
Abstract
Coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). To date, there is no effective therapeutic approach for treating SARS-CoV-2 infections. MicroRNAs (miRNAs) have been recognized to target the viral genome directly or indirectly, thereby inhibiting viral replication. Several studies have demonstrated that host miRNAs target different sites in SARS-CoV-2 RNA and constrain the production of essential viral proteins. Furthermore, miRNAs have lower toxicity, are more immunogenic, and are more diverse than protein-based and even plasmid-DNA-based therapeutic agents. In this review, we emphasize the role of miRNAs in viral infection and their potential use as therapeutic agents against COVID-19 disease. The potential of novel miRNA delivery strategies, especially EDV™ nanocells, for targeting lung tissue for treatment of SARS-CoV-2 infection is also discussed.
Similar content being viewed by others


Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a new member of the family Coronaviridae that mainly targets the respiratory system in humans, resulting in coronavirus disease 2019 (COVID-19). This virus was first discovered in late 2019 in Wuhan, the capital of China's Hubei Province, and it subsequently caused a pandemic [179] that infected more than 50.5 million people worldwide and killed more than 1.2 million by November 2020 [114]. A person infected with SARS-CoV-2 may be asymptomatic or have flu-like symptoms such as fever, dry cough, difficult or labored breathing, contusions, and lymphocytopenia [169]. Conventional vaccine development typically takes more than 15 years, commencing with a long discovery phase in which vaccine materials are constructed and preclinical investigations are performed. The development of vaccines for COVID-19 is following a faster timeline. Thanks to the information obtained from research on vaccines against Middle East respiratory syndrome-related coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV), the initial discovery stage was largely omitted. Available data were accepted, and phase I/II clinical trials were launched. With numerous clinical trials running in parallel, stage III clinical trials were started following the interim analysis of phase I/II findings. Based on World Health Organization (WHO) reports, as of January 15, 2021, 64 vaccine candidates were under clinical assessment, and 173 candidate vaccines are in preclinical stages. The vaccine platforms can be separated into ‘conventional methods (inactivated or live-virus vaccines), recombinant protein vaccines, vectored vaccines, and nucleic acid-based vaccines (RNA and DNA vaccines).
Nevertheless, these vaccines will need to be replaced in the future by novel vaccines that provide the same protective effect with more tolerable reactogenicity [33, 66]. Furthermore, the emergence of mutated strains of SARS-CoV-2 in South Africa, the United Kingdom, and other countries is of concern because some of these mutations lie in the viral receptor binding site for cell entry and enhance binding to angiotensin-converting enzyme 2 (ACE2). The continuous occurrence of mutations in the SARS-CoV-2 genome necessitates constant surveillance of the impact of these variations on vaccine coverage and preparation for the probability that mutations in the SARS-CoV-2 genome might make it necessary to change the vaccine strain.
In addition to the complexity of vaccine development, the trust and acceptance of the vaccines by the public is unclear and dynamic, making the achievement of herd immunity challenging. Other social problems with SARS-CoV-2 vaccination include the fact that minorities and lower-income and illiterate people are more susceptible than others to this infection [77]. These issues press researchers worldwide to develop novel, safe, and efficient preventive and therapeutic approaches.
Although vaccine development efforts are advancing well, secondary prevention and screening should be aimed at early disease detection and intervention before the onset of symptoms for those with subclinical or mild disease. It is notable that almost 80% of COVID-19 cases have mild signs [64]. Secondary prevention may therefore be particularly critical for containing COVID-19, since asymptomatic individuals may still be able to transmit the virus. Asymptomatic spread is a challenge for contact tracing and recognizing transmission chains. With COVID-19, the benefits of early detection testing seem to outweigh the consequences of potential false negatives; further follow-up testing and monitoring can be conducted if the suspicion index is high and other criteria are met [59]. For efficient secondary prevention, rapid establishment of isolation and quarantine facilities, treatment facilities, and decentralized diagnostic testing at the local level are essential.
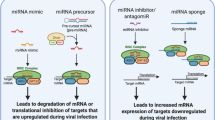
MicroRNAs (miRNAs) are short (about 20–23 nucleotides) single-stranded non-coding RNAs (ncRNA) that play a significant role in the posttranscriptional regulation of gene expression [4, 100]. In 1933, Lee et al. discovered an miRNA (lin-4) in the nematode Caenorhabditis elegans [72]. Since then, a huge variety of miRNAs with different functions have been found [101]. miRBase (http://www.mirbase.org/) is a website for all known miRNA sequences, annotation, nomenclature, and target anticipation data. In 2018, this site contained 38,589 entries, representing hairpin precursor miRNAs that express 48,885 mature miRNAs in 271 species [10, 43]. miRNAs play major regulatory roles in cell processes, such as proliferation, differentiation, growth, neoplastic transformation, and tissue regeneration [38, 133]. They also have a significant function in cell-cell communication and represent promising biomarkers and therapeutic tools for most diseases [104]. In addition, miRNAs exhibit fewer toxic effects and lower immunogenicity than protein-based drugs and even plasmid-DNA-based gene therapy. Current therapeutic applications of miRNAs involve two approaches: (1) inhibition of oncogenic miRNAs through the use of miRNA antagonists, such as antimiRs or antagomiRs, and (2) introduction of tumor suppressor miRNAs either via synthetic miRNA mimics or by stable and vector-based transfection of genes coding for miRNAs. miRNA antagonists are ss-RNA molecules of 21–23 nt that function via complementary base pairing with miRNAs [7, 22, 68]. AntimiRs containing cholesterol, conjugated via a 2′-O-methyl (2′-OMe) linkage, known as antigomiRs, are fully complementary to the mature miRNA sequence and contain various phosphorothioate moieties to enhance stability. Inhibition of miRNA by antimiRs requires optimization of the oligonucleotides for improved binding affinity, increased nuclease resistance, and more-efficient in vivo delivery. This optimization can be achieved by applying various chemical modifications, including unconjugated phosphorothioate antisense molecules with several further high-affinity 2′ sugar modifications such as 2′ O-methoxyethyl (2′ MOE) or a locked nucleic acid (LNA), which increase binding affinity [37].
The use of miRNA adds an extra level of complexity in comparison to using anti-miRs. This therapeutic strategy can potentially have side effects when new miRNAs are introduced into a cell. However, in vivo studies of toxic effects caused by miRNA mimics are still lacking [65]. Recent studies have demonstrated that miRNAs can target the genomes of RNA viruses to control the production of viral proteins and viral replication and modify viral pathogenesis. Furthermore, infection with an RNA virus can itself affect upregulation or downregulation of host cell miRNAs, causing downstream modifications of the transcriptome of the infected cell that promote the infection. However, up- or downregulation of miRNAs can inhibit viral replication [149], and it is therefore likely that human miRNAs may target important sites in the SARS-CoV-2 genome and inhibit virus replication. This comprehensive review emphasizes the role of miRNAs in viral infection and their potential therapeutic application in COVID-19. We also discuss a potential novel delivery strategy (EDV™ nanocells) for miRNAs to target specific tissues such as the lung.
miRNA biogenesis and function
Since miRNAs are not translated into proteins or oligopeptides, they are referred to as “noncoding RNAs” [102]. miRNA genes are transcribed by RNA polymerase II (Pol II) [9, 73, 106], and about 40% of miRNA genes are located within intron regions [13]. Transcription inside the nucleus generates pre-miRNAs and pro-miRNAs that form hairpin-like structures upon nuclear processing, resulting in molecules of 70-100 nucleotides. This process is catalyzed by a microprocessor complex (DGCR8 or Pasha, the catalytic RNAase III domain of Drosha). Pre-miRNA hairpins are exported from the nucleus to the cytosol by a RanGTP/exportin 5-dependent mechanism and cleaved by Dicer (RNase III) into a mature double-stranded ~22-nt miRNA/miRNA duplex in the cytoplasm [9, 30]. The active or mature single-stranded miRNAs bind to the RNA-induced silencing complex (RISC), which is composed of Argonaute-family proteins (Ago1 to Ago4 in humans) and TRBP [137]. The 5′ end of mature miRNAs possess seed sequences (about 8 nt) whose function is to recognize the mRNA [47]. A specific miRNA is capable of controlling hundreds of target genes. The repression mechanisms of miRNAs that target or regulate gene expression include (1) inhibition of elongation (mRNA target hybridization and degradation), (2) inhibition of translation (preventing the joining of 60S and 40S subunits), (3) premature termination of translation, and (4) co-translational protein degradation [31, 123]. miRNA functions in association with the miRISC. The core constituents of miRISC that are required for miRNA-dependent repression are the GW182 and Ago proteins [122] (Fig. 1).

The miRNA biogenesis pathway in cells. The Drosha, DGCR8, Exportin-5, Ran, and Dicer proteins are key constituents of miRNA biogenesis. Within the cytoplasm, the pre-miRNA loop is cleaved by a ternary complex formed by Dicer, producing small RNA duplexes (miRNA-miRNA*). The formation of immature RISC or pre-RISC occurs in a heat shock cognate 70 (Hsc-70)/ heat shock protein (Hsp90)-mediated process. The RISC binds the target mRNA via complementary binding of 6 to 8 base pairs of the miRNA, leading to gene silencing
miRNAs are unique tools in the diagnosis and treatment of malignancies, and this has generated great interest in other clinical applications [103, 112, 132]. miRNAs have crucial functions in various biological activities via post-transcriptional regulation [100, 128]. Altered expression of miRNAs is associated with human pathologies such as developmental abnormalities, cancer [160], inflammatory diseases [141], muscular and cardiovascular diseases [24, 35, 57], and viral infections [128].
The role of miRNAs in viral infection
Viral infections are associated with altered levels of host miRNAs. The effects of host miRNAs on viral infection can be exerted directly and indirectly. The direct impact of miRNAs on virus regulation occurs by directly targeting different regions of the viral RNA. The indirect effect involves modulating a cellular transcript encoding a host factor needed for one or more phases in the viral life cycle. However, virus infections may suppress the generation of miRNAs in an antiviral response. Viruses use the cellular machinery to express viral miRNAs (v-miRNAs), which were first discovered in Epstein-Barr virus (EBV) [20]. These v-miRNAs not only target their transcripts to regulate virus replication but also decrease the expression of host genes, generate environments conducive to their latency and replication, and elude clearance by the host immune response. Furthermore, virus infection can cause up- or downregulation of host miRNAs, thus inhibiting the immune response or promoting viral replication.
Viruses induce host miRNAs and control their alteration and functions
Host innate immune responses are the first line of protection against all pathogens. Natural killer cells, epithelial cells, monocytes, macrophages, dendritic cells, and granulocytes participate in the immune response [63, 71]. Cells of the innate immune system detect viral infection mostly via germline-encoded pattern recognition receptors (PRRs) present either on the cell surface or within distinct intracellular compartments. PRRs include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), and retinoic acid-inducible gene-1-like receptors (RLRs). Especially in the RLR-mediated pathway, the mitochondrion accelerates the signalling event through mitochondrial antiviral-signalling protein (MAVS), a downstream adaptor of RLRs located at the mitochondrial outer membrane (MOM) [45, 75]. Investigation of the roles of miRNAs in mitochondrial-mediated innate immunity showed that miR-302b and miR-372 affect the mitochondrial-mediated antiviral innate immune response by controlling mitochondrial function and metabolic demand. In dsRNA virus infection, upregulation of these miRNAs ultimately terminates the production of type I IFN (IFNα/β) and proinflammatory cytokines. These miRNAs are also involved in dynamin-related protein 1 (DRP1)-dependent mitochondrial fragmentation and disrupt mitochondrial metabolism by attenuating solute carrier family 25 member 12 (SLC25A12), a member of the SLC25 family [163]. Thus, inhibiting miR-302b and miR-372 activities might have an antiviral effect against RNA virus infection.
Recently, Mishra et al. reported that microRNA(miR)-30e-5p (miR-30e) is induced by hepatitis B virus (HBV) infection and acts as a master regulator for innate immune responses. Mechanistically, miR-30e targets multiple negative regulators such as SOCS1, SOCS3, ATG5, ATG12, TRIM38, TANK, and BECN1 of innate immune signalling pathways and increases innate immune responses [99].
Zhao et al. investigated differentially expressed microRNAs and found that miR-136 was overexpressed fivefold and exhibited potent antiviral effects in vitro against H5N1 influenza A virus, as well as vesicular stomatitis virus (VSV) in A549 human lung epithelial cells. Furthermore, 3′-untranslated region (UTR) reporter analysis revealed an miR-136 binding site in the 3′ UTR of IL-6. In addition, miR-136 acts as an immune agonist of retinoic acid-inducible gene 1 (RIG-I), thus triggering the accumulation of IL-6 and IFN-β in A549 cells. This work showed the dual function of miRNA-136 in regulating host antiviral innate immunity as well as suggesting a vital role for the miRNA-activated pathway in viral infection via pattern recognition receptors [174].
Chen et al. reported that upregulation of miR-146a in VSV infection could prevent transcription or translation of the RIG-I-related antiviral response by affecting TRAF6 (TNFR-associated factor 6) and IRAK1/2 (interleukin 1 receptor-associated kinase 1/2) [52]. Furthermore, it was demonstrated that miR-233 directly targets FOXO3 to control expression of type I IFN. The production of class I IFN triggered by VSV infection is responsible for the upregulation of miR-223 [19].
Liu et al. showed that VSV infection decreases miR-33/33* expression through the macrophage type I IFN receptor (IFNAR). They reported that the upregulation of miR-33/33* impaired the RIG-I pathway, increasing the viral load and lethality while attenuating type I IFN expression [79]. In addition, miR-33/33* significantly inhibited the mitochondrial MAVS by affecting adenosine monophosphate-activated protein kinase (AMPK), impeding the mitophagy-mediated elimination of damaged mitochondria, which is required for effective MAVS activation [79].
The IFN immune response induces an antiviral state in cells. However, it also leads to overexpression of some miRNAs that downregulate the nuclear factor kappa B (NF-κB) and IRF pathways [44]. For example, the expression of miR-221 was shown to be significantly increased in neural precursor cells (NPCs) infected with human cytomegalovirus (HCMV). miR-221 directly targeted the 3'-UTR of suppressor of cytokine signalling 1 (SOCS1) and inhibited its gene expression. The upregulation of miR-221 reduced the viral load by enhancing type I IFN production and induction of interferon-stimulated genes (ISGs). However, restoration of SOCS1 abrogated the antiviral activities of miR-221. Additionally, miR-221 controlled the phosphorylation and creation of NF-κB by inhibiting SOCS1 [162].
Feline herpesvirus 1 (FHV-1) infection leads to overexpression of miR-26a through a cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)-related pathway. miR-26a enhances the phosphorylation of the STAT1 and induces the activation of the type I IFN pathway by targeting the host SOCS5 directly, thereby suppressing viral replication [170].
Most viruses encode their own non-coding RNAs (ncRNAs). Like their host counterparts, viral ncRNAs associate with proteins that are crucial for their function, stability, or both. Various biological functions, including the regulation of viral replication, viral persistence, host immune evasion, and cellular transformation, have been attributed to viral ncRNAs [150]. Herpesvirus saimiri (HVS) encodes a short uracil (U)-rich ncRNA, HSUR 1, which has a sequence complementarity to miR-27. Binding to HSUR 1 induces cleavage of miR-27, and this downregulation triggers T cells during HVS infection [1, 14, 48].
HCMV generates an ncRNA called miRNA decay element (miRDE) that can bind and degrade miR-27 family members. This proviral function causes upregulation of the synthesis of viral DNA and virus production during lytic infection [97].
Small ncRNAs (sncRNA) generated by the H5N1 subtype of highly pathogenic avian influenza virus (HPAI) play essential roles in organ infection, especially in the brain and lungs. This virus replicates more in the lung and thus causes more damage in lung tissue than in brain tissue [178]. In an RNA sequencing study, changes in the global expression of the four main sncRNA groups (miRNAs, snoRNAs, snRNAs, and piRNAs) were compared between the brain and lung of a duck during a 120-h time course of infection with this HPAI strain. Major organ-specific changes were detected in the populations of miRNA, snoRNA, and piRNA even before infection, and there was considerable reprogramming of all sncRNA groups during infection, but less so in the brain. Pathway prediction analysis of miRNA targets showed enrichment of infection-, inflammation-, and apoptosis-associated pathways in the lung and enrichment of metabolism-related pathways (including tryptophan metabolism) in the brain [127].
Host cell miRNAs regulate virus replication
The expression levels of host miRNAs change in response viral infection. These up/downregulated miRNAs target the viral genome directly or indirectly to control viral replication and promote or inhibit the innate immune system and cell apoptosis in viral infection [176].
Recent studies have shown that host miRNAs can bind to many RNA viruses, thereby directly controlling their pathogenesis. (+)-strand RNA viral genome replication simulates cellular mRNAs, allowing direct binding of the miRNA to the viral RNA, and this mode of regulation might be similar to that of host mRNAs. The effect of these miRNAs is to inhibit the translation of the viral genome and consequently prevent viral replication [148]. For example, type I IFN-inducible miR-128 directly targets two sites in the transportin TNPO3 mRNA, considerably downregulating TNPO3 mRNA and protein expression levels. TNPO3 is a nuclear importer that is important for HIV-1 replication, and downregulation of this protein prevents viral replication [11]. Others have shown that miR-128 can inhibit HIV replication by directly targeting the 3′-UTR of the viral genome [110]. Some miRNAs, such as miR-323, hsa-miR-324-5p, miR-491, miR-3145, miR-654, and miR-485, inhibit influenza virus infection by directly targeting the main component of the viral polymerase complex, polymerase basic protein 1 (PB1) [58, 62, 69, 140]. A recent study showed that miR-188-3p could inhibit the replication of influenza A viruses (H1N1, H5N6, and H7N9) in A549 cells by directly targeting PB2 expression [27]. Overexpression of miR-296-5p was observed in enterovirus 71 (EV71)-infected human rhabdomyosarcoma (RD) and SK-N-Sh cells. miR-296-5p directly affects two capsid protein coding regions (VP1 and VP3) in the viral genome in response to viral infection. Furthermore, miR-23b can prevent EV71 translation and replication by targeting the VP1 region [51, 177]. Elsewhere, it was shown that miR-548g-3p binds directly to the stem loop A promoter element of the dengue virus (DENV) 5′-UTR, preventing the recruitment of the viral RNA-dependent RNA polymerase (NS5) to the viral genome and thus suppressing viral replication [155].
Some host miRNAs enhance RNA virus replication by targeting the viral genome directly. For example, miR-122 directly targets two different conserved sequences at the extreme 5′ end of the viral RNA, improving the stability of miR-122 and promoting the replication of hepatitis C virus (HCV) [96]. Another study demonstrated that miR-124-3p positively regulates Sindbis virus (SINV) by enhancing viral structural protein translation and virus production. Consequently, inhibition of this miR-124-3p can reduce viral reproduction. Furthermore, inhibition of miR-124 expression has also been shown to reduce chikungunya virus (CHIKV) replication in human cells [84].
Host miRNAs act as antiviral factors by targeting host mRNAs that express pro-viral proteins. In DENV-2 infection, miR-223 downregulates the microtubule destabilizing protein stathmin 1 (STMN1) in human EAhy926 cells, thus preventing viral replication [159]. miR-27b-3p in DF-1 cells increases the expression of chicken IFN-β, IRF3, and NF-κB by directly binding to SOCS3/6, preventing IBDV replication in host cells. Suppression of endogenous miR-27b-3p through its inhibitors suppresses IFN-β, IRF3, and NF-κB, leading to increased expression of SOCS3/6 and increased replication of IBDV [34].
ACE2, a negative regulator of angiotensin II in the renin-angiotensin system, has been reported to have a critical function in acute lung injury (ALI) caused by influenza virus infection. It has been shown that avian influenza virus subtype H5N1 induces the overexpression of miR-200c-3p. Further investigation also showed that miR-200c-3p targets the 3′-UTR of ACE2. The suppression of miR-200c-3p leads to ameliorated ALI induced by H5N1 viral infection in vivo [82].
Li et al. speculated that viral RNAs act as a sponge to sequester endogenous miRNAs within infected cells, thereby cross-regulating the stability and translational effectiveness of host mRNAs with shared miRNA response elements [74]. Host mRNAs and viral RNAs that are involved in crosstalk and reciprocal interactions are referred to as “competitive viral and host RNAs” (cvhRNAs). Exogenous viral RNAs harbour the same miRNA-binding sites as cellular RNAs and compete with host RNAs for the same miRNA pools in infected cells. The crosstalk mechanisms of cvhRNAs rely on an ability to sequester or degrade the common miRNAs by binding to viral RNAs as well as regulating the extent of desuppression of host mRNAs through downregulation of miRNAs. However, several recent studies have provided convincing evidence that host mRNAs and viral RNAs with common miRNA binding sequences mutually affect each other’s levels and activities by directly competing with the targeting miRNAs. Therefore, the relative abundance of viral vs. host RNAs, levels of common miRNAs, and the number of miRNA response elements might all affect cvhRNA interactions, based on a mathematical mass-action model for competitive endogenous RNA (ceRNA) networks. cvhRNAs play a role in infection with several viruses, including HBV, HCV, herpesvirus saimiri, murine cytomegalovirus, and HCMV [74, 138]. For example, pestiviruses hijack miR-17 and let-7 family members to enhance their replication. Both miRNAs directly target the 3′-UTR of the viral genome to stabilize bovine viral diarrhea virus (BVDV) RNA and increase its translation [129]. It has also been reported that miR-10a* (miR-10a-3p) interacts directly with the 3D-coding sequence (nt 6818–6941) of coxsackievirus B3 (CVB3), thereby increasing viral replication. Further in vivo studies are necessary to explain the function of miR-10a-3p in CVB3 infection, in which a posttranscriptional control mechanism appears to be involved [147]. The role of miR-122 in HCV infection is better understood [138]. miR-122 directly targets two locations in the 5′-UTR of HCV RNA, with this binding causing a slight increase in viral protein translation while protecting the genome from XRN1mediated degradation [50, 131]. miR-122 also competes with cellular poly(rC)-binding protein 2 (PCBP2) for binding to HCV genomic RNA and increases the efficiency of replication and packaging [94] (Table 1).
Exosomal miRNAs in infections with RNA viruses
Most miRNAs are intracellular; however, some are secreted from cells. In 2008, the first extracellular miRNA was described in maternal plasma, and circulating miRNAs were also reported in blood serum. Such miRNAs are detected in body fluids such as blood, urine, semen, cerebrospinal fluid (CSF), saliva, pleural effusions, and milk. Various possibilities for secretion of circulating miRNA have been suggested, involving apoptotic bodies, exosomes, microvesicles, protein complexes, and high-density lipoproteins [56, 168].
Exosomes are endocytic-membrane-derived vesicles (30–120 nm) that carry various biomolecules, such as proteins, lipids, mRNAs, and miRNAs, and they have vital functions in cell-to-cell signalling. They act as intermediaries in cell-to-cell communication to deliver miRNAs between cells. Exosomes are discharged into the extracellular space via fusion of multivesicular bodies with the cell membrane. The secreted exosomes supply their load to the recipient cells by integrating into the cell membrane [78, 130]. Since exosomes can carry viral miRNAs in addition to cellular miRNAs, they can be utilized to diagnose and treat viral infections [109] (Fig. 2). For example, in influenza virus infections, an elevated level of miR-483-3p has been found in exosomes of bronchoalveolar lavage fluid. miR-483-3p acts as a modulator of the RIG-I signalling pathway by directly targeting RNF5 and CD81. It was therefore concluded that this exosomal miRNA might mediate antiviral and inflammatory responses in influenza virus infections [91].

Exosomal miRNA biogenesis in viral infection. Specific viral proteins can alter miRNAs in exosomes. In addition, some miRNAs can affect host responses to the virus and either stimulate or suppress infection
Decreased expression of platelet-derived growth factor B (PDGF-B with a concomitant increase in miR-29b production was demonstrated in the basal ganglia region of the brains of morphine-dependent simian immunodeficiency virus (SIV)-infected macaques. In vitro results were verified in astrocytes treated with HIV Tat and morphine, resulting in an enhanced discharge of miR-29b in exosomes. Subsequent treatment of neuronal SH-SY5Y cells with exosomes from treated astrocytes led to reduced levels of PDGF-B, with a concomitant reduction in neuronal viability. It was discovered that PDGF-B is a target for miR-29b, as demonstrated by the fact that targeting miR-29 to the 3′-UTR of PDGF-B mRNA led to its translational suppression in SH-SY5Y cells [53].
Exosomal miR-155 has been shown to effectively inhibit enterovirus A71 infection by targeting phosphatidylinositol clathrin assembly protein (PICALM) in host cells [158]. Furthermore, exosomes released from EV71‐infected oral epithelial cells selectively packaged large amounts of miR‐30a, which was transferred to macrophages, targeting MyD88 and thereby reducing the expression of type I IFN in recipient cells and enhancing the replication of EV71 [153]. In CVB3 infection, miR-30a and miR-181a were also detected in serum exosomes [39].
Coronaviruses: SARS-CoV-2
Coronaviruses (CoVs) are enveloped, positive-sense, single-stranded RNA (+ssRNA) viruses that cause diverse illnesses in humans, animals, and birds [95, 105]. The family Coronaviridae is subdivided into four genera, Alpha-, Beta-, Gamma-, and Deltacoronavirus, and members of the genera Alphacoronavirus and Betacoronavirus are known to infect humans [3, 17, 46]. Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV), and SARS-CoV-2 are the main CoVs causing severe lower respiratory tract infection with significant mortality. Four other CoVs cause human diseases, namely, human coronavirus (HCoV)-OC43, HCoV-NL63, HCoV-HKU1, and HCoV-229E, which mostly cause mild, self-limiting upper respiratory tract infections such as the common cold [16, 55, 164]. Around the end of 2002, SARS-CoV appeared in southeast China and Hong Kong, causing more than 8000 confirmed cases worldwide, with about 800 fatalities. MERS-CoV, initially recognized in June 2012 in Saudi Arabia, has caused over 1,791 confirmed cases of infection in 27 countries, with a fatality rate of approximately 35% [166]. SARS-CoV-2 emerged in Wuhan, China, in December 2019 and resulted in the ICU admission of 26–33% of infected individuals, with a mortality rate of 4–15% [165]. The disease has spread worldwide, with more than 135 million confirmed cases and 2.9 million deaths across 216 countries, far more than caused by MERS-CoV and SARS-CoV-1 [114].
The genome size of CoVs ranges between 27 and 32 kb, making them the largest recognized RNA viruses [139]. All CoVs are similar in genome organization and expression. The structural proteins, including the nucleocapsid (N), membrane (M), envelope (E), and spike (S) proteins are encoded by ORFs located in the 3′ end. They are preceded by 16 nonstructural proteins (nsp1-16), which are encoded by open reading frame (ORF) 1a/b at the 5′ end [144]. The viral genome is used directly as an mRNA for translation of polyprotein 1a/1ab (pp1a/pp1ab) to generate the replication transcription complex (RTC) in double-membrane vesicles (DMVs). A nested set of subgenomic RNAs (sgRNAs) is synthesized by the RTC in a discontinuous manner. These negative‐strand sgRNAs, which have common 5′ leader and 3′‐terminal sequences, are used as templates for generating subgenomic mRNAs [23].
SARS-CoV-2 is genetically very similar to the bat CoV-RaTG13 (96.3%) [134, 172]. The SARS-CoV-2 genome contains 10 ORFs. The first ORF (ORF1a/b) codes for non-structural proteins 1–16. The remaining ORFs encode the structural proteins S, M, E, and N as well as accessory proteins [157] (Fig. 3). The viral surface (S) protein mediates entry of the virus into the host cell. To accomplish this, the S protein first binds to its receptor, ACE2, via its receptor-binding domain (RBD) and is proteolytically activated by host proteases. The S protein is normally cleaved in the host cell into S1 and S2 subunits, which remain noncovalently connected and embedded in the viral envelope [7]. The S1 subunit engages the cellular receptors, mediating viral attachment to the host cell surface, whereas the S2 subunit induces membrane fusion, a process that involves a rearrangement of characteristic elements called heptad repeats (HRs) in S2 to generate a stable 6-helix bundle fusion core [161] (Fig. 3).

A The structural features of SARS-CoV-2 and its main structural proteins. B Genetic structure of the SARS-CoV-2 S gene. SP, signal peptide; NTD, N-terminal domain; RBD, receptor-binding domain; SD1 and SD2, subdomains 1 and 2; S1/S2, S1/S2 protease cleavage site; S2′, S2′ protease cleavage site; FP, fusion peptide; HR1, heptad repeat 1; CH, central helix; CD, connector domain; HR2, heptad repeat 2; TM, transmembrane domain; CP, cytoplasmic tail
SARS-CoV-2 is internalized into the host cell via one of two distinct pathways: (1) the cell surface pathway, following activation by a serine protease such as TMPRSS2, or (2) the endocytic pathway within endosomal–lysosomal compartments, which involves processing by lysosomal cathepsins. The participation of each pathway in a certain cell type depends mainly on the expression of proteases, especially TMPRSS2. When TMPRSS2 is expressed, the initial entry pathway is chosen, while in the absence of this protease, the virus depends on the late pathway involving endocytosis and activation by cathepsin L. Subsequently, the virus most likely enters the cell in one of two ways: by direct fusion with the cytoplasmic membrane or by fusion with the endosomal membrane after receptor-mediated endocytosis and acidification of the endosome.
After internalization of the virus, the viral RNA is uncoated in the cytosol. ORF1a and ORF1ab are translated to generate pp1a and pp1ab, which are cleaved by the proteases encoded by ORF1a to produce the RNA replicase-transcriptase complex. This complex localizes to modified intracellular membranes derived from the rough endoplasmic reticulum (ER) in the perinuclear region, where it produces (–) RNAs. Full-length (–) RNA copies of the genome are generated and used as templates for synthesis of full-length (+) RNA genomes during replication. A subset of 7–9 subgenomic RNAs, including those encoding all of the structural proteins, is generated via discontinuous transcription. In this process, a nested set of subgenomic (–) RNAs is produced that vary in length at the 3′ end and contain the 5′ leader sequence, which is required for translation. These subgenomic (–) RNAs are then transcribed into subgenomic (+) mRNAs. Although the various subgenomic mRNAs might contain multiple ORFs, only the 5´-most ORF of each mRNA is translated to generate the structural proteins, which are assembled into the nucleocapsid and viral envelope at the ER–Golgi intermediate compartment, followed by the release of the nascent virion from the infected cells [28].
Role of miRNAs in CoV infection
Coronavirus replication can also be influenced by miRNAs. For example, porcine hemagglutinating encephalomyelitis virus (PHEV), a member of the genus Betacoronavirus that causes nervous system disorders in its host, has been reported to constitutively upregulate the expression of miR-10a-5p in host cells. Treatment with an miR-10a-5p mimic leads to enrichment of miR-10a-5p and a substantial decrease in PHEV replication, suggesting extensive negative control of RNA virus infection by miR-10a-5p [54]. It was also found that miR-21a-5p was significantly upregulated in the brains of mice, leading to negative regulation of Caskin1 (CASK-interactive protein1) through direct binding to the 3′-UTR of this gene. Since this miRNA promotes viral replication, the use of miR-21a-5p inhibitors could repress viral replication [86]. In addition, upregulation of miR-142a-3p in PHEV infection had a positive effect on viral proliferation by directly targeting the 3′-UTR of Rab3a-mRNA and suppressing its expression [40].
HCoV-OC43 is a member of the genus Betacoronavirus that causes viral infectious disease in the upper respiratory tract (common cold). It has been reported that the nucleocapsid protein of OC43 triggers potentiation of NF-κB activation. During infection, nucleocapsid proteins induce upregulation of miR-9, which inhibits the NF-κB pathway via binding to the 3-UTR of NFKB1 mRNA (nuclear factor kappa B subunit 1), decreasing its expression [70].
Transmissible gastroenteritis coronavirus (TGEV) is a member of the genus Alphacoronavirus that infects pigs and causes endoplasmic reticulum (ER) stress and induces production of IFN-I. Inositol-requiring enzyme 1α (IRE1α), an ER transmembrane sensor, increases virus proliferation through downregulation of miR-30a-5p. This miRNA commonly enhances the antiviral function of IFN-I by binding directly to JAK-signal transducer and activator of transcription (STAT), the inhibitor of SOCS1 and SOCS3. The viral infection reduces the abundance of miR-30a-5p and increases SOCS1 and SOCS3 expression [88].
Furthermore, circular RNA (circRNA)-EZH2 inhibits TGEV infection in the IPEC-J2 cell line by increasing NF-κB expression via binding to miR-22. miR-22 binds directly to HK2 and IL-6 during TGEV infection. Inhibition of HK2 increases TGEV-induced mitochondrial permeability transition pore (mPTP) opening whilst leaving the NF-κB pathway unaffected. Inhibiting IL-6 also increases TGEV-induced mPTP opening and suppresses the NF-κB pathway. Likewise, suppressing NF-κB enhances TGEV-induced mPTP opening [175].
In avian infectious bronchitis virus (IBV) infection, upregulated expression of miR-146a-5p promotes the primary step of viral replication. Ectopic expression of miR-200 caused the downregulation of IRAK2 and tumour necrosis factor receptor superfamily member 18 (TNFRSF18) by targeting the 3'-UTRs of these genes [81].
In an in silico study using an miRNA database, it was demonstrated that miRNAs, including hsa-miR-628-5p, -7974, -208a-3p, -510-3p, -3934-5p,-6804-3p, -18a-3p, -548ax, -4474 5p, -6865-5p, and 342-3p, may have an inhibitory effect on MERS-CoV replication [49]. In SARS-CoV-1 infection, bronchoalveolar stem cells (BASCs; CD34+ Sca-1+ CD45- PE-CAM2-) are the most important cell types in the bronchoalveolar duct junction (BADJ) that support viral replication. The encoded miR-17*, miR-574-5p, and miR-214 are increased in this stem cell line when SARS infection occurs. These miRNAs inhibit viral replication by binding to the mRNA encoding the S protein. In addition, the N and S coding regions targeted by the miRNAs co-opt downregulated miR-223 and miR-98, respectively, within BASCs to regulate the different phases of BASC differentiation, activation of inflammatory chemokines, and downregulation of ACE2, promoting effective viral transmission and replication within BASCs and continuous decay of lung tissue [92] (Table 2).
miRNAs inhibit SARS-CoV-2 infection by direct binding to the viral genome
To obtain deep insight into miRNAs expressed by SARS-CoV and SARS-CoV-2, a series of in silico tools were applied to predict miRNAs generated by these viruses. The miRNAFold algorithm was first applied to predict loops in viral genomes, since hairpin loops generate the precursors of small RNAs. The algorithm predicted about 573 and 575 hairpin loops in the SARS-CoV forward and reverse genome, respectively, and 574 and 550 hairpin loops in the SARS-CoV-2 forward and reverse genome sequence, respectively. Since not all hairpin structures lead to functional miRNAs, the miRBoost algorithm was applied to identify hairpin loops that are more likely to yield functional miRNAs. Remarkably, it was found that, out of 65 potential miRNA sequences in the SARS-CoV-2 forward genome sequence that were identified by miRBoost, 56 were recognized by mirBase as matching human-specific miRNAs. Likewise, out of 64 miRNAs predicted in the SARS-CoV-2 reverse genome sequence, 39 matched human-specific miRNAs.
Furthermore, the algorithm identified 66 and 33 predicted miRNAs out of a repertoire of 89 and 79 in the forward and reverse SARS-CoV genome sequence, respectively, that matched human-specific miRNAs. Next, all of the available SARS-CoV and SARS-CoV-2 genome sequences were compared to identify any virus-encoded miRNAs common to all of the sequences. Contrary to expectation, however, no virus-encoded miRNAs were detected that were common to all reverse and forward genome sequences of SARS-CoV-2 and SARS-CoV isolates. Utilizing this strategy, about 263-196 host miRNAs were recognized that could bind to any forward or reverse sequence of SARS-CoV-2 RNA. It was found that, compared to SARS-CoV, SARS-CoV-2 genomes possessed binding sites for a larger number of host miRNAs, which might be related to the lower pathogenicity of SARS-CoV-2 in healthy people. Correspondingly, although the genomes of SARS-CoV and SARS-CoV-2 differed in the number of miRNA targets, there was a strong resemblance among the virus-encoded noncoding RNAs in both genomes, suggesting a likely overlap between the pathogenesis of SARS-CoV and SARS-CoV-2 and the plausibility of developing novel therapeutic strategies that might effectively control both viral infections [116].
Other studies using in silico analysis methods showed that all SARS-CoV-2 genes (S, M, N, ORF1ab, ORF3a, ORF8, ORF7a, and ORF10) except E and ORF6, are targeted by several human miRNAs. For example, hsa-miR-203b-3p, which has already been shown to suppress influenza A virus replication [171], was predicted to target ORF1ab and ORF3a. Although hsa-miR-148a-3p targets ORF8 to prevent interspecies transmission and replication, it was also found to target the S, E, M, and ORF1a genes in SARS-CoV [93]. hsa-let-7c-5p is predicted to target ORF1ab in SARS-CoV-2, while it has been found to be involved in suppression of H1N1 influenza A virus by targeting its M1 gene [89].
The protein encoded by ORF6 of SARS-CoV 1 represses the type I IFN pathway by inhibiting nuclear transport of STAT1 in the presence of INF-β. Thus, hsa-miR-190a-5p might target the ORF6 gene to the overwhelm the immune system escape during SARS-CoV-2 infection [29]. As a result, using a mimic of this miRNA as an inhibitor might also affect COVID-19 (Fig. 4). Some human miRNAs targeting the SARS-CoV-2 genome can also be effective in other CoVs, such as MERS-CoV, SARS-CoV, and human coronavirus NL63 (HCoV-NL63). For example, miR-4259 is predicted to recognize MERS-CoV and SARS-CoV-2, binding to the N coding region in the SARS-CoV-2 genome. Other miRNAs such as miR-1181, miR-1307-3p, miR-146b-3p, and miR-1229-5p are predicted to recognize both SARS-CoV-2 and SARS-CoV, and hsa-miR-1229-5p is predicted to recognize both SARS-CoV-2 and HCoV-NL63, targeting the ORF3a gene region in the SARS-CoV-2 viral genome [29, 83]. The differences in miRNA targets between SARS-CoV-2 and other human CoVs suggests a possible role in their distinct clinical features [83].

Homo sapiens miRNAs (hsa-miR-) have been identified to bind to sites in SARS-CoV-2 RNA encoding structural proteins and non-structure proteins. Reproduced with permission [29]
Indirect effects of miRNAs on SARS-CoV-2 infection
SARS-CoV-2 infection causes variations in the expression levels of host miRNAs. For example, in lung epithelial cells infected with SARS-CoV-2, the expression levels of hsa-let-7a-3p, hsa-miR-135b-5p, hsa-miR-16-2-3p, and hsa-miR-1275 decrease, while those of hsa-miR-155-3p and hsa-miR-139-5p increase. As a result, decreasing the expression level of host miRNAs is likely to increase the susceptibility of the respiratory epithelium to infection. On the other hand, enhancing the expression levels of host miRNAs may provide protection against viral infection and replication [26].
Viral infections lead to a high abundance of viral RNA, and the increased number of potential miRNA binding sites could represent a very effective defense by decreasing the levels of cellular miRNA throughout the initial phases of infection. Furthermore, pathogenic human CoVs could inhibit host-specific miRNAs and repress the immune reaction or suppress induction of unfolded protein response (UPR)-dependent apoptosis. SARS-CoV-2 genomic RNA can decrease the effective host miRNA level by functioning as an miRNA sponge, thereby accelerating viral replication and inhibiting immune responses. For example, a SARS-CoV-2-mediated decrease in host miR-495-5p and miR-34a-3p rates could enhance X-box binding protein transcription factor and binding immunoglobulin protein expression, respectively, by enhancing the endoplasmic reticulum folding capacity and increasing survival. SARS-CoV-2 can also affect the mTOR and autophagy pathways by regulating miR-376b-3p. Some of these miRNAs, such as miR-376a-3p, miR-99b-5p, miR-10a-5p, miR-99b-5p, miR-548av-5p, and miR-376a-3p, have also been suggested to regulate immune responses [8].
Viruses have strategies to increase their replication, such as hijacking host RNA helicases. RIG-I/Ddx58 receptors possess a helicase domain, which interacts with viral nsp13 and commences the viral life cycle. The expression of Ddx58 is increases immensely in SARS-CoV-2-infected cells, which might also be one of the possible causes of the enhancement of viral replication. Also, Ddx58 is involved in processes of miRNA biogenesis and mRNA splicing; therefore, its overexpression would trigger reprogramming of miRNA splicing events leading to negative regulation of miRNAs, such as miR-124-3p, which has a predicted recognition site in the 3′-UTR of Ddx58. Hence, upregulation of miR-124-3p would trigger Ddx58 degradation, thereby leading to reduced viral replication. RIG-I/Ddx58 also upregulates Stat1 (TF), which, upon phosphorylation, associates with IFN regulatory factor 9 and Stat2 to produce heterotrimers and commence the transcription of ISGs to exert antiviral effects. Furthermore, the virus might activate cytosolic translocation of Drosha to neutralize ISG-mediated antiviral effects, which could reprogram splicing events to yield other lncRNAs and circRNAs that could act as a sponge for miR-124-3p and hamper its ability to degrade Ddx58 [6].
Lu et al. showed that miR-200c suppresses ACE2 expression in both human and rat cardiomyocytes, and therefore, miRNAs can be exploited for prevention of cardiovascular problems in SARS-CoV-2 infection [85].
ACE2 levels on cell membranes are controlled by ADAM17, which increases the shedding of the protein. Notch signalling negatively regulates ADAM17 and positively regulates furin through the transcription of miRNA-145. Thus, inhibition of γ-secretase, which suppresses Notch activation, might be a promising strategy for inhibiting SARS-CoV-2 entry into the cells by decreasing furin expression and enhancing ADAM17-meidiated shedding. In addition, miRNA-145, which negatively regulates ADAM17, targets Jagged1/Notch1 signalling in vascular smooth muscle cells. Therefore, the use of an antagomir of miR-145 might be an alternative approach for upregulation of ADAM17 [121].
The kidney manifestations of SARS-CoV-2 infection include acute kidney injury, collapsing glomerulonephritis, podocyte apoptosis, interstitial nephritis, and progressive chronic kidney disease. Batle et al. revealed that acute kidney injury is one of the crucial manifestations of SARS-CoV-2 infection. Proinflammatory mediators play a vital role in these manifestations. ACE2, a crucial proinflammatory mediator in acute kidney injury or glomerular disorders related to COVID-19, is upregulated by miRNA expression. Certain miRNAs, in particular those associated with ACE2 expression, are involved in the augmented levels of proinflammatory circulatory mediators. Numerous miRNAs are implicated in ACE2 expression, with most of them being affected in other organs, while miR-125b and miR-18 are mainly expressed in the kidney. At present, only antimir-18 has solid data showing it to be a silencer of ACE2 expression. miRNA-based therapy, particularly with antimiR-18 and antimir-125b, is a novel potential ACE2-targeting therapeutic choice for nephropathy related to COVID-19. Additional studies of the effects of antimiR-18 and antimiR-125b are still needed [156].
miR-5197-3p has been shown to interact efficiently with the genomic RNA (gRNA) of MERS-CoV, SARS-CoV, and SARS-CoV-2. A recent study has shown that critical miRNAs, including miR-5197-3p, miR-4778-3p, and miR-6864-5p, interact with complete complementary miRNA (cc-miR) and have therapeutic potential owing to their binding affinity for the gRNA of SARS-CoV-2. A complementary miRNA based on the complete miR-5197-3p sequence might have considerable therapeutic value due to its affinity for the gRNA of SARS-CoV-2 and lack of undesired effects on human genes [5] (Table 3).
Strategies for designing miRNA mimics and antagonists
The miRNA mimic technology (miR-Mimic) is a pioneering approach for gene silencing. The nonnatural double-stranded RNA generated in this approach is designed to possess a 5′ end bearing a motif partially complementary to a selected sequence in the 3′-UTR that is unique to the target gene. Once present in the cell, this piece of RNA, which simulates an endogenous miRNA, can bind specifically to its target gene and cause posttranscriptional suppression of the gene. The primary stage of producing an miRNA mimic is to recognize a stretch of sequence in the 3′-UTR that is unique to the gene of interest (target mRNA), similar to designing an siRNA [107]. However, unlike the full complementarity between an siRNA and its target in any area of the gene, an miRNA mimic base-pairs only partially with the target sequence in the 3′-UTR. The size of the sequence should be long enough for the miRNA to function, which is at least 8 nt, and ideally > 14 nt [154].
miRNA mimics are unstable and have less durability in cells. In a recent study, Nogimori et al. found that miRNA mimics are quickly destroyed via a mechanism different from Tudor-staphylococcal/micrococcal-like nuclease (TSN)-mediated miRNA decay, which destroys endogenous miRNAs. Instead, the newly recognized 2′-5′-oligoadenylate synthetase (OAS)/RNase L was identified as the main element responsible for the degradation of miRNA mimics in human cells. The authors propose that the OAS1 identifies the miRNA mimic and generates 2′-5′-oligoadenylates (2–5A), causing the triggering of latent endoribonuclease RNase L to destroy the miRNA mimic. A small-molecule suppressor that inhibits RNase L can block degradation of miRNA mimics [111].
In another study, to suppress oncogenic KRAS in pancreatic ductal adenocarcinoma (PDAC) cells, Ferino et al. developed ss-miR-216b mimics with unlocked nucleic acid (UNA) modifications to increase their nuclease resistance. Variants of ss-miR-216b mimics were prepared with and without a 5ʹ phosphate group. The main characteristic of UNA is the lack of a C2ʹ-C3ʹ bond in the ribose moiety, an alteration that increases the flexibility of the RNA strand. A single UNA modification in the center of an RNA/RNA duplex can reduce the melting temperature (Tm) by 5–10 °C; however, when the UNA modification is located close to the duplex terminal, it leads to a relatively low drop to a Tm of 1–3 °C [42].
Anti-miRNA oligonucleotides (antagomirs, also known as anti-miRs) are synthetically designed molecules that are applied to neutralize miRNAs. They can be applied as further control and for treating certain cellular disorders via hybridization to miRNA and a steric blocking mechanism. Understanding the miRNA sequences involved in various disorders, especially viral infections, can allow us to apply anti-miRs to interrupt pathways that cause the up/downregulation of cell proteins that generate signs of diseases. During anti-miRs design, essential modifications to optimize binding affinity, improving nuclease resistance, and in vivo delivery must be considered.
Most studies on anti-miR design strategies have focused on chemically modifying the sugar or phosphodiester backbone of the oligonucleotide to enhance the thermodynamic stability and nuclease resistance of the miRNA/anti-miR duplex. For instance, phosphorothioate (PS) bonds and 2′-O-methyl alterations of anti-miRs have been demonstrated to increase nuclease resistance. Locked nucleic acid (LNA) alterations, which pre-organize the sugar in a 3′-endo pucker, allow miRNAs to be targeted with higher affinity.
As the active fragment of a miRNA in a cell is dependent on an Ago protein, structures of Ago proteins connected to RNA provide a potential starting point for further anti-miR optimization. Numerous crucial aspects of miRNA recognition within hAgo2 have been identified, including targeting the 5′ end of the miRNA strand in the middle (MID) domain and 3′ end in the PAZ (Piwi/Argonaute/Zwille) domain, which allows cradling of the miRNA-target duplex. One attractive structural aspect of human Ago2 is a small solvated pocket within the L2 and MID domains of hAgo2, which allows specific binding to adenosine in the target strand at nucleotide position 1 (t1A). As miRNAs connect anti-miRs by target strand recognition, the t1 nucleotide position matches the 3′ end of a typical anti-miR.
Pham et al. developed a unique anti-miR production method based on the structure of hAgo2 with bound miRNA-target RNA duplex to enhance the potency of anti-miRs. They carried out copper-catalyzed alkyne/azide cycloaddition reactions to produce t1-triazole-altered anti-miRs and tested their activity in cell-based anti-miR assays. Numerous triazole-altered anti-miRs were found to be more potent than anti-miRs bearing adenosine at the t1 location. This ester modification was found to enhance the potency of two different anti-miR sequences. Furthermore, triazole-altered anti-miRs displayed improved 3′-exonuclease resistance [117].
A major obstacle to the use of anti-miRNAs in vivo is the need to deliver them to the location where the target gene is located. In order to make them more resistant to degradation and to facilitate their entry into cells, miRNAs are encapsulated into various delivery systems or conjugated to artificial carriers.
LNA, an altered antisense oligonucleotide that can bind the miRNA seed region, has been suggested as a promising type of anti-miR. LNA seed family inhibitors can inhibit entire miRNA families as well as individual specific miRNAs [2].
2′-O-methyl (2′-OMe)-4′-thioRNA is a hybrid type of chemically modified oligonucleotide that shows significant binding affinity to complementary RNAs and strong resistance to destruction by nucleases. Takahashi et al. assessed 2′-OMe-4′-thioribonucleosides for chemical modification of anti-miRs. Optimization of the modification pattern using various chemically altered anti-miRs that were complementary to a mature miR-21 showed that a uniformly 2′-OMe-4′-thioribonucleoside–modified antimiR was the most potent. Further research revealed that phosphorothioate modification contributed to long-lasting miR-122 inhibition through the 2′-OMe-4′-thioribonucleoside–modified anti-miR. Also, anti-miRs that were systemically administered to mice using a liposomal delivery system, YSK05-MEND, were delivered to the liver, and effective suppression of miR-122 function was achieved at a lower dose in vivo [145].
The 2′-O-Me modification, as well as the 2′-O-methoxyethyl (2′-MOE) and 2′-fluoro (2′-F) modifications, affect the 2′ position of the sugar moiety, whereas LNA is a class of bicyclic RNA analogues in which the furanose ring in the sugar-phosphate backbone is chemically locked in an RNA-simulating N-type (C3′-endo) conformation through the formation of a 2′-O,4′-C methylene bridge. These alterations provide resistance against nucleases and improve the targeting of anti-miR oligonucleotides to their cognate miRNAs. LNA in particular has the strongest effect on binding to the complementary RNA, with an increase in the duplex melting temperature (Tm) of 2–8 °C per LNA monomer when compared to unmodified duplexes. Another key feature of the LNA is that the monomers can twist the sugar conformation of flanking DNA nucleotides from an S-type (C2′-endo) toward an N-type sugar pucker in LNA-altered DNA oligonucleotides [143].
miRNA delivery approaches for efficient treatment of SARS-CoV-2 infection
miRNAs play a significant role in the development and prognosis of various diseases and are potential candidates for improving novel therapeutic approaches. Instability, high probability of destruction by nucleases, and immunotoxicity have led to special attention being paid to safe and effective miRNA delivery strategies [22, 108, 115, 173].
Two important delivery strategies, local and systemic, have been considered for miRNA delivery, though the lack of proper release mechanisms is a roadblock to their utility. Effective gene silencing has been achieved through local administration of miRNAs, which results in higher bioavailability. Local delivery of miRNAs also results in lower cytotoxicity than systemic delivery. Systemic miRNA delivery activates the innate immune response, causing unexpected effects and adverse reactions. Following systemic injection, a steep drop occurs in the concentration of miRNA antagonists or mimics in the lung tissue [22, 108, 115, 173]. Valuable efforts have been made towards modifying the delivery of miRNA-based therapies to overcome the obstacles of in vivo delivery systems.
Several viral vectors, including retroviruses, lentiviruses, and adenoviruses, have been used for gene transfer into many different cell types both in vitro and in vivo. Despite promising results in various pre-clinical and clinical studies, their strong immunogenicity and the risk of insertional mutagenesis are major obstacles to the clinical translation of viral vectors [22, 108, 115, 173]. Recently, various nonviral delivery systems, including polymeric nanoparticles, lipid nanocapsules, inorganic nanoparticles, etc., have been designed and investigated to deliver different therapeutic molecules, especially miRNAs [15, 41, 146].
LNPs have been recognized as a highly promising platform for producing vaccines [36]. Since mRNA is highly unstable and can induce an innate immune response upon injection, it has not been traditionally used as a therapeutic agent. Furthermore, mRNA requires a carrier system to pass the plasma membranes of target cells. Thus, LNP-based delivery systems have been designed to encapsulate mRNAs coding for MAbs.
This RNA-therapy platform stabilizes the mRNA and can be administered repeatedly, resulting in sustained production of antibodies while escaping the effect of the innate immunity against exogenous RNA. Moreover, LNPs improve mucosal and cellular absorption and increase biocompatibility. Positively charged LNPs cause electrostatic absorption to the negatively charged mucosal membranes, reducing their clearance via the mucosal cilia [124].
Nanotechnology-based strategies offer feasible solutions to the delivery challenge by trafficking the vaccine to appropriate cellular populations and subcellular sites. While synthetic nanodelivery systems such as polymeric nanoparticles and cationic liposomes have been utilized to deliver DNA vaccines through plasma membranes, targeted formulations could further improve the plasmid nuclear translocation of DNA. Nanotechnology platforms such as dendrimers, liposomes, polysaccharide particles, and cationic nanoemulsions have been used to enhance the stability and transfer of mRNA-based vaccines [136]. For example, Pfizer and Moderna used LNPs for the delivery of mRNA, which may enable cytoplasmic delivery via fusogenic mechanisms [120, 126]. An mRNA-based technology platform, RNActive®, has been used to develop a SARS-CoV-2 vaccine containing sequence-optimized mRNA coding for a stabilized form of the S protein loaded in LNPs. No vaccine-related serious side effects were observed [67].
A self-amplifying RNA (saRNA) expressing the SARS-CoV-2 spike protein loaded within an LNP as a vaccine showed high and dose-dependent SARS-CoV-2 specific antibody titers in mouse sera, as well as robust neutralization of both a wild-type virus and a pseudovirus. saRNA LNP immunization induced a Th1-biased response in mice. A strong cellular response, as characterized by IFN-γ production, was observed upon re-stimulation with SARS-CoV-2 peptides. This potent LNP-encapsulated saRNA vaccine can be injected using a normal needle and syringe and does not require electroporation equipment, perhaps allowing more extensive vaccination against COVID-19 [98].
Cationic lipids might be fabricated from commercially available products, such as Lipofectamine®, because of their hydrophilic head and a hydrophobic tail. Numerous investigations have demonstrated the applicability of cationic liposomes as a delivery system for delivering miRNA in vivo. Presently, many types of cationic lipids are available for nucleic acid medicine delivery. Nevertheless, the key downside of using cationic lipids is low delivery efficiency. More recently, novel lipids have been produced to solve this problem, and novel tools have been established for creating lipid nanocomplexes. Polyethylene glycol (PEG), a frequently applied functional group, was grafted to cationic lipids to inhibit phagocytosis of the reticuloendothelial system when administrated systemically [36].
Liposomes have been commonly applied as cell transfection reagents and vaccine adjuvants [167]. The interest in liposomal vaccine delivery systems has markedly increased. The effectiveness of this method has been shown, and subsequent human experiments are underway [21] (Fig. 5). Ju et al. have demonstrated the use of carbon dots to deliver LNA-based inhibitors for specific inhibition of viral miRNAs, which prevented the reproduction of Kaposi’s sarcoma-associated herpesvirus (KSHV)-related primary effusion lymphoma (PEL) cells. KSHV has essential functions in controlling the proliferation and survival of virus-induced cancer cells. Particularly, an amalgamation of Cdots-LNAs to knock down the levels of miR-K12-1, miR-K12-4, and miR-K12-11 of KSHV induces apoptosis and suppresses the increase in PEL cells [60]. Karlsen and Brinchmann used a liposome delivery system for transfection of human articular chondrocytes (hAC) and mesenchymal stem cells with amiR-145, which caused off-target immunological effects mediated by RIG-I. An immune response was also observed with blunt-ended and 2-nucleotide 3′ overhang versions of synthetic miR-145 lacking a 5′ppp cap. Importantly, exposure to liposomes alone caused overexpression of immune genes such as RIG-I [61].

Different delivery systems for mRNA. LNPs are generated via the self-assembly of an ionizable cationic lipid. Different nanoparticles of these cationic lipids (such as 1,2- dioleoyloxy-3-trimethylammoniumpropane [DOTAP] or dioleoyl phosphatidylethanolamine [DOPE]) are formulated with subtle modifications (such as cationic lipids + cholesterol nanoparticle, cationic lipids + cholesterol + PEG-LNP), where cholesterol and PEG-lipid are added to enhance stability. Other nanodelivery systems include protamine (cationic peptide) nanoliposomes (sized about 100 nm), PEG-lipid functionalized dendrimer nanoparticles (about 200 nm in size), positively charged oil-in-water (O/W) cationic nanoemulsion (about 120 nm in size), polyethyleneimine nanoparticles (about 100−300 nm in size), and cationic polymer (chitosan) nanoparticles (about 300−600 nm in size) [18]
Ohno et al. developed a bionanocapsule delivery system for the exogenous release of miR-93 in HBV-infected hepatocytes. This nanosystem comprising HBV envelope L proteins restored MICA (MHC class I polypeptide-related sequence A) protein expression levels in the cell culture supernatant. The findings of that study suggested that the rescued suppression of soluble MICA protein levels by miRNA93 targeted to HBV-infected hepatocytes applying bionanocapsules may be beneficial for inhibiting HBV-induced HCC by altering the expression of deregulated miRNA93 [113].
Various viral and nonviral vaccine platforms have been used to develop efficient SARS-CoV-2 vaccines. The China-based CanSinoBIO's Convidicea (Ad5-nCoV) vaccine is a genetically engineered vaccine candidate with a replication-defective adenovirus type 5 as a vector for expression of the SARS-CoV-2 S protein [135]. INO-4800, a DNA plasmid encoding S protein, was delivered in the host intradermally or intramuscularly by an electropermeabilization method, which uses an electric field for enhancing the permeability of cell membranes [12]. The LV-SMENP-DC vaccine was made by modifying DCs with lentiviral vectors expressing SARS-CoV-2 minigene SMENP and immune-modulatory genes to activate cytotoxic T cells [87, 142].
Exploiting genetically engineered viral vectors for miRNA delivery can increase the risk of integrating viral DNA into the host DNA. Liposomes may be toxic and produce side effects in normal organs and tissues. Furthermore, liposomes may have low resistance to immune responses [25]. Van Zandwijk et al. developed a nanoparticle-based delivery method as an alternative to liposomes utilizing EnGeneIC Delivery Vehicle (EDV) packaging. After loading with miR-16-5p mimics, EDVs were coated with bispecific antibodies [80, 151]. Upon targeting receptors present on the membranes of non-small-cell lung carcinoma (NSCLC) cells, EDV undergoes a process of endocytosis. This delivery system has shown effective delivery of the miRNAs to induce adaptive immune responses [90] and might be highly effective for delivering therapeutic miRNAs against SARS-CoV-2 infection. In this approach, a bacterially derived EDV™ nanocell platform and specific antibodies can be used for targeted therapy of lung cells infected with SARS-CoV-2. To accomplish this, an anti-ACE2 antibody is incorporated into the EDV™ nanocells containing SARS-CoV-2 RNA-targeting miRNAs. An empty EDV™ nanocell with a diameter of 400 nm can load about 1 million different SARS-CoV-2 RNA-targeting miRNAs. Using an anti-ACE2 antibody, EDV might bind to the ACE2 receptors present on the membranes of infected cells and undergo a process of endocytosis, resulting in miRNAs targeting viral RNA and inhibition of SARS-CoV-2 infection (Fig. 6). Following promising data from in vitro studies on appropriate cell line models of COVID-19, such as Vero E6 cells, the efficiency of miRNAs loaded into anti-ACE2-antibody-conjugated EDV™ nanocells can be evaluated for inhibition of SARS-CoV-2 infection in infected human ACE2 transgenic mice and rhesus macaques. Human ACE2 transgenic mice showed weight loss, virus replication in the lungs, and interstitial pneumonia after SARS-CoV-2 infection [8], and infected macaques had high viral loads in the upper and lower respiratory tract, pathologic evidence of viral pneumonia, and humoral and cellular immune responses. Rhesus macaques were used to evaluate the therapeutic efficacy of remdesivir, DNA vaccine candidates expressing the S protein, and adenovirus-vectored vaccines [20, 153, 160, 170].

3D illustration of EnGeneIC's bacterially derived EDV™ nanocell platform for possibly inhibiting SARS-CoV-2 infection
Conclusion
Scientists are developing efficient treatment and preventive strategies for COVID-19 infection. Using the knowledge that has been gained about SARS-CoV-2, including the structure of the viral genome, characteristics of the structural and non-structure proteins, its target cells, its mechanism of replication in host cells, and the identity of its receptor, different strategies can be tailored for the treatment of COVID-19. Exploiting therapeutic molecules such as miRNAs with high therapeutic efficiency and high diversity might be one effective solution. miRNAs have been shown to have different functions in different coronaviruses, such as activating the immune system and directly binding to CoV RNA to inhibit CoV replication. CoV infections also lead to up- and downregulation of host cell miRNAs. It was revealed that miR-6729-5p targets the S and N genes, as well as miR-2052; miR-3127-5p targets the ORF1A and S genes in SARS-CoV-2 RNA. However, some miRNAs target only one site in the viral RNA. For example, miR-447b only targets the S protein gene and miR-325, and miR-34a-5p only targets the M protein gene.
Some host cell miRNAs involved in SARS-CoV-2 infection have a similar role in MERS, SARS, and HCoV-NL63 infection, indicating the high potential of using only one miRNA to treat different viral infections. In addition, miR-200c-3, by binding to the 3′-UTR of ACE2, causes a decrease in ACE2 levels, resulting in increased ALI or ARDS in H5N1 infection and SARS. Thus, using such miRNA mimics as an inhibitor might be efficient in treating SARS-CoV-2 infections (Fig. 7). Poor intracellular delivery and disruptive naked miRNAs aggregation within endosomes lead to inefficient gene silencing. These shortcomings diminish the clinical efficacy of miRNA delivery. Thus, efficient delivery strategies such as EDV™ nanocells might provide a safe and targeted delivery system for miRNA mimics and inhibitors to target infected tissues such as the lung and inhibit SARS-CoV-2 replication. Nevertheless, it should be noted that the application of miRNAs for targeted therapies is still in its early phases, and only a small number of miRNAs have been employed in clinical trials.

The function of miRNAs in SARS-CoV-2 infection. miRNAs directly target viral RNAs at several sites and inhibit viral replication. Also, indirect effects include modulation of the expression of host factors that are essential for one or more phases of the viral life cycle
References
Abedini M, Zhang C (2020) Performance assessment of concrete and steel material models in ls-dyna for enhanced numerical simulation, a state of the art review. Arch Computat Methods Eng. https://doi.org/10.1007/s11831-020-09483-5
Abu-Izneid T, AlHajri N, Ibrahim AM, Javed M, Salem KM, Pottoo FH, Kamal MA (2020) Micro-RNAs in the regulation of immune response against SARS COV-2 and other viral infections. J Adv Res. https://doi.org/10.1016/j.jare.2020.11.013
Adams MJ, Lefkowitz EJ, King AM, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR (2016) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Adv Virol 161:2921–2949
Andalib S, Talebi M, Sakhinia E, Farhoudi M, Sadeghi-Bazargani H, Motavallian A, Pilehvar-Soltanahmadi Y (2013) Multiple sclerosis and mitochondrial gene variations: a review. J Neurol Sci 330:10–15
Arisan ED, Dart A, Grant GH, Arisan S, Cuhadaroglu S, Lange S, Uysal-Onganer P (2020) The prediction of miRNAs in SARS-CoV-2 genomes: hsa-miR databases identify 7 Key miRs linked to host responses and virus pathogenicity-related KEGG pathways significant for comorbidities. Viruses 12:614
Arora S, Singh P, Dohare R, Jha R, Syed MA (2020) Unravelling host-pathogen interactions: ceRNA network in SARS-CoV-2 infection (COVID-19). Gene 762:145057
Bader AG, Brown D, Winkler M (2010) The promise of microRNA replacement therapy. Can Res 70:7027–7030
Bartoszewski R, Dabrowski M, Jakiela B, Matalon S, Harrod KS, Sanak M, Collawn JF (2020) SARS-CoV-2 may regulate cellular responses through depletion of specific host miRNAs. Am J Physiol Lung Cell Mol Physiol 319:L444–L455
Berindan-Neagoe I, Monroig PdC, Pasculli B, Calin GA (2014) MicroRNAome genome: a treasure for cancer diagnosis and therapy. Cancer J Clin 64:311–336
Bhaskaran M, Mohan M (2014) MicroRNAs: history, biogenesis, and their evolving role in animal development and disease. Vet Pathol 51:759–774
Bochnakian A, Zhen A, Zisoulis DG, Idica A, KewalRamani VN, Neel N, Daugaard I, Hamdorf M, Kitchen S, Lee K (2019) Interferon-inducible MicroRNA miR-128 modulates HIV-1 replication by targeting TNPO3 mRNA. J Virol 93:e00364–00319
Yu J, Tostanoski LH, Peter L, Mercado NB, McMahan K, Mahrokhian SH, Nkolola JP, Liu J, Li Z, Chandrashekar A (2020) DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369:806–811
Cai X, Hagedorn CH, Cullen BR (2004) Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10:1957–1966
Cazalla D, Yario T, Steitz JA (2010) Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328:1563–1566
Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee S-S (2017) Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Therapy Nucleic Acids 8:132–143
Chan JF, Li KS, To KK, Cheng VC, Chen H, Yuen K-Y (2012) Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J Infect 65:477–489
Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen K-Y (2015) Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev 28:465–522
Chauhan G, Madou MJ, Kalra S, Chopra V, Ghosh D, Martinez-Chapa SO (2020) Nanotechnology for COVID-19: therapeutics and vaccine research. ACS Nano 14:7760–7782
Chen L, Song Y, He L, Wan X, Lai L, Dai F, Liu Y, Wang Q (2016) MicroRNA-223 promotes type I interferon production in antiviral innate immunity by targeting forkhead box protein O3 (FOXO3). J Biol Chem 291:14706–14716
Chen L, Zhou Y, Li H (2018) LncRNA, miRNA and lncRNA-miRNA interaction in viral infection. Virus Res 257:25–32
Chen WC, Huang L (2005) Non-viral vector as vaccine carrier. Adv Genet 54:315–337
Chen Y, Gao D-Y, Huang L (2015) In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv Drug Deliv Rev 81:128–141
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. https://doi.org/10.1002/jmv.25681
Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C (2007) MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol 170:1831–1840
Chira S, Jackson CS, Oprea I, Ozturk F, Pepper MS, Diaconu I, Braicu C, Raduly L-Z, Calin GA, Berindan-Neagoe I (2015) Progresses towards safe and efficient gene therapy vectors. Oncotarget 6:30675
Chow JT-S, Salmena L (2020) Prediction and analysis of SARS-CoV-2-targeting MicroRNA in human lung epithelium. Genes 11:1002
Cui H, Zhang C, Zhao Z, Zhang C, Fu Y, Li J, Chen G, Lai M, Li Z, Dong S (2020) Identification of cellular microRNA miR-188-3p with broad-spectrum anti-influenza a virus activity. Virol J 17:12
de Wit E, van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523
Demirci MDS, Adan A (2020) Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. PeerJ 8:e9369
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the microprocessor complex. Nature 432:231–235
Di Leva G, Garofalo M, Croce CM (2014) MicroRNAs in cancer. Annu Rev Pathol 9:287–314
Dickey LL, Worne CL, Glover JL, Lane TE, O’Connell RM (2016) MicroRNA-155 enhances T cell trafficking and antiviral effector function in a model of coronavirus-induced neurologic disease. J Neuroinflammation 13:240
Dong Y, Dai T, Wei Y, Zhang L, Zheng M, Zhou F (2020) A systematic review of SARS-CoV-2 vaccine candidates. Signal Transduct Target Ther 5:1–14
Duan X, Zhao M, Li X, Gao L, Cao H, Wang Y, Zheng SJ (2020) gga-miR-27b-3p enhances type I interferon expression and suppresses infectious bursal disease virus replication via targeting cellular suppressors of cytokine signaling 3 and 6 (SOCS3 and 6). Virus Res. https://doi.org/10.1016/j.virusres.2020.197910
Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S (2007) Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci 104:17016–17021
El-Nabi SH, Elhiti M, El-Sheekh M (2020) A new approach for COVID-19 treatment by micro-RNA. Med Hypoth 143:110203
Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R (2006) miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3:87–98
Etheridge A, Lee I, Hood L, Galas D, Wang K (2011) Extracellular microRNA: a new source of biomarkers. Mut Res Fundam Mol Mechan Mutagen 717:85–90
Fan K, Li M, Cui F, Feng F, Kong L, Zhang F, Hao H, Yin M, Liu Y (2019) Altered exosomal miR-181d and miR-30a related to the pathogenesis of CVB3 induced myocarditis by targeting SOCS3. Eur Rev Med Pharmacol Sci 23:2208–2215
Fan P, Guan J, He W, Lv X, Hu S, Lan Y, Zhao K, Gao F, Li F, Fan G (2020) miR-142a-3p promotes the proliferation of porcine hemagglutinating encephalomyelitis virus by targeting Rab3a. Adv Virol 165:345–354
Farajzadeh R, Zarghami N, Serati-Nouri H, Momeni-Javid Z, Farajzadeh T, Jalilzadeh-Tabrizi S, Sadeghi-Soureh S, Naseri N, Pilehvar-Soltanahmadi Y (2018) Macrophage repolarization using CD44-targeting hyaluronic acid–polylactide nanoparticles containing curcumin. Artif Cells Nanomed Biotechnol 46:2013–2021
Ferino A, Miglietta G, Picco R, Vogel S, Wengel J, Xodo LE (2018) MicroRNA therapeutics: design of single-stranded miR-216b mimics to target KRAS in pancreatic cancer cells. RNA Biol 15:1273–1285
Finnegan EF, Pasquinelli AE (2013) MicroRNA biogenesis: regulating the regulators. Crit Rev Biochem Mol Biol 48:51–68
Forster SC, Tate MD, Hertzog PJ (2015) MicroRNA as type I interferon-regulated transcripts and modulators of the innate immune response. Front Immunol 6:334
Gantier MP (2010) New perspectives in MicroRNA regulation of innate immunity. J Interferon Cytokine Res 30:283–289
Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ (2006) Nidovirales: evolving the largest RNA virus genome. Virus Res 117:17–37
Gozuacik D, Akkoc Y, Ozturk DG, Kocak M (2017) Autophagy-regulating microRNAs and cancer. Front Oncol 7:65
Guo YE, Riley KJ, Iwasaki A, Steitz JA (2014) Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol Cell 54:67–79
Hasan MM, Akter R, Ullah M, Abedin M, Ullah G, Hossain M (2014) A computational approach for predicting role of human microRNAs in MERS-CoV genome. Adv Bioinform 2014. https://doi.org/10.1155/2014/967946
Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, Jünemann C, Niepmann M (2008) microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27:3300–3310
Ho B-C, Yang P-C, Yu S-L (2016) MicroRNA and pathogenesis of enterovirus infection. Viruses 8:11
Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X (2009) MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol 183:2150–2158
Hu G, Yao H, Chaudhuri A, Duan M, Yelamanchili SV, Wen H, Cheney P, Fox HS, Buch S (2012) Exosome-mediated shuttling of microRNA-29 regulates HIV Tat and morphine-mediated neuronal dysfunction. Cell Death Dis 3:e381–e381
Hu S, Li Z, Lan Y, Guan J, Zhao K, Chu D, Fan G, Guo Y, Gao F, He W (2020) MiR-10a-5p-Mediated syndecan 1 suppression restricts porcine hemagglutinating encephalomyelitis virus replication. Front Microbiol 11:105
Huang C-C, Chang L-J, Tsai Y-Y, Hung C-C, Fang M-Y, Su Y-N, Chen H-F, Chen S-U (2013) A feasible strategy of preimplantation genetic diagnosis for carriers with chromosomal translocation: using blastocyst biopsy and array comparative genomic hybridization. J Formos Med Assoc 112:537–544
Huang YK, Yu JC (2015) Circulating microRNAs and long non-coding RNAs in gastric cancer diagnosis: An update and review. World J Gastroenterol 21:9863–9886
Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT (2007) Altered microRNA expression in human heart disease. Physiol Genom 31:367–373
Ingle H, Kumar S, Raut AA, Mishra A, Kulkarni DD, Kameyama T, Takaoka A, Akira S, Kumar H (2015) The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci Signal 8:ra126–ra126
Jiang D, Chen F-X, Zhou H, Lu Y-Y, Tan H, Yu S-J, Yuan J, Liu H, Meng W, Jin Z-B (2020) Bioenergetic crosstalk between mesenchymal stem cells and various ocular cells through the intercellular trafficking of mitochondria. Theranostics 10:7260
Ju E, Li T, Liu Z, da Silva SR, Wei S, Zhang X, Wang X, Gao S-j (2020) Specific inhibition of viral microRNAs by carbon dots-mediated delivery of locked nucleic acids for therapy of virus-induced cancer. ACS Nano 14:476-487
Karlsen TA, Brinchmann JE (2013) Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol Ther 21:1169–1181
Khongnomnan K, Makkoch J, Poomipak W, Poovorawan Y, Payungporn S (2015) Human miR-3145 inhibits influenza A viruses replication by targeting and silencing viral PB1 gene. Exp Biol Med 240:1630–1639
Kitazawa H, Villena J (2014) Modulation of respiratory TLR3-anti-viral response by probiotic microorganisms: lessons learned from Lactobacillus rhamnosus CRL1505. Front Immunol 5:201
Koralnik IJ, Tyler KL (2020) COVID-19: a global threat to the nervous system. Ann Neurol 88:1–11
Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang H-W, Chang T-C, Vivekanandan P, Torbenson M, Clark KR (2009) Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137:1005–1017
Krammer F (2020) SARS-CoV-2 vaccines in development. Nature 586:516–527
Rauch S, Roth N, Schwendt K, Fotin-Mleczek M, Mueller SO, Petsch B (2021) mRNA-based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus-neutralising antibodies and mediates protection in rodents. NPJ Vaccines 6:1–9
Krützfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M (2007) Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 35:2885–2892
Kumar A, Kumar A, Ingle H, Kumar S, Mishra R, Verma MK, Biswas D, Kumar NS, Mishra A, Raut AA (2018) MicroRNA hsa-miR-324-5p suppresses H5N1 virus replication by targeting the viral PB1 and host CUEDC2. J Virol 92:e01057–e01018
Lai FW, Stephenson KB, Mahony J, Lichty BD (2014) Human coronavirus OC43 nucleocapsid protein binds microRNA 9 and potentiates NF-κB activation. J Virol 88:54–65
Lanier LL (2008) Evolutionary struggles between NK cells and viruses. Nat Rev Immunol 8:259–268
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23:4051–4060
Li C, Hu J, Hao J, Zhao B, Wu B, Sun L, Peng S, Gao GF, Meng S (2014) Competitive virus and host RNAs: the interplay of a hidden virus and host interaction. Protein Cell 5:348–356
Li Y, Shi X (2013) MicroRNAs in the regulation of TLR and RIG-I pathways. Cell Mol Immunol 10:65–71
Liau NP, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, Callaghan K, Nicola NA, Kershaw NJ, Babon JJ (2018) The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun 9:1–14
Lin C, Tu P, Beitsch LM (2021) Confidence and receptivity for COVID-19 vaccines: a rapid systematic review. Vaccines 9:16
Lin J, Li J, Huang B, Liu J, Chen X, Chen XM, Xu YM, Huang LF, Wang XZ (2015) Exosomes: novel biomarkers for clinical diagnosis. Scientific World J. https://doi.org/10.1155/2015/657086
Liu D, Tan Q, Zhu J, Zhang Y, Xue Y, Song Y, Liu Y, Wang Q, Lai L (2019) MicroRNA-33/33* inhibit the activation of MAVS through AMPK in antiviral innate immunity. Cell Mol Immunol. https://doi.org/10.1038/s41423-019-0326-x
Liu G, Ren G, Zhao L, Cheng L, Wang C, Sun B (2017) Antibacterial activity and mechanism of bifidocin A against Listeria monocytogenes. Food Control 73:854–861
Liu H, Yang X, Zhang ZK, Zou WC, Wang HN (2018) miR-146a-5p promotes replication of infectious bronchitis virus by targeting IRAK2 and TNFRSF18. Microb Pathog 120:32–36
Liu Q, Du J, Yu X, Xu J, Huang F, Li X, Zhang C, Li X, Chang J, Shang D (2017) miRNA-200c-3p is crucial in acute respiratory distress syndrome. Cell Discovery 3:1–17
Liu Z, Wang J, Ge Y, Xu Y, Guo M, Mi K, Xu R, Pei Y, Zhang Q, Luan X (2021) SARS-CoV-2 encoded microRNAs are involved in the process of virus infection and host immune response. J Biomed Res 35:216–227
López P, Girardi E, Mounce BC, Weiss A, Chane-Woon-Ming B, Messmer M, Kaukinen P, Kopp A, Bortolamiol-Becet D, Fendri A (2020) High-throughput fluorescence-based screen identifies the neuronal microRNA miR-124 as a positive regulator of alphavirus infection. J Virol 94:e02145–02119
Lu D, Chatterjee S, Xiao K, Riedel I, Wang Y, Foo R, Bär C, Thum T (2020) MicroRNAs targeting the SARS-CoV-2 entry receptor ACE2 in cardiomyocytes. J Molec Cell Cardiol 148:46–49
Lv X, Zhao K, Lan Y, Li Z, Ding N, Su J, Lu H, Song D, Gao F, He W (2017) miR-21a-5p contributes to porcine hemagglutinating encephalomyelitis virus proliferation via targeting CASK-interactive protein1 in vivo and vitro. Front Microbiol 8:304
Lythgoe MP, Middleton P (2020) Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends Pharmacol Sci 41:363–382
Ma Y, Wang C, Xue M, Fu F, Zhang X, Li L, Yin L, Xu W, Feng L, Liu P (2018) The coronavirus transmissible gastroenteritis virus evades the type I interferon response through IRE1α-mediated manipulation of the microRNA miR-30a-5p/SOCS1/3 axis. J Virol 92:e00728–00718
Ma YJ, Yang J, Fan XL, Zhao HB, Hu W, Li ZP, Yu GC, Ding XR, Wang JZ, Bo XC (2012) Cellular micro RNA let-7c inhibits M1 protein expression of the H1N1 influenza A virus in infected human lung epithelial cells. J Cell Mol Med 16:2539–2546
MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, Haasdyk JE, Dickson K-A, Brahmbhatt VN, Pattison ST (2007) Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 11:431–445
Maemura T, Fukuyama S, Sugita Y, Lopes TJ, Nakao T, Noda T, Kawaoka Y (2018) Lung-derived exosomal miR-483-3p regulates the innate immune response to influenza virus infection. J Infect Dis 217:1372–1382
Mallick B, Ghosh Z, Chakrabarti J (2009) MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PloS One. https://doi.org/10.1371/journal.pone.0007837
Mallick B, Ghosh Z, Chakrabarti J (2009) MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PloS one 4:e7837
Masaki T, Arend KC, Li Y, Yamane D, McGivern DR, Kato T, Wakita T, Moorman NJ, Lemon SM (2015) miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 17:217–228
Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66:193–292
Mata M, Neben S, Majzoub K, Carette J, Ramanathan M, Khavari PA, Sarnow P (2019) Impact of a patient-derived hepatitis C viral RNA genome with a mutated microRNA binding site. PLoS pathogens 15:e1007467
McCaskill J, Praihirunkit P, Sharp PM, Buck AH (2015) RNA-mediated degradation of microRNAs: A widespread viral strategy? RNA Biol 12:579–585
McKay PF, Hu K, Blakney AK, Samnuan K, Brown JC, Penn R, Zhou J, Bouton CR, Rogers P, Polra K (2020) Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat Commun 11:1–7
Mishra R, Bhattacharya S, Rawat BS, Kumar A, Kumar A, Niraj K, Chande A, Gandhi P, Khetan D, Aggarwal A (2020) MicroRNA-30e-5p has an integrated role in the regulation of the innate immune response during virus infection and systemic lupus erythematosus. Iscience 23:101322
Mohammadian F, Abhari A, Dariushnejad H, Nikanfar A, Pilehvar-Soltanahmadi Y, Zarghami N (2016) Effects of chrysin-PLGA-PEG nanoparticles on proliferation and gene expression of miRNAs in gastric cancer cell line. Iranian J Cancer Preven 9:e4190
Mohammadian F, Abhari A, Dariushnejad H, Zarghami F, Nikanfar A, Pilehvar-Soltanahmadi Y, Zarghami N (2016) Upregulation of Mir-34a in AGS gastric cancer cells by a PLGA-PEG-PLGA chrysin nano formulation. Asian Pac J Cancer Prev 16:8259–8263
Mohammadian F, Pilehvar-Soltanahmadi Y, Mofarrah M, Dastani-Habashi M, Zarghami N (2016) Down regulation of miR-18a, miR-21 and miR-221 genes in gastric cancer cell line by chrysin-loaded PLGA-PEG nanoparticles. Artif Cells Nanomed Biotechnol 44:1972–1978
Mohammadian F, Pilehvar-Soltanahmadi Y, Alipour S, Dadashpour M, Zarghami N (2017) Chrysin alters microRNAs expression levels in gastric cancer cells: possible molecular mechanism. Drug Res 67:509–514
Mohammadian F, Pilehvar-Soltanahmadi Y, Zarghami F, Akbarzadeh A, Zarghami N (2017) Upregulation of miR-9 and Let-7a by nanoencapsulated chrysin in gastric cancer cells. Artif Cells Nanomed Biotechnol 45:1201–1206
Mohd HA, Al-Tawfiq JA, Memish ZA (2016) Middle East respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir. Virol J 13:87
Mohr AM, Mott JL (2015) Overview of microRNA biology. Semin Liver Dis 35:3–11
Mousazadeh H, Pilehvar-Soltanahmadi Y, Dadashpour M, Zarghami N (2020) Cyclodextrin based natural nanostructured carbohydrate polymers as effective non-viral siRNA delivery systems for cancer gene therapy. J Controll Release 330:1046-1070
Muthiah M, Park IK, Cho CS (2013) Nanoparticle-mediated delivery of therapeutic genes: focus on miRNA therapeutics. Expert Opin Drug Deliv 10:1259–1273
Nahand JS, Mahjoubin-Tehran M, Moghoofei M, Pourhanifeh MH, Mirzaei HR, Asemi Z, Khatami A, Bokharaei-Salim F, Mirzaei H, Hamblin MR (2020) Exosomal miRNAs: Novel players in viral infection. Epigenomics 12:353–370
Nathans R, Chu C-y, Serquina AK, Lu C-C, Cao H, Rana TM (2009) Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell 34:696–709
Nogimori T, Furutachi K, Ogami K, Hosoda N, Hoshino S-i (2019) A novel method for stabilizing microRNA mimics. Biochem Biophys Res Commun 511:422–426
Norouzi M, Yasamineh S, Montazeri M, Dadashpour M, Sheervalilou R, Abasi M, Pilehvar-Soltanahmadi Y (2019) Recent advances on nanomaterials-based fluorimetric approaches for microRNAs detection. Mater Sci Eng 104:110007
Ohno M, Otsuka M, Kishikawa T, Shibata C, Yoshikawa T, Takata A, Muroyama R, Kowatari N, Sato M, Kato N (2014) Specific delivery of microRNA93 into HBV-replicating hepatocytes downregulates protein expression of liver cancer susceptible gene MICA. Oncotarget 5:5581
Dexter D, Simons D, Kiyaga C, Kapata N, Ntoumi F, Kock R, Zumla A (2020) Mitigating the effect of the COVID-19 pandemic on sickle cell disease services in African countries. Lancet Haematol 7:e430–e432
Peng B, Chen Y, Leong KW (2015) MicroRNA delivery for regenerative medicine. Adv Drug Deliv Rev 88:108–122
Jafarinejad-Farsangi S, Jazi MM, Rostamzadeh F, Hadizadeh M (2020) High affinity of host human microRNAs to SARS-CoV-2 genome: An in silico analysis. Non-coding RNA Res 5:222–231
Pham KM, Suter SR, Lu SS, Beal PA (2020) Ester modification at the 3′ end of anti-microRNA oligonucleotides increases potency of microRNA inhibition. Bioorgan Med Chem 29:115894
Pontecorvi G, Bellenghi M, Ortona E, Carè A (2020) microRNAs as new possible actors in gender disparities of Covid‐19 pandemic. Wiley Online Library 230:e13538
Ramaiah MJ (2020) mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Reports 20:100765
Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC (2017) Modified mRNA vaccines protect against Zika virus infection. Cell 168:1114-1125.e1110
Rizzo P, Dalla Sega FV, Fortini F, Marracino L, Rapezzi C, Ferrari R (2020) COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res Cardiol 115:31
Roberts AP, Lewis AP, Jopling CL (2011) The role of microRNAs in viral infection. Prog Mol Biol Transl Sci 102:101–139
Romero-Cordoba SL, Salido-Guadarrama I, Rodriguez-Dorantes M, Hidalgo-Miranda A (2014) miRNA biogenesis: biological impact in the development of cancer. Cancer Biol Ther 15:1444–1455
Ruiz-Hitzky E, Darder M, Wicklein B, Ruiz-Garcia C, Martín-Sampedro R, Del Real G, Aranda P (2020) Nanotechnology Responses to COVID-19. Adv Healthcare Mater 9:2000979
Safdar M, Khan MS, Karim AY, Omar SA, Smail SW, Saeed M, Zaheer S, Ali M, Ahmad B, Tasleem M (2021) SNPs at 3′ UTR of APOL1 and miR-6741-3p target sites associated with kidney diseases more susceptible to SARS-COV-2 infection: in silco and in vitro studies. Mamm Genome:1–12
Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Vormehr M, Baum A, Pascal K, Quandt J, Maurus D (2020) COVID-19 vaccine BNT162b1 elicits human antibody and TH 1 T cell responses. Nature 586:594–599
Samir M, Vidal RO, Abdallah F, Capece V, Seehusen F, Geffers R, Hussein A, Ali AA, Bonn S, Pessler F (2020) Organ-specific small non-coding RNA responses in domestic (Sudani) ducks experimentally infected with highly pathogenic avian influenza virus (H5N1). RNA Biol 17:112–124
Sankaranarayanan R, Palani SN, Kumar A, AS PS, Tennyson J, (2020) Prediction and experimental confirmation of banana bract mosaic virus encoding miRNAs and their targets. ExRNA 2:5
Scheel TK, Luna JM, Liniger M, Nishiuchi E, Rozen-Gagnon K, Shlomai A, Auray G, Gerber M, Fak J, Keller I (2016) A broad RNA virus survey reveals both miRNA dependence and functional sequestration. Cell Host Microbe 19:409–423
Schwarzenbach H (2015) The clinical relevance of circulating, exosomal miRNAs as biomarkers for cancer. Expert Rev Mol Diagn 15:1159–1169
Sedano CD, Sarnow P (2014) Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe 16:257–264
Sheervalilou R, Ansarin K, Fekri Aval S, Shirvaliloo S, Pilehvar-soltanahmadi Y, Mohammadian M, Zarghami N (2016) An update on sputum Micro RNA s in lung cancer diagnosis. Diagn Cytopathol 44:442–449
Sheervalilou R, Shahraki O, Hasanifard L, Shirvaliloo M, Mehranfar S, Lotfi H, Pilehvar-Soltanahmadi Y, Bahmanpour Z, Zadeh SS, Nazarlou Z (2020) Electrochemical Nano-biosensors as Novel Approach for the Detection of Lung Cancer-related MicroRNAs. Curr Mol Med 20:13–35
Sheervalilou R, Shirvaliloo M, Dadashzadeh N, Shirvalilou S, Shahraki O, Pilehvar-Soltanahmadi Y, Ghaznavi H, Khoei S, Nazarlou Z (2020) COVID-19 under spotlight: A close look at the origin, transmission, diagnosis, and treatment of the 2019-nCoV disease. J Cell Physiol 235:8873–8924
Shi Y, Wang N, Zou Q (2020) Progress and challenge of vaccine development against 2019 novel coronavirus (2019-nCoV). Zhonghua yu Fang yi xue za zhi [Chinese Journal of Preventive Medicine] 54:E029–E029
Shin MD, Shukla S, Chung YH, Beiss V, Chan SK, Ortega-Rivera OA, Wirth DM, Chen A, Sack M, Pokorski JK (2020) COVID-19 vaccine development and a potential nanomaterial path forward. Nat Nanotechnol 15:646–655
Simonson B, Das S (2015) MicroRNA Therapeutics: the Next Magic Bullet? Mini Rev Med Chem 15:467–474
Slonchak A, Shannon RP, Pali G, Khromykh AA (2016) Human microRNA miR-532-5p exhibits antiviral activity against West Nile virus via suppression of host genes SESTD1 and TAB3 required for virus replication. J Virol 90:2388–2402
Sola I, Almazan F, Zuniga S, Enjuanes L (2015) Continuous and discontinuous RNA synthesis in coronaviruses. Ann Rev Virol 2:265–288
Song L, Liu H, Gao S, Jiang W, Huang W (2010) Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J Virol 84:8849–8860
Sonkoly E, Wei T, Janson PC, Sääf A, Lundeberg L, Tengvall-Linder M, Norstedt G, Alenius H, Homey B, Scheynius A (2007) MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PloS one 2:e610
Sorbera L, Graul A, Dulsat C (2020) Taking aim at a fast-moving target: targets to watch for SARS-CoV-2 and COVID-19. Drugs Future 45:1–6
Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S (2012) Inhibition of microRNA function by antimiR oligonucleotides. Silence 3:1
Su S, Wong G, Shi W, Liu J, Lai AC, Zhou J, Liu W, Bi Y, Gao GF (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 24:490–502
Takahashi M, Yamada N, Hatakeyama H, Murata M, Sato Y, Minakawa N, Harashima H, Matsuda A (2013) In vitro optimization of 2′-OMe-4′-thioribonucleoside–modified anti-microRNA oligonucleotides and its targeting delivery to mouse liver using a liposomal nanoparticle. Nucleic Acids Res 41:10659–10667
Tavakoli F, Jahanban-Esfahlan R, Seidi K, Jabbari M, Behzadi R, Pilehvar-Soltanahmadi Y, Zarghami N (2018) Effects of nano-encapsulated curcumin-chrysin on telomerase, MMPs and TIMPs gene expression in mouse B16F10 melanoma tumour model. Artif Cells Nanomed Biotechnol 46:75–86
Tong L, Lin L, Wu S, Guo Z, Wang T, Qin Y, Wang R, Zhong X, Wu X, Wang Y (2013) MiR-10a* up-regulates coxsackievirus B3 biosynthesis by targeting the 3D-coding sequence. Nucleic Acids Res 41:3760–3771
Trobaugh DW, Gardner CL, Sun C, Haddow AD, Wang E, Chapnik E, Mildner A, Weaver SC, Ryman KD, Klimstra WB (2014) RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 506:245–248
Trobaugh DW, Klimstra WB (2017) MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol Med 23:80–93
Tycowski KT, Guo YE, Lee N, Moss WN, Vallery TK, Xie M, Steitz JA (2015) Viral noncoding RNAs: more surprises. Genes Dev 29:567–584
van Zandwijk N, Pavlakis N, Kao S, Clarke S, Lee A, Brahmbhatt H, MacDiarmid J, Pattison S, Leslie F, Huynh Y (2015) P1: 02 Mesomir 1: a phase i study of targomirs in patients with refractory malignant pleural mesothelioma (mpm) and lung cancer (NSCLC). Ann Oncol 26:ii16
Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Chien-an AH (2008) Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem 283:21540–21549
Wang Y, Zhang S, Song W, Zhang W, Li J, Li C, Qiu Y, Fang Y, Jiang Q, Li X (2020) Exosomes from EV71‐infected oral epithelial cells can transfer miR‐30a to promote EV71 infection. Oral Dis 26:778:788
Wang Z (2011) The guideline of the design and validation of MiRNA mimics. MicroRNA and Cancer. Methods Mol Biol 676:211–223
Wen W, He Z, Jing Q, Hu Y, Lin C, Zhou R, Wang X, Su Y, Yuan J, Chen Z (2015) Cellular microRNA-miR-548g-3p modulates the replication of dengue virus. J Infect 70:631–640
Widiasta A, Sribudiani Y, Nugrahapraja H, Hilmanto D, Sekarwana N, Rachmadi D (2020) Potential role of ACE2-related microRNAs in COVID-19-associated nephropathy. Non-coding RNA Res 5:153–166
Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B 10:766-788
Wu J, Gu J, Shen L, Fang D, Zou X, Cao Y, Wang S, Mao L (2019) Exosomal MicroRNA-155 Inhibits Enterovirus A71 Infection by Targeting PICALM. Int J Biol Sci 15:2925
Wu N, Gao N, Fan D, Wei J, Zhang J, An J (2014) miR-223 inhibits dengue virus replication by negatively regulating the microtubule-destabilizing protein STMN1 in EAhy926 cells. Microbes Infect 16:911–922
Wu Z, Yuan Q, Yang C, Zhang X, Qi P, Huang H, Ma Z (2020) Downregulation of oncogenic gene TGFβR2 by miRNA-107 suppresses non-small cell lung cancer. Pathol Res Practice 216:152690
Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, Qi F, Bao L, Du L, Liu S (2020) Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 30:343–355
Yan B, Ma H, Jiang S, Shi J, Yang Z, Zhu W, Kong C, Chen L, Yan H, Ma C (2019) microRNA-221 restricts human cytomegalovirus replication via promoting type I IFN production by targeting SOCS1/NF-κB pathway. Cell Cycle 18:3072–3084
Yasukawa K, Kinoshita D, Yaku K, Nakagawa T, Koshiba T (2020) The microRNAs miR-302b and miR-372 regulate mitochondrial metabolism via the SLC25A12 transporter, which controls MAVS-mediated antiviral innate immunity. J Biol Chem 295:444–457
Yi H (2020) 2019 novel coronavirus is undergoing active recombination. Clin Infect Dis 71:884–887
Young BE, Ong SWX, Kalimuddin S, Low JG, Tan SY, Loh J, Ng OT, Marimuthu K, Ang LW, Mak TM, Lau SK, Anderson DE, Chan KS, Tan TY, Ng TY, Cui L, Said Z, Kurupatham L, Chen MI, Chan M, Vasoo S, Wang LF, Tan BH, Lin RTP, Lee VJM, Leo YS, Lye DC (2020) Epidemiologic Features and Clinical Course of Patients Infected With SARS-CoV-2 in Singapore. JAMA 323:1488–1494
Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, Lu G, Wu Y, Yan J, Shi Y (2017) Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8:15092
Zavari-Nematabad A, Alizadeh-Ghodsi M, Hamishehkar H, Alipour E, Pilehvar-Soltanahmadi Y, Zarghami N (2017) Development of quantum-dot-encapsulated liposome-based optical nanobiosensor for detection of telomerase activity without target amplification. Anal Bioanal Chem 409:1301–1310
Zen K, Zhang CY (2012) Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev 32:326–348
Zhang J-j, Dong X, Cao Y-y, Yuan Y-d, Yang Y-b, Yan Y-q, Akdis CA, Gao Y-d (2020) Clinical characteristics of 140 patients infected with SARS‐CoV‐2 in Wuhan, China. Allergy 75:1730–1741
Zhang J, Li Z, Huang J, Yin H, Tian J, Qu L (2020) miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling. Viruses 12:2
Zhang S, Li J, Li J, Yang Y, Kang X, Li Y, Wu X, Zhu Q, Zhou Y, Hu Y (2018) Up-regulation of microRNA-203 in influenza A virus infection inhibits viral replication by targeting DR1. Sci Rep 8:1–15
Zhang Y-Z, Holmes EC (2020) A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 181:223–227
Zhang Y, Wang Z, Gemeinhart RA (2013) Progress in microRNA delivery. J Controll Release 172:962–974
Zhao L, Zhu J, Zhou H, Zhao Z, Zou Z, Liu X, Lin X, Zhang X, Deng X, Wang R (2015) Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci Rep 5:14991
Zhao X, Ma X, Guo J, Mi M, Wang K, Zhang C, Tang X, Chang L, Huang Y, Tong D (2019) Circular RNA CircEZH2 suppresses transmissible gastroenteritis coronavirus-induced opening of mitochondrial permeability transition pore via targeting MiR-22 in IPEC-J2. Int J Biol Sci 15:2051
Zheng B, Zhou J, Wang H (2020) Host microRNAs and exosomes that modulate influenza virus infection. Virus Res 279:197885
Zheng Z, Ke X, Wang M, He S, Li Q, Zheng C, Zhang Z, Liu Y, Wang H (2013) Human microRNA hsa-miR-296-5p suppresses enterovirus 71 replication by targeting the viral genome. J Virol 87:5645–5656
Zhu S, Wang X, Zheng Z, Zhao X-E, Bai Y, Liu H (2020) Synchronous measuring of triptolide changes in rat brain and blood and its application to a comparative pharmacokinetic study in normal and Alzheimer’s disease rats. J Pharmaceut Biomed Analy 185:113263
Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z, Yu J, Kang M, Song Y, Xia J (2020) SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med 382:1177–1179
Acknowledgements
The authors would like to thank the “Cellular and Molecular Medicine Institute, Urmia University of Medical Sciences,” for their kind cooperation.
Funding
No funding was received.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any authors.
Additional information
Handling Editor: Zhenhai Chen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hu, J., Stojanović, J., Yasamineh, S. et al. The potential use of microRNAs as a therapeutic strategy for SARS-CoV-2 infection. Arch Virol 166, 2649–2672 (2021). https://doi.org/10.1007/s00705-021-05152-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05152-5