
Abstract
In this study, we hypothesized that deregulation in the maintenance of the pool of coenzyme A (CoA) may play a crucial role in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Specific deletion of Acot12 (Acot12−/−), the major acyl-CoA thioesterase, induced the accumulation of acetyl-CoA and resulted in the stimulation of de novo lipogenesis (DNL) and cholesterol biosynthesis in the liver. KEGG pathway analysis suggested PPARα signaling as the most significantly enriched pathway in Acot12−/− livers. Surprisingly, the exposure of Acot12−/− hepatocytes to fenofibrate significantly increased the accumulation of acetyl-CoA and resulted in the stimulation of cholesterol biosynthesis and DNL. Interaction analysis, including proximity-dependent biotin identification (BioID) analysis, suggested that ACOT12 may directly interact with vacuolar protein sorting-associated protein 33A (VPS33A) and play a role in vesicle-mediated cholesterol trafficking and the process of lysosomal degradation of cholesterol in hepatocytes. In summary, in this study, we found that ACOT12 deficiency is responsible for the pathogenesis of NAFLD through the accumulation of acetyl-CoA and the stimulation of DNL and cholesterol via activation of PPARα and inhibition of cholesterol trafficking.
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease and occurs when fat accumulates due to factors that are closely associated with obesity and type 2 diabetes mellitus. Nonalcoholic steatohepatitis (NASH) is an extreme form of NAFLD that can eventually lead to cirrhosis and hepatocellular cancer. Even though NAFLD is an important risk factor and the leading cause of liver diseases, there is still no effective pharmaceutical treatment. To identify potential targets for NAFLD for therapeutic treatment, it is necessary to unravel and understand the pathophysiological mechanism of NAFLD.
The liver acts as the main regulator of lipid homeostasis by synthesizing fatty acids, oxidizing lipids, storing excess triglycerides (TGs), or taking up and releasing lipoproteins from/to other interacting organs, such as adipose tissue. The major function of adipose tissue is the storage and release of TGs and free fatty acids (FFAs) through lipogenesis and lipolysis, respectively1. Previously, it has been suggested that approximately 60% of hepatic TG comes from FFA pools of adipose tissue, and the rest comes from either de novo lipogenesis (DNL) or imbalanced nutrients by unhealthy diets2. Increased FFA flux impairs insulin sensitivity and promotes DNL by upregulating the expression levels of sterol regulatory element-binding protein 1c (SREBP-1c) or PPARγ3,4 and stimulates hepatic inflammation through activation of the NF-κB signaling pathway and resident hepatic macrophages known as kupffer cells5. Although dysregulation in the interaction between adipose tissue and the liver in NAFLD is well described, the molecular and pathological mechanisms of NAFLD are not fully understood due to the multifaceted nature of NAFLD.
Hepatic cholesterol can be regulated by several alternative metabolic pathways: efflux into the blood in the form of very-low-density lipoprotein or via ATP binding cassette transporter 1 (ABCA1), uptake through bile via ATP binding cassette subfamily G member 5 (ABCG5)/ABCG8, and/or deposition as cholesterol esters or substrates for bile acid synthesis. When the cellular cholesterol level is low, cholesterol synthesis and uptake are induced by HMG-CoA reductase (HMGCR) and low-density lipoprotein receptor (LDLR), respectively. Intracellular cholesterol synthesis could also be regulated by the transcription factor SREBP-2 in the endoplasmic reticulum (ER) membrane. Under low cholesterol conditions, SREBP-2 is processed by proteases and travels into the nucleus to upregulate genes involved in cholesterol syntheses, such as LDLR and HMGCR. On the other hand, at high cholesterol levels, genes involved in cholesterol efflux, such as ABCA1, are increased to reverse cellular cholesterol accumulation.
Recent data suggest that the disturbance of hepatic cholesterol homeostasis and the accumulation of free cholesterol (FC) could cause NAFLD6. A significant increase in the level of cholesterol, as well as increased levels of TG and FFAs, has been observed in the liver and plasma of NAFLD patients compared to normal patients7,8. Deregulation of hepatic genes related to cholesterol anabolism, catabolism, and efflux, leading to increased hepatic cholesterol levels, has been reported in patients with NAFLD9. Moreover, consumption of a high-fat diet with cholesterol led to the development of NAFLD and was accompanied by severe histopathological features, including fibrosis and steatosis10. However, the regulatory mechanism and responsible factors contributing to cholesterol accumulation in NAFLD remain to be investigated.
Acetyl-CoA is a key intermediate metabolite that is generated through the breakdown of carbohydrates and fatty acids and resynthesized into TG, glucose, or cholesterol. Acetyl-CoA is regulated by several main key enzymes, such as acetyl-CoA synthetase (ACS), adenosine triphosphate (ATP)-citrate lyase (ACLY), citrate synthase (CS), pyruvate dehydrogenase (PDH), acyl-CoA thioesterase (ACOT), acetyl-CoA carboxylase (ACC), and/or acetyl-CoA acetyltransferase (ACAT). ACOTs that hydrolyze a variety of coenzyme A (CoASH) thioesters, including fatty acyl- and acetyl-CoAs, are known to provide important biological and pathological responses by regulating the intracellular CoA pool11,12. Loss of ACOT7 in neurons leads to neurological dysfunction and neurodegeneration due to an increased level of long-chain FFAs13, and ACOT11 in brown adipose tissue reduces energy expenditure by hydrolyzing acyl-CoA from lipolysis of LDs in response to adrenergic stimuli14. ACOT11-null mice develop resistance to diet-induced obesity and hepatic inflammation steatosis by improving glucose homeostasis15. However, to date, the biological and pathological functions of various ACOTs remain poorly understood, and emerging evidence suggests the important role of ACOTs in maintaining metabolic homeostasis and diverse biological responses in the liver.
In this study, we explored the important role of ACOT12, an acyl-CoA thioesterase primarily expressed in the liver, kidney, and small intestine in the pathogenesis of NAFLD. We found that deficiency of ACOT12 resulted in the stimulation of DNL and cholesterol synthesis in the liver through the accumulation of acetyl-CoA and activation of peroxisome proliferator-activated receptor (PPAR) α as well as the inhibition of cholesterol trafficking.
Materials and methods
Human and animal study
All human studies included patients with NAFLD diagnosed at Hanyang University Hospital, Seoul, Korea, and fourteen normal patients and 61 NAFLD patient samples were selected for this study. The Institutional Review Board of Hanyang University Hospital approved the human biopsy collections (HY-IRB 2017-03-002-002), and written informed consent was obtained from all patients. All animal studies were performed following approval from the Wonkwang University Animal Care and Use Committee and were in compliance with the institutional guidelines. Eight-week-old male C57BL/6N mice were fed ad libitum a normal chow diet (NCD), a high-fat diet (HFD; 60% kcal from fat; Research Diets Inc.), or a high fat-high-cholesterol diet (HFHCD; 40% kcal from fat with added 1.25% cholesterol (D12108C, Research Diets Inc.) for the indicated times. Genetically obese mice (ob/ob) were purchased from Central Lab. Animal Inc. (Seoul, Korea) and fed an HFD ad libitum. In simvastatin administration experiments, 10 mg/kg simvastatin (S6196, Sigma) was injected intraperitoneally daily under HFHCD feeding. In fenofibrate administration experiments, 100 mg/kg fenofibrate (F6020, Sigma) dissolved in corn oil was orally administered daily. All animals fasted for 12 h (overnight) before sacrifice, and blood was collected to isolate serum for biochemical analysis. The tissue was immediately fixed for staining and IHC study, and the rest of the tissue was rapidly kept frozen at −80 °C for subsequent analysis.
Generation of ACOT12 knockout mice
Acot12 KO mice were generated using the RNA-guided endonuclease (RGEN) method. Acot12 exon 1-specific guide RNA (50 ng/μl) and Cas9 mRNA (50 ng/μl) were injected into the cytoplasm of C57BL/6N mouse zygotes and transferred into C57BL/6N pseudopregnant female mice. To identify Acot12 mutant mice, PCR analysis was performed using genomic DNA from live F0 mice. Finally, we obtained Acot12 mutant mice with the deletion of 14 base pairs. The following primer pair was designed to amplify a 140-bp PCR product from the wild-type allele or 126-bp RGEN-induced mutant allele: forward 5′- AGCCAGGACGATGGAGTCGA-3′, reverse 5′-GGTGTCCATCCACTTGAGCA-3′.
Histological analysis
Tissues were fixed with 10% (v/v) neutral buffered formalin. One part of each liver was embedded in a paraffin block, and the other part was frozen with Tissue-Tek O.C.T. Compound (Sakura) after cryoprotection with 30% sucrose. Paraffin-embedded livers were sliced into 5-μm sections and stained with hematoxylin and eosin (H&E) for immunostaining in this study. Frozen liver sections were cut into 5-μm sections and stained with Oil red O (Sigma) in 60% isopropanol or 0.25 mg/ml filipin. Paraffin-embedded inguinal white adipose tissue was sliced into 5-μm sections and stained with H&E or CellMask Orange Plasma membrane Stain (Thermo Fisher) with DAPI for nuclear staining. Adipocyte area and adipocyte counting were measured using ImageJ software in 6–8 different fields and are presented in a bar graph. For immunostaining, sections were incubated with the following primary antibodies: ACOT12 (MBS273137, Mybiosource), FASN (GTX109833, GeneTex), SCD1 (ab19862, Abcam), PPARα (ab8934, Abcam), and HA-tag (3724, Cell Signaling). Positive staining was visualized by detection with DAB Peroxidase (HRP) Substrate (SK-4105, Vector Laboratories), and counterstaining with hematoxylin. Histological images were acquired with a light microscope (Thermo Fisher).
Lentivirus production and injection into the mouse tail vein
To prepare lentiviral particles for injection, 293T cells were transfected with pLenti-GIII-CMV or pLenti-GIII-CMV-Acot12-HA using a third-generation packaging system mix (Abm) according to the manufacturer’s protocol. After 2 days, 20 ml of virus-containing medium was collected and concentrated with a Lenti-X concentrator (Takara) according to the manufacturer’s protocol. Mice were then injected six times (in 2 weeks) by tail vein injection with 100 μl of the concentrated virus.
Isolation and culture of primary hepatocytes and primary adipocytes from the stromal vascular fraction (SVF)
Hepatocytes were isolated using the collagenase perfusion technique from 8-week-old male Acot12+/+ mice and Acot12−/− mice anesthetized by isoflurane. The hepatocytes were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose (4.5 g/L) (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 U/ml penicillin–streptomycin, and 0.1 μM dexamethasone in a 5% CO2 atmosphere at 37 °C. Since the expression level of Acot12 is continuously decreased after plating16, all primary hepatocytes were sampled within 2 days after plating. Mouse stromal vascular fraction (SVF) cells were isolated from white adipose tissue of Acot12+/+ or Acot12−/− mice using a previously reported method17. Briefly, iWAT was collected from Acot12+/+ or Acot12−/− mice and digested in a collagenase medium. Digested iWAT was centrifuged, and mature adipose cells and SVF cells were separated. Resuspended SVF cells were plated on collagen-coated plates and incubated with 1 μM rosiglitazone in the differentiation medium for adipocyte differentiation.
Cell culture and fluorescence microscopy
HeLa cells and AML12 cells were cultured in DMEM-high glucose (4.5 g/L) (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 100 U/ml penicillin–streptomycin (pen–strep) in a 5% CO2 atmosphere at 37 °C. The NCTC clone 1469 normal mouse liver cell line was cultured in DMEM high glucose supplemented with 10% horse serum and 100 U/ml pen–strep in a 5% CO2 atmosphere at 37 °C. Empty vector (pLKO.1) or pLKO-shAcot12 packaged lentivirus particles were transfected into AML12 cells for 24 h. Sodium acetate (20 mM) was added 24 h after transfection and then fixed using 4% PFA. Primary hepatocytes isolated from Acot12+/+ and Acot12−/− mice have treated with sodium acetate buffer or 50 μM fenofibrate and transfected with empty vector (pcDNA 3.1) or pcDNA-Ppara using Lipofectamine 3000 (L3000015, Thermo Fisher). Fixed AML12 cells and primary hepatocytes were incubated using 2 μM BODIPY493/503 (D3922, Thermo Fisher) for neutral lipid staining and counterstained with DAPI. For intracellular cholesterol staining of AML12 cells and primary hepatocytes, 0.05 mg/ml filipin in phosphate-buffered saline (PBS) with 10% fetal bovine serum was stained overnight after 1.5 mg/ml glycine incubation. NCTC clone 1469 cells were treated with 50 μg/ml water-soluble cholesterol for 24 h and then fixed in 4% PFA. Fixed NCTC clone 1469 cells were permeabilized with 0.1% Triton X-100 in PBS. Primary anti-ACOT12 antibody (1:100 dilution) and FITC-conjugated rabbit secondary antibody (1:200 dilution) were used for immunocytochemistry. EGFP-tagged VPS33A and TagRFP-tagged ACOT12 expression plasmid DNA were transfected into HeLa cells using Lipofectamine 3000 reagent, and LysoTracker (L12492, Thermo Fisher) and BODIPY cholesterol (24618, Cayman Chemical) was used for focal contact. Images were acquired with a light microscope in an EVOS FL Auto imaging system (Thermo Fisher). Relative fluorescence intensity was measured by ImageJ software to determine ACOT12 expression and the LysoTracker-BODIPY cholesterol merge ratio.
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Acot12+/+ and Acot12−/− mice were intraperitoneally injected with glucose (2 g/kg) or insulin (0.75 U/kg) (I-034, Sigma) after overnight fasting (16 h) or 4 h fasting, and blood glucose levels were measured at the indicated time points using a glucometer and glucose test strips.
Immunoblotting
Twenty milligrams of liver tissue were homogenized with RIPA buffer (Cell Signaling) supplemented with PMSF protease inhibitor (Thermo Fisher) for whole lysates. Fifty micrograms of protein were run on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, transferred to nitrocellulose membranes, and then incubated with the following primary antibodies: ACOT12 (MBS273137, Mybiosource), GAPDH (AP0066, Bioworld), and PPARα (ab8934, Abcam). The blotted nitrocellulose membranes were developed using horseradish peroxidase (HRP)-conjugated secondary antibodies (α-Rabbit 1:2000 dilution) purchased from Bethyl Laboratories, and the immunoreactive proteins were detected with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher).
qRT-PCR
Total RNA was isolated from liver tissue using RNAiso Plus (TaKaRa) according to the manufacturer’s instructions. Then, 1 μg of RNA was reverse transcribed using 5× All-In-One RT Master Mix (with AccuRT Genomic DNA Removal Kit) (G492, Abm). Target mRNA levels were amplified using gene-specific primers (Supplementary Table 1) with Step-One-Plus real-time PCR systems (Applied Biosystems). All PCRs were performed in triplicate. The relative expression level of each gene was normalized to the 18S rRNA expression level.
RNA-seq analysis
RNA sequencing was performed using an Illumina HiSeq4000 instrument, and libraries were quantified using quantitative real-time PCR (qPCR). RNA library quality was checked using an Agilent Technologies 2100 Bioanalyzer. The raw data were calculated as fragments per kilobase of transcript per million mapped reads of each sample using cufflinks software. Data were transformed logarithmically and normalized using the quantile normalization method.
Statistical analysis
The results are expressed as the mean ± SEM. The mean values for RNA levels and biochemical data from two experimental groups were compared by unpaired two-tailed Student’s t test. When more than two treatment groups were compared, one- or two-way ANOVA for multiple comparisons was used in most studies, as noted in the figure legends. Statistical tests with P < 0.05 were considered significant, and significance is denoted as *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. All tests used GraphPad Prism software (GraphPad).
Results
ACOT12 is closely related to nonalcoholic fatty liver disease
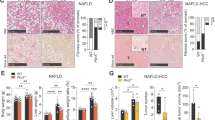
To identify the most relevant and responsible genes of the acetyl-CoA metabolic process (GO: 0006084) underlying the pathogenesis of NAFLD, we performed in silico analysis using Gene Expression Omnibus (GEO, GSE48452)18 data of human patient liver biopsy of normal liver, NASH, and steatosis (Fig. 1a). Among key regulators in acetyl-CoA metabolism, the expression level of ACOT12 was the most significantly altered in both steatosis and NASH compared to normal liver tissue, and its expression gradually changed during the progression of NAFLD (Fig. 1b and Supplementary Fig. 1). A previous report suggested that ACOT12 is closely associated with the metastasis of hepatocellular carcinoma (HCC) and survival of HCC patients19. Overall survival was significantly reduced in HCC patients with low expression levels of ACOT12. In addition, The Cancer Genome Atlas (TCGA) database of HCC patients showed that a low expression level of ACOT12 reduced the survival rate (Fig. 1c). Here, we found an increased expression level of ACOT12 in fatty liver patients with no alcohol consumption compared to normal patients, suggesting the possible involvement of ACOT12 in NAFLD (Fig. 1d). To further confirm the involvement of ACOT12 in NAFLD, wild-type mice or genetically obese mice (ob/ob) were fed an HFD or HFHCD to induce fatty liver and samples stained with H&E and Oil red O (Supplementary Fig. 2a). Significantly increased levels of hepatic TG, total cholesterol (TC), FFAs, and acetyl-CoA were observed in HFD or HFHCD mice or HFD ob/ob mice compared to NCD mice (Fig. 1e). The expression level of ACOT12 was dramatically increased in the livers of HFD and HFHCD mice compared to the livers of mice fed an NCD (Fig. 1f). Consistent with the analysis of human GEO data, among genes in acetyl-CoA metabolism, ACOT12 was one of the most significantly increased genes in the liver of HFD or HFHCD-Wt mice or HFD ob/ob mice (Fig. 1g and Supplementary Fig. 2b).

a Transcription level of 35 enzymes related to acetyl-CoA metabolism in the GSE48452 dataset. b Transcription level of ACOT12 in the GSE48452 dataset. c The Kaplan–Meier plot of HCC patient survival with no alcohol consumption in the high-ACOT12 and low-ACOT12 groups. d Representative image of immunohistochemical staining of ACOT12 from normal (n = 14) and steatotic patient (n = 61) liver biopsies. e Hepatic triglyceride (TG), total cholesterol (TC), free fatty acid (FFA), and acetyl-CoA levels in NCD, HFD, HFHCD, and ob/ob mice (n = 3 mice per group). The diet chow strategy is presented as a bar graph. f Representative immunohistochemical staining of ACOT12 from normal chow diet (NCD)-, high-fat diet (HFD)-, and high-fat high-cholesterol diet (HFHCD)-fed wild-type mice and HFD ob/ob mouse livers. Scale bars, 100 μm. g Transcription level of Acot12 in NCD, HFD, HFHCD, and ob/ob mice (n = 3 per group). All data are presented as the mean ± SEM. Statistical differences among three or more groups were performed using one-way ANOVA, followed by Dunnett’s multiple comparison test (f, g) or Fisher’s LSD post hoc test (b). n.s = nonsignificant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Acot12 −/− mice have increased lipid and cholesterol synthesis independent of a high-fat diet
To fully understand the role of Acot12 in the pathogenesis of NAFLD, Acot12 knockout (KO, Acot12−/−) mice were generated using RGEN-induced mutant alleles in the Acot12 gene, and the efficiency of Acot12 KO was confirmed by immunoblotting and immunostaining (Supplementary Fig. 3). To gain insight into the underlying mechanism in ACOT12 deficiency-induced NAFLD, 8-week-old Acot12+/+ and Acot12−/− mice were challenged with NCD, HFD, and HFHCD for 8 weeks. Interestingly, impaired glucose tolerance and insulin resistance were observed in NCD Acot12−/− mice without a significant increase in body weight compared to NCD Acot12+/+ mice, but the severity of impaired glucose tolerance and insulin resistance was significantly greater in HFD or HFHCD Acot12−/− mice (Fig. 2a, b, and Supplementary Fig. 4a, b). Increased liver weight and mild hepatomegaly were observed in HFD and HFHCD Acot12−/− mice compared to NCD Acot12+/+ mice (Fig. 2c, d). Moreover, NAFLD features, such as increased levels of fat, hepatic TG, TC, FFA, and acetyl-CoA accumulation, were observed in all Acot12−/− mouse diet groups fed an HFD, and HFHCD was more pronounced (Fig. 2e, f, Supplementary Fig. 4c, d, Supplementary Fig. 5 and Supplementary Fig. 6). To identify gene groups associated with steatotic livers of HFD Acot12−/− mice, we performed gene set enrichment analysis of RNA sequencing data using HFD Acot12+/+ and Acot12−/− livers and found dysregulation of lipid homeostasis genes in Acot12−/− livers (Supplementary Fig. 7a). The expression levels of genes in the DNL and mevalonate pathways were significantly increased in HFD Acot12−/− mice compared to HFD Acot12+/+ mice (Supplementary Fig. 7b, c).

Male C57BL/6N wild-type mice (Acot12+/+) and Acot12−/− mice were fed an NCD, HFD, or HFHCD for 8 weeks. a Body weight of Acot12+/+ mice fed an NCD or Acot12−/− mice fed an NCD, HFD, or HFHCD (n = 4). b Oral glucose tolerance test (GTT) and insulin tolerance test (ITT). The glucose value in the area under the curve (AUC) was measured for both the GTT and ITT in all Acot12+/+ or Acot12−/− mice. c Photographs of the liver of an NCD-fed Acot12+/+ mouse and NCD-, HFD-, and HFHCD-fed Acot12−/− mouse livers. Scale bars = 1 cm (lower). d Liver weight (left) and liver weight to body weight (right). e Representative liver staining of hematoxylin and eosin (H&E), Oil Red O (ORO), and filipin. Scale bars, H&E, ORO; 100 μm, filipin; 200 μm. f Hepatic levels of triglyceride (TG), total cholesterol (TC), free fatty acid (FFA), and acetyl-CoA in NCD Acot12+/+ or NCD, HFD, and HFHCD Acot12−/− liver (n = 4). All data are presented as the mean ± SEM. Statistical differences among three or more groups were performed using one-way ANOVA (b, d, f) or two-way ANOVA (a), followed by Dunnett’s multiple comparison test. n.s. = nonsignificant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we investigated whether the exogenous introduction of Acot12 could rescue the accumulation of lipids and cholesterol (Supplementary Fig. 8a). Intravenous injection of pLenti-GIII-CMV-Acot12-HA (HA-Acot12) into HFHCD Acot12+/+ mice significantly decreased the ratio of liver weight to body weight and rescued the impaired glucose and insulin tolerance (Supplementary Fig. 8b, c). Hepatic fat and cholesterol accumulation were dramatically decreased by ACOT12 overexpression (Fig. 3a and Supplementary Fig. 8d). Increased levels of hepatic TG, cholesterol, acetyl-CoA, and malonyl CoA as well as the levels of genes involved in lipogenesis and cholesterol synthesis induced by HFHCD were significantly reduced by the overexpression of ACOT12 (Fig. 3b, c). Moreover, impaired glucose and insulin tolerance in HFHCD Acot12−/− mice were rescued by intravenous injection of HA-Acot12 (Supplementary Fig. 9a–c). Hepatic accumulation of LDs, TGs, cholesterol, acetyl-CoA, and malonyl CoA (Fig. 3d, e) and the expression levels of genes in lipogenesis and cholesterol biosynthesis (Fig. 3f) were also markedly decreased by the restoration of ACOT12 in HFHCD Acot12−/− mice. Administration of simvastatin, a widely used cholesterol-lowering drug that competitively inhibits HMGCR, also inhibited the accumulation of LDs and TGs in HFHCD Acot12−/− mice (Supplementary Fig. 10).

a Eight-week-old normal C57BL/6 male mice fed an HFHCD for 14 weeks were injected with lentivirus containing control vector (lenti-con) or HA-tagged Acot12 vector (HA-Acot12) after 8 weeks of HFHCD feeding (n = 4). Representative liver image of immunostaining of HA-tag, H&E, Sirius Red, Oil Red O, and filipin staining in C57BL/6 mice injected with lentiviruses containing HA-Acot12 or control vector (lenti-con). A magnified image shows the HA-negative site and HA-positive site in HA-Acot12-injected mice. Scale bars, 200 μm. b Hepatic TG, TC, acetyl-CoA, and malonyl CoA levels of lenti-con- or HA-Acot12-injected 14-week HFHCD Acot12+/+ mice. c Expression level of genes related to lipogenesis (upper) and cholesterol metabolism (lower) in lenti-con- or HA-Acot12-injected 14-week HFHCD Acot12+/+ mouse livers (n = 4). d Eight-week-old male Acot12−/− mice injected with lentivirus containing lenti-con or HA-Acot12. After injection, both mouse groups were fed an HFHCD for 8 weeks (n = 4). Representative liver image of immunostaining of HA and H&E, Sirius Red, Oil red O, and filipin staining in HFHCD Acot12−/− mice injected with HA-Acot12 or lenti-con. Scale bars, 200 μm. e Hepatic TG, TC, acetyl-CoA, and malonyl-CoA levels of lenti-con- or HA-Acot12-injected Acot12−/− mice fed an HFHCD for 8 weeks. f Expression level of genes related to lipogenesis (upper) and cholesterol metabolism (lower) in lenti-con- or HA-Acot12-injected 8-week HFHCD Acot12−/− mouse livers (n = 4). All data are presented as the mean ± SEM. Statistical differences between two groups were determined using unpaired two-tailed Student’s t test (b, e) or multiple comparison tests followed by the Holm–Sidak method. c, f *P < 0.05; **P < 0.01; ****P < 0.0001.
Increased acetyl-CoA accumulation in Acot12 −/− mice leads to upregulation of lipid and cholesterol synthesis
As acetyl-CoA is a precursor to fat biosynthesis, we asked whether the increased level of hepatic acetyl-CoA due to ACOT12 deficiency is directly associated with the increased lipid and cholesterol synthesis observed in Acot12−/− mice. To test this hypothesis, we depleted Acot12 in mouse hepatic AML12 cells by treating them with an Acot12-specific short hairpin RNA expression vector (shAcot12) (Supplementary Fig. 11a). Similar to our observations in Acot12−/− mice, the depletion of Acot12 expression in AML12 cells increased the cellular levels of acetyl-CoA, TG, and TC and the accumulation of intracellular lipids and cholesterol (Supplementary Fig. 11b–d). The expression of genes involved in lipogenesis and cholesterol synthesis was also significantly increased by Acot12 silencing (Supplementary Fig. 11e).
To evaluate whether excess acetyl-CoA could affect the synthesis of hepatic fatty acids and cholesterol, AML12 cells were treated with acetate. We observed a dose-dependent increase in acetyl-CoA levels upon acetate treatment in AML12 cells (Supplementary Fig. 12a). The expression level of Acot12 also increased in a dose-dependent manner with acetate treatment (Supplementary Fig. 12b). In addition, the cellular levels of TG and TC and the expression levels of genes involved in lipogenesis and cholesterol biosynthesis increased with increasing concentrations of acetyl-CoA (Supplementary Fig. 12c–e). These data suggested that acetyl-CoA accumulation or overload by Acot12 deficiency could stimulate the synthesis of lipids and cholesterol.
ACOT12 binds with VPS33A and regulates the cholesterol trafficking to the lysosomes process
ACOT12, also known as STARD15, consists of esterase domains and a C-terminal steroidogenic acute regulatory-related lipid transfer (START) domain20. Fifteen STARD proteins have been found in mammals and are known to play a role in transferring cholesterol and lipids between cellular organelles. STARD1 moves cholesterol to the mitochondrial inner membrane, and STARD3 (MLN64) conveys cholesterol from the ER to endosomes21,22. Moreover, STARD4 binds with intracellular FC, and STARD5 and STARD6 contribute to cholesterol trafficking23,24,25. In Acot12−/− primary hepatocytes, accumulation of cellular cholesterol as assessed by filipin staining was observed (Fig. 4a), and exposure of primary hepatocytes to water-soluble cholesterol-induced the expression level of ACOT12 (Fig. 4b). Moreover, treatment of Acot12−/− primary hepatocytes with BODIPY cholesterol-induced cholesterol accumulation (Fig. 4c), suggesting the possible involvement of ACOT12 in cholesterol trafficking.

a Representative filipin staining of Acot12+/+ and Acot12−/− primary hepatocytes. Scale bars, 25 μm. b Representative immunocytochemical staining for ACOT12 in the NCTC clone 1469 normal mouse liver cell line with or without 50 μg/ml water-soluble cholesterol treatment. Scale bars, 100 μm. Fluorescence intensity was measured in five randomly collected images by ImageJ and is visualized as a bar graph (right). c Treatment of BODIPY cholesterol in Acot12+/+ and Acot12−/− primary hepatocytes. Scale bars, 20 μm. d Diagram of the location and function of ACOT12-interacting proteins as assessed by BioID. e Merged image of RFP-ACOT12 and EGFP-VPS33A. Scale bars, 20 μm. f Representative merged image of EGFP-fused VPS33A (green, EGFP-VPS33A) and LysoTracker staining (red). Scale bars, 20 μm. g Merged and magnified images (a’ and b’) of TagRFP-fused ACOT12 (red, RFP-ACOT12) and BODIPY cholesterol (green, BD-Chol). Scale bars, 100 μm. h Representative image of LysoTracker and BD-Chol in Acot12+/+ and Acot12−/− primary hepatocytes. Nuclei were stained by Hoechst 33342. Scale bars, 50 μm. Fluorescence intensity was measured in ten randomly collected images by ImageJ and is visualized as a bar graph (right). All data are presented as the mean ± SD. Statistical differences between the two groups were determined using an unpaired two-tailed Student’s t test. **P < 0.01.
To gain greater insight into the function and role of ACOT12 in cholesterol homeostasis, we performed proximity-dependent biotin identification (Bio-ID) analysis to identify possible interacting partners. Analysis of Bio-ID showed that ACOT12 interacted predominately with endosomal and lysosomal proteins (Fig. 4d). One such lysosomal protein is the vesicle trafficking protein vacuolar protein sorting-associated protein 33A (VPS33A), which is known to play a role in vesicle-mediated protein trafficking to lysosomal compartments. To examine whether ACOT12 may interact with VPS33A, we examined its subcellular localization in HeLa cells by confocal fluorescence microscopy and found that ACOT12 colocalized on VPS33A-positive structures (Fig. 4e) and that VPS33A partially colocalized on lysosomes (Fig. 4f). As VPS33A is localized on both lysosomes and endosomes, these images suggest that ACOT12 may be localized on lysosomes.
The homotypic fusion and vacuole protein sorting (HOPS) complex composed of six VPS subunits, including VPS33A, is required for the trafficking of internalized cholesterol by mediating endosome–lysosome fusion. To investigate whether ACOT12 is required for cholesterol trafficking within the endosome–lysosome pathway, we examined TagRFP-fused ACOT12 (RFP-Acot12) with BODIPY cholesterol and found that they colocalize with each other (Fig. 4g). Next, we examined whether Acot12 KO cells can disrupt cholesterol trafficking by examining the subcellular localization of BODIPY cholesterol in Acot12−/− primary hepatocytes. In wild-type primary hepatocytes, the majority of lysosomes contained cholesterol (Fig. 4h: Acot12+/+). However, the colocalization of BODIPY cholesterol and lysosomes was lost in Acot12−/− primary hepatocytes, suggesting that Acot12 may be required for the trafficking of cholesterol through the endosome–lysosome pathway.
ACOT12 deficiency induces overactivation of PPARα that leads to the accumulation of hepatic acetyl-CoA and cholesterol
To identify a key signaling pathway responsible for the accumulation of lipids and cholesterol in Acot12−/− livers, RNA sequencing was performed, and the enrichment of biological pathways was assessed by KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis. KEGG pathway analysis suggested PPARα as the most significantly enriched signaling pathway in HFD Acot12−/− livers (Fig. 5a, b). Introduction of shAcot12 into murine alpha mouse liver 12 (AML12) cells significantly increased the expression level of Ppara (Supplementary Fig. 13a). Furthermore, the expression level of PPARα was also significantly increased in HFD Acot12−/− livers (Fig. 5c and Supplementary Fig. 13b). In silico analysis using GSE48452 indicated increased levels of PPARα in NASH patients (Fig. 5d). Consistent with this, we also found an increased level of PPARα in HFD Acot12+/+ livers (Supplementary Fig. 13c). Consistent with the Acot12−/− mouse liver data, PPARα protein levels were inversely proportional to the protein levels of ACOT12 in the fatty liver patient samples (Fig. 5e, f).

a KEGG pathway analysis of RNA sequencing data of HFD Acot12−/− livers compared with Acot12+/+ livers. b Gene set analysis of RNA sequencing data of HFD Acot12−/− livers. c Immunostaining of PPARα in 8-week HFD-fed Acot12+/+ and Acot12−/− livers. The magnified image shows representative PPARα-positive sites in HFD-fed Acot12+/+ and Acot12−/− mouse livers. Scale bars, 200 μm. d Gene set analysis of human liver biopsy of a normal, NASH, steatosis patient-related GEO dataset (GSE48452). e Representative image of immunohistochemical staining of PPARα in liver biopsies from patients with steatosis with high ACOT12 expression (n = 18) and patients with steatosis with low ACOT12 expression (n = 18). f Histological score of immunohistochemical staining of ACOT12 and PPARα in liver biopsies of steatosis patients. High and low expression of ACOT12 sorted by H-score of 200. g Levels of TG and TC in Acot12+/+ and Acot12−/− mouse primary hepatocytes with or without acetate. h Transcription level of Ppara in Acot12+/+ and Acot12−/− mouse primary hepatocytes with or without acetate. i Expression level of lipogenesis genes in Acot12+/+ and Acot12−/− mouse primary hepatocytes with or without acetate. All data are presented as the mean ± SEM. Statistical differences between two groups were determined using unpaired two-tailed Student’s t test (f); statistical differences among four groups were performed using one-way ANOVA, followed by the Holm–Sidak multiple comparison test (g, h); statistical differences in individual gene expression among four groups were performed using two-way ANOVA, followed by Tukey’s multiple comparison test (i). n.s. nonsignificant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Several studies suggested that newly synthesized fatty acids and other lipid metabolites could act as ligands for PPARα. Long-chain fatty acids generated de novo by fatty acid synthase (FASN) could serve as ligands for PPARα in the liver26. High malonyl-CoA induces the accumulation of long-chain fatty acyl-CoAs by inhibiting carnitine palmitoyltransferase-1 (CPT-1) and results in the activation of PPARα in the mouse heart27. Several saturated fatty acids, such as palmitate (16:0), myristate (14:0), and stearate (18:0), also bind to PPARα and act as ligands for PPARα, similar to other unsaturated fatty acids28. To investigate whether high acetyl-CoA induced by ACOT12 deficiency could induce the activation of PPARα, primary hepatocytes isolated from Acot12+/+ and Acot12−/− livers were exposed to acetate for acetyl-CoA overload. With acetate treatment, the cellular levels of acetyl-CoA, TGs, and FFAs (Fig. 5g) and the expression levels of PPARα (Fig. 5h) and lipogenic genes (Fig. 5i) were significantly increased in Acot12−/− primary hepatocytes.
Previous studies have suggested that the activation of PPARα could improve hepatic steatosis, inflammation, and fibrosis by enhancing fatty acid oxidation29,30. However, in ACOT12 deficiency, an increased expression level of PPARα worsens the symptoms of NAFLD through the accumulation of lipids and cholesterol possibly converted from acetyl-CoA. To confirm the vicious action of PPARα under ACOT12 deficiency in NAFLD, primary hepatocytes isolated from Acot12+/+ and Acot12−/− livers were transfected with lentiviral vectors containing Ppara (Supplementary Fig. 14a). The cellular levels of acetyl-CoA, FFAs, cholesterol (Supplementary Fig. 14b–e), DNL, and cholesterol synthesis (Supplementary Fig. 14f, g) were significantly increased in Acot12−/− primary hepatocytes but not in Acot12+/+ primary hepatocytes. These data suggested that increased acetyl-CoA by ACOT12 deficiency produces lipids that activate PPARα, which stimulates DNL.
To further test whether PPARα was upregulated in Acot12−/− hepatocytes to counter the cellular increase in lipids, we treated the cells with fenofibrate, a known PPARα agonist. As expected in Acot12+/+ hepatocytes, fenofibrate treatment increased PPARα expression and acetyl-CoA levels. However, we found that the activation of PPARα by fenofibrate failed to reduce FFAs or TC but instead resulted in a greater increase in cellular lipids and cholesterol in the Acot12−/− cells (Supplementary Fig. 15a–e). Moreover, the expression levels of genes involved in DNL and cholesterol synthesis were also significantly increased upon exposure to fenofibrate in Acot12−/− primary hepatocytes but not in wild-type primary hepatocytes (Supplementary Fig. 15f, g). To validate our findings in cultured primary hepatocytes, we treated Acot12−/− mice on an HFHCD with fenofibrate for 6 weeks (100 mg/kg/day). Similar to the cultured cells, the fenofibrate-treated HFHCD Acot12+/+ mice showed evidence of decreased accumulation of LDs and cholesterol and a significant decrease in liver weight, hepatic TGs, and acetyl-CoA compared to untreated animals. On the other hand, in fenofibrate-administered HFHCD Acot12−/− mice, the accumulation of hepatic LDs and cholesterol was distinctly increased even though PPARα was successfully increased by fenofibrate administration (Fig. 6a–e). Unlike Acot12+/+ mice, fenofibrate administration to Acot12−/− mice induced the expression level of DNL and cholesterol synthesis genes with the activation of PPARα (Fig. 6f, g and Supplementary Fig. 16).

a Representative H&E staining of Acot12+/+ and Acot12−/− mouse livers administered vehicle (corn oil) or fenofibrate (FF, 100 mg/kg). n = 3–4 per mice group. Scale bars, 100 μm. b Representative ORO staining in Acot12+/+ and Acot12−/− mouse livers administered vehicle or FF. Scale bars, 100 μm. c Representative filipin staining in Acot12+/+ and Acot12−/− mouse livers administered vehicle or FF. Scale bars, 100 μm. d Liver weight and the ratio of liver weight to body weight in vehicle- or FF-administered HFHCD Acot12+/+ or Acot12−/− mice. e Hepatic TG, TC, FFA, and acetyl-CoA levels in Acot12+/+ and Acot12−/− mouse livers administered vehicle or FF. f Expression levels of Acot12 and Ppara in Acot12+/+ and Acot12−/− mouse livers administered vehicle or FF. g Expression levels of cholesterol biosynthesis genes in Acot12+/+ and Acot12−/− mouse livers administered vehicle or FF (n = 3–4 mice per group). h Expression levels of lipogenic genes in Acot12+/+ and Acot12−/− mouse livers administered vehicle or FF (n = 3–4 mice per group). All data are presented as the mean ± SEM. Statistical differences among four groups were performed using one-way ANOVA (d, e, g, h) followed by Tukey’s multiple comparison test or multiple t test, followed by Holm–Sidak’s multiple comparison test (f). n.s. nonsignificant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
ACOT12 deficiency induces NAFLD independently from lipolysis of adipose tissue
To determine the time of pathological onset of fatty liver by ACOT12 deficiency, we analyzed adipose and liver tissue in HFD Acot12+/+ and Acot12−/− mice at 2, 4, 6, and 8 weeks. In HFD Acot12−/− mice, the accumulation of hepatic fat (Supplementary Fig. 17a) and increased levels of hepatic TG and TC (Supplementary Fig. 17b) were observed after 4 weeks of HFD feeding, and adipose tissue hypertrophy was not significantly changed (Supplementary Fig. 17c, d).
Adipose tissue plays an important role in energy metabolism and contributes to the development of NAFLD. However, the pathological onset of fatty liver in Acot12−/− mice suggests the possibility that hepatic fat accumulation may not be influenced by an increase in adipose tissue. To investigate whether fatty liver induced by ACOT12 deficiency occurred independent of adipose tissue, primary hepatocytes isolated from Acot12+/+ and Acot12−/− mice were co-cultured with primary adipocytes isolated from adipose-derived SVF (adSVF) cells from the inguinal white adipose tissue (iWAT) of either Acot12+/+ or Acot12−/− mice. Acot12−/− primary hepatocytes showed significantly more lipid droplet (LD) accumulation than Acot12+/+ cells regardless of whether primary adipocytes were cocultured (Supplementary Fig. 18a). In fact, no difference was observed between Acot12−/− hepatocytes cocultured with Acot12+/+ and Acot12−/− primary adipocytes. Moreover, the expression level of lipogenic genes was also significantly increased in Acot12−/− primary hepatocytes cocultured with Acot12+/+ primary adipocytes without alteration of the expression level of lipid transport genes (Supplementary Fig. 18b, c).
Discussion
As a central regulator of lipid homeostasis, the liver is responsible for the biosynthesis of fatty acids and exports them to other tissues as energy substrates. The disruption of hepatic lipid homeostasis by an imbalance between lipid acquisition and lipid disposal due to deregulation of circulating lipid uptake, DNL, FAO, and/or lipid export may cause the retention of fat and result in the development of NAFLD. It is well-known that FAs released from adipose tissues are closely associated with fatty liver disease31. Lipolysis in adipose tissue, i.e., the hydrolysis of TGs by adipose TG lipase (ATGL) and monoacylglycerol lipase (MGL) results in the release of FAs into the circulation and leads to fat accumulation in the liver32. DNL is an integrated metabolic pathway that includes glycolysis, the biosynthesis of saturated fatty acids followed by desaturation, and the formation of TG33. Hepatic DNL starts with the conversion of acetyl-CoA into malonyl-CoA by acetyl-CoA carboxylase (ACC) and uses malonyl-CoA as a substrate to produce 16-carbon palmitic acyl-CoA by FASN, which is responsible for the building of new fatty acids. Pharmacologic inhibition or genetic inactivation of hepatic ACC has been predicted to be an attractive approach to the reduction of NAFLD. Mice with targeted mutations that activate ACC constitutively demonstrated histological and clinical signs of NAFLD, whereas liver-directed ACC inhibition reduced hepatic steatosis by suppressing hepatic DNL34. Despite the close association between DNL and NAFLD, surprisingly little is known about the sources of acetyl-CoA, fuel in this process.
In normal liver, acetyl-CoA is oxidized in the tricarboxylic acid cycle to form CO2 or is condensed in the ketogenic pathway to form acetoacetate or β-hydroxybutyrate35,36. NAFLD patients showed dramatic differences in the metabolic fate of acetyl-CoA and elevated rates of pyruvate carboxylase flux, gluconeogenesis, EGP, and oxygen consumption37,38,39. Here, we demonstrated that the development of NAFLD in Acot12−/− mice closely reflected the histopathology of human NAFLD by increased DNL and cholesterol biosynthesis through the accumulation of acetyl-CoA. Moreover, coculture of Acot12−/− hepatocytes and Acot12+/+ adipocytes showed increased DNL in Acot12−/− hepatocytes, and inhibition of lipolysis did not affect hepatic FFAs, TGs, TC, or DNL in Acot12−/− livers. These data suggested that fatty liver in Acot12−/− mice might result from increased hepatic DNL but not from lipolysis of adipocytes.
Two well-known transcription factors, PPARs, members of the nuclear hormone receptor superfamily and sterol-regulatory element-binding proteins (SREBPs), a subgroup of basic-helix–loop–helix–leucine zipper (bHLH-LZ) transcription factors, play a crucial role in lipid homeostasis, such as fatty acid synthesis and cholesterol metabolism40. In particular, PPARα is highly expressed in tissues with high FAO rates, such as liver, heart, skeletal muscle, and brown adipose tissue, and regulates peroxisomal and mitochondrial fatty acid catabolism. PPARα deficiency results in severe steatosis and hepatitis, whereas treatment with the PPARα agonist Wy-14,643 protects against steatosis and steatohepatitis in methionine- and choline-deficient diet-fed mice29,41. A large variety of long-chain fatty acids, eicosanoids, and synthetic compounds have been shown to serve as endogenous PPARα ligands and activators42,43. Fibrates have been defined as exogenous PPARα ligands, which are widely used to reduce hepatic fat in animal experiments44. However, recent clinical and preclinical studies with PPARα agonists such as fenofibrate and clofibrate suggested no significant improvement in NAFLD activity45 but rather showed that PPARα activation increases hepatic DNL and cholesterol accumulation46,47. In this study, we suggested that accumulated acetyl-CoA could act as an endogenous ligand for PPARα and activate the anabolic synthesis of cholesterol and DNL. Moreover, our BioID analysis suggested the possible interaction between ACOT12 and VPS33A, a core component of the class C core vacuole/endosome tethering complex and the HOPS complex known to play a role in vesicle-mediated protein trafficking to lysosomal compartments. The HOPS complex plays a role in the transport and processing of cholesterol48,49. This finding suggested the involvement of ACOT12 in the trafficking of cholesterol by interacting with VPS33A and could serve as a new model of intracellular cholesterol trafficking.
In summary, in this study, we found that ACOT12 deficiency may be responsible for the pathogenesis of NAFLD through the accumulation of cholesterol in the liver and suggest that Acot12 could be a promising therapeutic therapy for controlling NAFLD.
Data and materials availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Nielsen, T. S., Jessen, N., Jorgensen, J. O., Moller, N. & Lund, S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J. Mol. Endocrinol. 52, R199–R222 (2014).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 115, 1343–1351 (2005).
Ji, C., Chan, C. & Kaplowitz, N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J. Hepatol. 45, 717–724 (2006).
Moran-Salvador, E. et al. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. Faseb J. 25, 2538–2550 (2011).
Boden, G. et al. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 54, 3458–3465 (2005).
Min, H. K. et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 15, 665–674 (2012).
Puri, P. et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 50, 1827–1838 (2009).
Neuschwander-Tetri, B. A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52, 774–788 (2010).
Cheung, O. et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 48, 1810–1820 (2008).
Ichimura, M. et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol. Res. 45, 458–469 (2015).
Hunt, M. C., Siponen, M. I. & Alexson, S. E. The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. Biochim. Biophys. Acta 1822, 1397–1410 (2012).
Tillander, V., Alexson, S. E. H. & Cohen, D. E. Deactivating fatty acids: acyl-CoA thioesterase-mediated control of lipid metabolism. Trends Endocrinol. Metab. 28, 473–484 (2017).
Ellis, J. M., Wong, G. W. & Wolfgang, M. J. Acyl coenzyme A thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol. Cell. Biol. 33, 1869–1882 (2013).
Adams, S. H. et al. BFIT, a unique acyl-CoA thioesterase induced in thermogenic brown adipose tissue: cloning, organization of the human gene and assessment of a potential link to obesity. Biochem. J. 360, 135–142 (2001).
Zhang, Y. et al. Targeted deletion of thioesterase superfamily member 1 promotes energy expenditure and protects against obesity and insulin resistance. Proc. Natl Acad. Sci. USA 109, 5417–5422 (2012).
Horibata, Y., Ando, H., Itoh, M. & Sugimoto, H. Enzymatic and transcriptional regulation of the cytoplasmic acetyl-CoA hydrolase ACOT12. J. Lipid Res. 54, 2049–2059 (2013).
Liu, L. et al. Isolation of mouse stromal vascular cells for monolayer culture. Methods Mol. Biol. 1566, 9–16 (2017).
Starmann, J. et al. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PloS ONE 7, e46584 (2012).
Lu, M. et al. ACOT12-dependent alteration of acetyl-CoA drives hepatocellular carcinoma metastasis by epigenetic induction of epithelial-mesenchymal transition. Cell Metab. 29, 886–900 e885 (2019).
Swarbrick, C. M. et al. Structural basis for regulation of the human acetyl-CoA thioesterase 12 and interactions with the steroidogenic acute regulatory protein-related lipid transfer (START) domain. J. Biol. Chem. 289, 24263–24274 (2014).
Wilhelm, L. P. et al. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 36, 1412–1433 (2017).
Miller, W. L. Mechanism of StAR’s regulation of mitochondrial cholesterol import. Mol. Cell. Endocrinol. 265-266, 46–50 (2007).
Rodriguez-Agudo, D. et al. Intracellular cholesterol transporter StarD4 binds free cholesterol and increases cholesteryl ester formation. J. Lipid Res. 49, 1409–1419 (2008).
Rodriguez-Agudo, D. et al. StarD5: an ER stress protein regulates plasma membrane and intracellular cholesterol homeostasis. J. Lipid Res. 60, 1087–1098 (2019).
LaVoie, H. A. et al. STARD6 is expressed in steroidogenic cells of the ovary and can enhance de novo steroidogenesis. Exp. Biol. Med. 239, 430–435 (2014).
Chakravarthy, M. V. et al. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1, 309–322 (2005).
Campbell, F. M. et al. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J. Biol. Chem. 277, 4098–4103 (2002).
Xu, H. E. et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 3, 397–403 (1999).
Ip, E., Farrell, G., Hall, P., Robertson, G. & Leclercq, I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 39, 1286–1296 (2004).
Pawlak, M., Lefebvre, P. & Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 62, 720–733 (2015).
Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 142, 711–725 e716 (2012).
Fuchs, C. D., Claudel, T. & Trauner, M. Role of metabolic lipases and lipolytic metabolites in the pathogenesis of NAFLD. Trends Endocrinol. Metab. 25, 576–585 (2014).
Ameer, F., Scandiuzzi, L., Hasnain, S., Kalbacher, H. & Zaidi, N. De novo lipogenesis in health and disease. Metabolism 63, 895–902 (2014).
Goedeke, L. et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology 68, 2197–2211 (2018).
Martinez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020).
Grabacka, M., Pierzchalska, M., Dean, M. & Reiss, K. Regulation of ketone body metabolism and the role of PPARalpha. Int. J. Mol. Sci. 17, 2093 (2016).
Foufelle, F. & Ferre, P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem. J. 366, 377–391 (2002).
Sunny, N. E., Parks, E. J., Browning, J. D. & Burgess, S. C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 14, 804–810 (2011).
Fletcher, J. A. et al. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 5, e127737 (2019).
Shimano, H. & Sato, R. SREBP-regulated lipid metabolism: convergent physiology—divergent pathophysiology. Nat. Rev. Endocrinol. 13, 710–730 (2017).
Ip, E. et al. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 38, 123–132 (2003).
Oswal, D. P. et al. Divergence between human and murine peroxisome proliferator-activated receptor alpha ligand specificities. J. Lipid Res. 54, 2354–2365 (2013).
Tachibana, K. et al. Discovery of peroxisome proliferator-activated receptor alpha (PPARalpha) activators with a ligand-screening system using a human PPARalpha-expressing cell line. J. Biol. Chem. 293, 10333–10343 (2018).
van der Veen, J. N. et al. Fenofibrate, but not ezetimibe, prevents fatty liver disease in mice lacking phosphatidylethanolamine N-methyltransferase. J. Lipid Res. 58, 656–667 (2017).
Laurin, J. et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology 23, 1464–1467 (1996).
Knight, B. L. et al. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 389, 413–421 (2005).
Huang, Z. et al. Activation of peroxisome proliferator-activated receptor-alpha in mice induces expression of the hepatic low-density lipoprotein receptor. Br. J. Pharmacol. 155, 596–605 (2008).
Balderhaar, H. J. & Ungermann, C. CORVET and HOPS tethering complexes—coordinators of endosome and lysosome fusion. J. Cell Sci. 126, 1307–1316 (2013).
van der Kant, R. et al. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 126, 3462–3474 (2013).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) [2019R1A2C2002157 and 2021R1I1A3041149].
Author information
Authors and Affiliations
Contributions
Conceptualization: Eun-Jung Jin. Data curation: Sujeong Park. Funding acquisition: Eun-Jung Jin. Investigation: Sujeong Park, In-Jeoung Baek, Jinsoo Song, Kyu Yun Jang, Chang Yeob Han, Dae Won Jun, Peter K. Kim, and Brian Raught. Writing original draft: Eun-Jung Jin.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, S., Song, J., Baek, IJ. et al. Loss of Acot12 contributes to NAFLD independent of lipolysis of adipose tissue. Exp Mol Med 53, 1159–1169 (2021). https://doi.org/10.1038/s12276-021-00648-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00648-1
This article is cited by
-
Genetic risk factors for severe and fatigue dominant long COVID and commonalities with ME/CFS identified by combinatorial analysis
Journal of Translational Medicine (2023)
