S, Zou W, Cochran EJ et al (1997) Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat Med 3:1009–1015. https://doi.org/10.1038/nm0997-100
Cochran EJ, Bennett DA, Cervenáková L, Kenney K, Bernard B, Foster NL et al (1996) Familial Creutzfeldt-Jakob disease with a five-repeat octapeptide insert mutation. Neurology 47:727–733. https://doi.org/10.1212/wnl.47.3.727
Collinge J, Palmer MS, Sidle KC, Gowland I, Medori R, Ironside J et al (1995) Transmission of fatal familial insomnia to laboratory animals. Lancet 346:569–570. https://doi.org/10.1016/s0140-6736(95)91405-6
Cracco L, Appleby BS, Gambetti P (2018) Fatal familial insomnia and sporadic fatal insomnia. Handb Clin Neurol 153:271–299. https://doi.org/10.1016/B978-0-444-63945-5.00015-5
Eigenbrod S, Frick P, Giese A, Schelzke G, Zerr I, Kretzschmar HA (2011) Comprehensive neuropathologic analysis of genetic prion disease associated with the E196K mutation in PRNP reveals phenotypic heterogeneity. J Neuropathol Exp Neurol 70:192–200. https://doi.org/10.1097/NEN.0b013e31820cd8a4
Gabizon R, Telling G, Meiner Z, Halimi M, Kahana I, Prusiner SB (1996) Insoluble wild-type and protease-resistant mutant prion protein in brains of patients with inherited prion disease. Nat Med 2:59–64. https://doi.org/10.1038/nm0196-59
Ghetti B, Piccardo P, Zanusso G (2018) Dominantly inherited prion protein cerebral amyloidoses—a modern view of Gerstmann-Sträussler-Scheinker. Handb Clin Neurol 153:243–269. https://doi.org/10.1016/B978-0-444-63945-5.00014-3
Goldfarb LG, Brown P, Haltia M, Cathala F, McCombie WR, Kovanen J et al (1992) Creutzfeldt-Jakob disease cosegregates with the codon 178Asn PRNP mutation in families of European origin. Ann Neurol 31:274–281. https://doi.org/10.1002/ana.410310308
Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P et al (1992) Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 258:806–808. https://doi.org/10.1126/science.1439789
Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA (2004) Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol 108:476–484. https://doi.org/10.1007/s00401-004-0913-4
Grasbon-Frodl E, Schmalzbauer R, Weber P, Krebs B, Windl O, Zerr I et al (2004) A novel three extra-repeat insertion in the prion protein gene (PRNP) in a patient with Creutzfeldt-Jakob disease. Neurogenetics 5:249–250. https://doi.org/10.1007/s10048-004-0196-x
Hainfellner JA, Parchi P, Kitamoto T, Jarius C, Gambetti P, Budka H (1999) A novel phenotype in familial Creutzfeldt-Jakob disease: prion protein gene E200K mutation coupled with valine at codon 129 and type 2 protease-resistant prion protein. Ann Neurol 45:812–816. https://doi.org/10.1002/1531-8249(199906)45:6%3c812::AID-ANA20%3e3.0.CO;2-2
Haltia M, Kovanen J, Van Crevel H, Bots GT, Stefanko S (1979) Familial Creutzfeldt-Jakob disease. J Neurol Sci 42:381–389. https://doi.org/10.1016/0022-510x(79)90171-0
Head MW, Bunn TJR, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS et al (2004) Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: U.K. cases 1991–2002. Ann Neurol 55:851–859. https://doi.org/10.1002/ana.20127
Hill AF, Joiner S, Beck JA, Campbell TA, Dickinson A, Poulter M et al (2006) Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain 129:676–685. https://doi.org/10.1093/brain/awl013
Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD et al (1989) Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature 338:342–345. https://doi.org/10.1038/338342a0
Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D et al (1991) Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med 324:1091–1097. https://doi.org/10.1056/NEJM199104183241604
Imbriani P, Marfia GA, Marciani MG, Poleggi A, Pocchiari M, Puoti G et al (2016) Heidenhain variant in two patients with inherited V210I Creutzfeldt-Jakob disease. Int J Neurosci 126:381–383. https://doi.org/10.3109/00207454.2015.1047017
Jarius C, Kovacs GG, Belay G, Hainfellner JA, Mitrova E, Budka H (2003) Distinctive cerebellar immunoreactivity for the prion protein in familial (E200K) Creutzfeldt-Jakob disease. Acta Neuropathol 105:449–454. https://doi.org/10.1007/s00401-002-0664-z
Jaumain E, Quadrio I, Herzog L, Reine F, Rezaei H, Andréoletti O et al (2016) Absence of evidence for a causal link between bovine spongiform encephalopathy strain variant L-BSE and known forms of sporadic creutzfeldt-jakob disease in human PrP transgenic mice. J Virol 90:10867–10874. https://doi.org/10.1128/JVI.01383-16
Jeffrey M, McGovern G, Sisó S, González L (2011) Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol 121:113–134. https://doi.org/10.1007/s00401-010-0700-3
Kaski DN, Pennington C, Beck J, Poulter M, Uphill J, Bishop MT et al (2011) Inherited prion disease with 4-octapeptide repeat insertion: disease requires the interaction of multiple genetic risk factors. Brain 134:1829–1838. https://doi.org/10.1093/brain/awr079
Kobayashi A, Asano M, Mohri S, Kitamoto T (2007) Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 282:30022–30028. https://doi.org/10.1074/jbc.M704597200
Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T (2010) Experimental verification of a traceback phenomenon in prion infection. J Virol 84:3230–3238. https://doi.org/10.1128/JVI.02387-09
Kobayashi A, Mizukoshi K, Iwasaki Y, Miyata H, Yoshida Y, Kitamoto T (2011) Co-occurrence of types 1 and 2 PrP(res) in sporadic Creutzfeldt-Jakob disease MM1. Am J Pathol 178:1309–1315. https://doi.org/10.1016/j.ajpath.2010.11.069
Kobayashi A, Iwasaki Y, Otsuka H, Yamada M, Yoshida M, Matsuura Y et al (2013) Deciphering the pathogenesis of sporadic Creutzfeldt-Jakob disease with codon 129 M/V and type 2 abnormal prion protein. Acta Neuropathol Commun 1:74. https://doi.org/10.1186/2051-5960-1-74
Kobayashi A, Matsuura Y, Mohri S, Kitamoto T (2014) Distinct origins of dura mater graft-associated Creutzfeldt-Jakob disease: past and future problems. Acta Neuropathol Commun 2:32. https://doi.org/10.1186/2051-5960-2-32
Kobayashi A, Teruya K, Matsuura Y, Shirai T, Nakamura Y, Yamada M et al (2015) The influence of PRNP polymorphisms on human prion disease susceptibility: an update. Acta Neuropathol 130:159–170. https://doi.org/10.1007/s00401-015-1447-7
Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J et al (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci U S A 100:4784–4789. https://doi.org/10.1073/pnas.2627989100
Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C et al (2005) Genetic prion disease: the EUROCJD experience. Hum Genet 118:166–174. https://doi.org/10.1007/s00439-005-0020-1
Kovacs GG, Seguin J, Quadrio I, Höftberger R, Kapás I, Streichenberger N et al (2011) Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol 121:39–57. https://doi.org/10.1007/s00401-010-0713-y
Kovacs GG, Molnár K, Keller E, Botond G, Budka H, László L (2012) Intraneuronal immunoreactivity for the prion protein distinguishes a subset of E200K genetic from sporadic Creutzfeldt-Jakob Disease. J Neuropathol Exp Neurol 71:223–232. https://doi.org/10.1097/NEN.0b013e318248aa70
Krebs B, Lederer RM, Windl O, Grasbon-Frodl EM, Zerr I, Kretzschmar HA (2005) Creutzfeldt-Jakob disease associated with an R148H mutation of the prion protein gene. Neurogenetics 6:97–100. https://doi.org/10.1007/s10048-004-0208-x
Ladogana A, Kovacs GG (2018) Genetic Creutzfeldt-Jakob disease. Handb Clin Neurol 153:219–242. https://doi.org/10.1016/B978-0-444-63945-5.00013-1
Manetto V, Medori R, Cortelli P, Montagna P, Tinuper P, Baruzzi A et al (1992) Fatal familial insomnia: clinical and pathologic study of five new cases. Neurology 42:312–319. https://doi.org/10.1212/wnl.42.2.312
Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ et al (1999) Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med 340:1630–1638. https://doi.org/10.1056/NEJM199905273402104
Mastrianni JA, Capellari S, Telling GC, Han D, Bosque P, Prusiner SB et al (2001) Inherited prion disease caused by the V210I mutation: transmission to transgenic mice. Neurology 57:2198–2205. https://doi.org/10.1212/wnl.57.12.2198
May WW, Itabashi HH, De Jong RN (1968) Creutzfeldt-Jakob disease. II. Clinical, pathologic, and genetic study of a family. Arch Neurol 19:137–149. https://doi.org/10.1001/archneur.1968.00480020023002
Mead S, Poulter M, Beck J, Webb TE, Campbell TA, Linehan JM et al (2006) Inherited prion disease with six octapeptide repeat insertional mutation–molecular analysis of phenotypic heterogeneity. Brain 129:2297–2317. https://doi.org/10.1093/brain/awl226
Mead S, Webb TE, Campbell TA, Beck J, Linehan JM, Rutherfoord S et al (2007) Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology 69:730–738. https://doi.org/10.1212/01.wnl.0000267642.41594.9d
Mead S, Lloyd S, Collinge J (2019) Genetic factors in mammalian prion diseases. Annu Rev Genet 53:117–147. https://doi.org/10.1146/annurev-genet-120213-092352
Medori R, Tritschler HJ, LeBlanc A, Villare F, Manetto V, Chen HY et al (1992) Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 326:444–449. https://doi.org/10.1056/NEJM199202133260704
Melis M, Molari A, Floris G, Vascellari S, Balestrino L, Ladogana A et al (2020) Genetic Creutzfeldt-Jakob disease in Sardinia: a case series linked to the PRNP R208H mutation due to a single founder effect. Neurogenetics 21:251–257. https://doi.org/10.1007/s10048-020-00618-1
Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF et al (2016) Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 8:322ra9. https://doi.org/10.1126/scitranslmed.aad5169
Minikel EV, Vallabh SM, Orseth MC, Brandel JP, Haïk S, Laplanche JL et al (2019) Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology 93:e125–e134. https://doi.org/10.1212/WNL.0000000000007745
Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J et al (1994) Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA 91:2839–2842. https://doi.org/10.1073/pnas.91.7.2839
Montagna P, Cortelli P, Avoni P, Tinuper P, Plazzi G, Gallassi R et al (1998) Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol 8:515–520. https://doi.org/10.1111/j.1750-3639.1998.tb00172.x
Nemani SK, Xiao X, Cali I, Cracco L, Puoti G, Nigro M et al (2020) A novel mechanism of phenotypic heterogeneity in Creutzfeldt-Jakob disease. Acta Neuropathol Commun 8:85. https://doi.org/10.1186/s40478-020-00966-x
Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G et al (2006) Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2:e12. https://doi.org/10.1371/journal.ppat.0020012
Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B et al (2004) Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem 279:16797–16804. https://doi.org/10.1074/jbc.M313220200
Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P et al (2007) A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest 87:1103–1112. https://doi.org/10.1038/labinvest.3700676
Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P et al (2014) Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis 20:2006–2014. https://doi.org/10.3201/eid2012.140548
Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D et al (1989) Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet 1:51–52. https://doi.org/10.1016/s0140-6736(89)91713-3
Padovani A, D’Alessandro M, Parchi P, Cortelli P, Anzola GP, Montagna P et al (1998) Fatal familial insomnia in a new Italian kindred. Neurology 51:1491–1494. https://doi.org/10.1212/wnl.51.5.1491
Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB et al (1995) Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann Neurol 38:21–29. https://doi.org/10.1002/ana.410380107
Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39:767–778. https://doi.org/10.1002/ana.410390613
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. https://doi.org/10.1002/1531-8249(199908)46:2%3c224::AID-ANA12%3e3.0.CO;2-W
Parchi P, Capellari S, Chin S, Schwarz HB, Schecter NP, Butts JD et al (1999) A subtype of sporadic prion disease mimicking fatal familial insomnia. Neurology 52:1757–1763. https://doi.org/10.1212/wnl.52.9.1757
Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B et al (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A 97:10168–10172. https://doi.org/10.1073/pnas.97.18.10168
Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A, Ghetti et al (2009) Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol 118:659–671. https://doi.org/10.1007/s00401-009-0585-1
Parchi P, Notari S, Weber P, Schimmel H, Budka H, Ferrer I et al (2009) Inter-laboratory assessment of PrPSc typing in Creutzfeldt-Jakob disease: a Western blot study within the NeuroPrion Consortium. Brain Pathol 19:384–391. https://doi.org/10.1111/j.1750-3639.2008.00187.x
Parchi P, Cescatti M, Notari S, Schulz-Schaeffer WJ, Capellari S, Giese A et al (2010) Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 133:3030–3042. https://doi.org/10.1093/brain/awq234
Parchi P, de Boni L, Saverioni D, Cohen ML, Ferrer I, Gambetti P et al (2012) Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol 124:517–529. https://doi.org/10.1007/s00401-012-1002-8
Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A (2005) Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 4:805–814. https://doi.org/10.1016/S1474-4422(05)70225-8
Prusiner SB (1998) Prions. Proc Natl Acad Sci U S A 95:13363–13383. https://doi.org/10.1073/pnas.95.23.13363
Ritchie DL, Barria MA, Peden AH, Yull HM, Kirkpatrick J, Adlard P et al (2017) UK Iatrogenic Creutzfeldt-Jakob disease: investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches. Acta Neuropathol 133:579–595. https://doi.org/10.1007/s00401-016-1638-x
Roeber S, Krebs B, Neumann M, Windl O, Zerr I, Grasbon-Frodl EM et al (2005) Creutzfeldt-Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17-kDa prion protein fragment. Acta Neuropathol 109:443–448. https://doi.org/10.1007/s00401-004-0978-0
Roeber S, Grasbon-Frodl EM, Windl O, Krebs B, Xiang W, Vollmert C et al (2008) Evidence for a pathogenic role of different mutations at codon 188 of PRNP. PLoS ONE 3:e2147. https://doi.org/10.1371/journal.pone.0002147
Rossi M, Saverioni D, Di Bari M, Baiardi S, Lemstra AW, Pirisinu L et al (2017) Atypical Creutzfeldt-Jakob disease with PrP-amyloid plaques in white matter: molecular characterization and transmission to bank voles show the M1 strain signature. Acta Neuropathol Commun 5:87. https://doi.org/10.1186/s40478-017-0496-7
Rossi M, Baiardi S, Parchi P (2019) Understanding prion strains: evidence from studies of the disease forms affecting humans. Viruses 11:309. https://doi.org/10.3390/v11040309
Sánchez-Valle R, Yagüe J, Turón A, Aróstegui JI, Nos C, Rey MJ et al (2012) Inherited prion disease with 4-octapeptide repeat insertion linked to valine at codon 129. Brain 135:e212. https://doi.org/10.1093/brain/awr358
Saverioni D, Notari S, Capellari S, Poggiolini I, Giese A, Kretzschmar HA et al (2013) Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J Biol Chem 288:27972–27985. https://doi.org/10.1074/jbc.M113.477547
Skworc KH, Windl O, Schulz-Schaeffer WJ, Giese A, Bergk J, Nagele A et al (1999) Familial Creutzfeldt-Jakob disease with a novel 120-bp insertion in the prion protein gene. Ann Neurol 46:693–700. https://doi.org/10.1002/1531-8249(199911)46:5%3c693::AID-ANA3%3e3.0.CO;2-Z
Takeuchi A, Mohri S, Kai H, Tamaoka A, Kobayashi A, Mizusawa H et al (2019) Two distinct prions in fatal familial insomnia and its sporadic form. Brain Commun 1:fcz05. https://doi.org/10.1093/braincomms/fcz045
Tateishi J, Brown P, Kitamoto T, Hoque ZM, Roos R, Wollman R et al (1995) First experimental transmission of fatal familial insomnia. Nature 376:434–435. https://doi.org/10.1038/376434a0
Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R et al (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082. https://doi.org/10.1126/science.274.5295.2079
Tiple D, Poleggi A, Mellina V, Morocutti A, Brusa L, Iani C et al (2019) Clinicopathological features of the rare form of Creutzfeldt-Jakob disease in R208H–V129V PRNP carrier. Acta Neuropathol Commun 7:47. https://doi.org/10.1186/s40478-019-0699-1
Vorberg I, Buschmann A, Harmeyer S, Saalmüller A, Pfaff E, Groschup MH (1999) A novel epitope for the specific detection of exogenous prion proteins in transgenic mice and transfected murine cell lines. Virology 255:26–31. https://doi.org/10.1006/viro.1998.9561
Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10:e1003990. https://doi.org/10.1371/journal.ppat.1003990
Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I et al (2010) Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68:162–172. https://doi.org/10.1002/ana.22094
Acknowledgements
We are indebted to all physicians and family members who provided clinical information or pathological material regarding the patients studied. We wish to thank Benedetta Carlà, Silvia Piras, Barbara Polischi, Diane Kofskey, Rose Marie Richardson, and Brenda Duprè, for their valuable technical assistance, and Francine Epperson for her help in collecting the medical records for some of the patients. The human tissue of UK cases examined in this study was provided by the MRC Edinburgh Brain Bank. We would also like to thank the staff of the National Prion Disease Pathology Surveillance Center, who helped with many of the U.S. cases included in the study. This work was financially supported by the Italian Ministry of Health grant “Ricerca Corrente” to PP and AL, the University of Bologna grants “RFO” to PP and, in part, by NIH grants K99AG068359 to IC, and P01P01AI07774 to PG.
Funding
Open access funding provided by Alma Mater Studiorum - Università di Bologna within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
PP and SC conceived the overall project. BSA, MB, IC, PG, EG, AG, BG, JH, AL, JM, DLR, VR, OW, and PP provided tissue specimens and key clinical information, and interpretations. SB, SC, and PP revised all clinical data. SB and PP revised neuropathological data. SC supervised genetic analysis. MR and AM performed protein studies. SB performed the statistical analysis. SB and PP prepared the manuscript. All authors helped to interpret the results and edit the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Financial interests: BSA received funding through CDC, NIH, Ionis Pharmaceutical and Alector; the other authors declare they have no conflict of interest. Non-financial interests: BSA is member of the board of directors for the CJD Foundation; the other authors declare they have no conflict of interest.
Ethical approval
The study was conducted according to the revised Declaration of Helsinki and Good Clinical Practice guidelines. All medical information was collected as part of national surveillance programs for prion diseases and approved by the local Institutional Review Board for each country/participating center (CE-ISS 09/266, May 2009). All patients’ data and samples were coded to protect their identities. Written informed consent was given by study participants or the next of kin.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baiardi, S., Rossi, M., Mammana, A. et al. Phenotypic diversity of genetic Creutzfeldt–Jakob disease: a histo-molecular-based classification. Acta Neuropathol 142, 707–728 (2021). https://doi.org/10.1007/s00401-021-02350-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02350-y