
Abstract
Obesity, diabetes mellitus, hypertension and cardiovascular disease are risk factors for chronic kidney disease (CKD) and kidney failure. Chronic, low-grade inflammation is recognized as a major pathogenic mechanism that underlies the association between CKD and obesity, impaired glucose tolerance, insulin resistance and diabetes, through interaction between resident and/or circulating immune cells with parenchymal cells. Thus, considerable interest exists in approaches that target inflammation as a strategy to manage CKD. The initial phase of the inflammatory response to injury or metabolic dysfunction reflects the release of pro-inflammatory mediators including peptides, lipids and cytokines, and the recruitment of leukocytes. In self-limiting inflammation, the evolving inflammatory response is coupled to distinct processes that promote the resolution of inflammation and restore homeostasis. The discovery of endogenously generated lipid mediators — specialized pro-resolving lipid mediators and branched fatty acid esters of hydroxy fatty acids — which promote the resolution of inflammation and attenuate the microvascular and macrovascular complications of obesity and diabetes mellitus highlights novel opportunities for potential therapeutic intervention through the targeting of pro-resolution, rather than anti-inflammatory pathways.
Key points
-
The role of inflammation in the pathogenesis of diabetes and obesity-associated kidney disease is increasingly appreciated; cytokines and pro-inflammatory lipids have important roles as drivers of inflammation and in the pathogenesis of impaired glucose tolerance, insulin resistance and diabetes.
-
The initiation and resolution of inflammation occurs via a coordinated host response, involving pro-inflammatory and anti-inflammatory or pro-resolving mediators, which are produced at the site of organ injury in a temporally controlled manner.
-
The discovery of endogenously generated lipid mediators that promote the resolution of inflammation and attenuate microvascular and macrovascular complications of diabetes and obesity highlights potential opportunities for therapeutic intervention.
-
‘Resolution pharmacology’ is a novel therapeutic paradigm that seeks to make targeted use of endogenous pro-resolving mediators to treat chronic inflammation, such as occurs in diabetic kidney disease.
Similar content being viewed by others
Introduction
In 2019, an estimated 463 million people were living with diabetes mellitus, and this figure is predicted to rise to 700 million by 2045 (ref.1). The escalating prevalence of diabetes is paralleled by rising levels of obesity (with ~650 million adults classified as obese in 2016), and increasing morbidity and mortality associated with chronic microvascular complications, cardiovascular disease and other conditions frequently associated with diabetes, including infections, dementia and cancer. The finding that reported improvements in the epidemiology of chronic complications of diabetes from 1990 to 2010 were superseded by a resurgence of complications between 2010 and 2015 is cause for concern2. Given the prevalence of diabetes and obesity, population growth and increased lifespan, the impact of diabetes and its complications on resource allocations and quality of life cannot be overemphasized.
Obesity, diabetes hypertension and cardiovascular disease are major risk factors for the development and progression of chronic kidney disease (CKD)3. Although studies clearly indicate that obesity-associated CKD can be driven by diabetes and hypertension, we now appreciate that obesity is also an independent risk factor for CKD. Both obesity and diabetes-associated CKD share common initiating events that involve interactions between metabolic and haemodynamic factors, which lead to the activation of common intracellular signalling pathways and the production of pro-inflammatory cytokines, growth factors and lipids. The key role of inflammation in the macrovascular and microvascular complications of metabolic disease has led to considerable interest in exploring this pathogenic factor as a therapeutic target.
Inflammation is a critical component of appropriate host defence. Several highly conserved pathways orchestrate its onset and progression to ensure an effective and adequately controlled pro-inflammatory response to safely remove pathogens and damaged tissue components and facilitate tissue repair. It is vital that the magnitude and duration of such responses are tightly regulated. Chronic, unresolved inflammation can result in scarring, reduced function and eventual organ failure, and is implicated in many prevalent diseases including atherosclerosis, diabetes, obesity and their associated complications in which the inflammatory response is typically provoked by endogenous stimuli in a process known as sterile inflammation. Under physiological conditions, the effective resolution of inflammation is controlled by specific mediators4. This class of endogenous molecules includes lipid mediators that are produced locally at the site of organ injury and act to limit excessive leukocyte infiltration and pro-inflammatory signals. Furthermore, they stimulate innate immune cells to kill microbes and enhance the resolution of inflammation and tissue repair by regulating macrophage function, including the phagocytic clearance of apoptotic cells at inflammatory foci and accelerating drainage into the lymphatics. Emerging insights into the unique chemical structures, receptors and bioactions of specialized pro-resolving lipid mediators (SPMs) has led to an unprecedented appreciation of the ‘off switches’ that dictate responses to inflammation in response to pathogens and sterile provocation.
Although the components that drive the inflammatory response are relatively well defined and frequently identified as targets for therapeutic intervention via inhibition of enzyme activity, receptor antagonism, antibody neutralization or other anti-inflammatory strategies, the exploitation of endogenous resolution pathways for therapeutic gain has been less well studied. However, targeting of these endogenous resolution pathways may hold promise and may avoid some of the detrimental effects associated with direct targeting of inflammatory molecules, especially in the context of compromised host defence and/or long term therapeutic modalities. Here, we review the role of endogenous lipid mediators in the resolution of inflammation and explore how insights into their function could identify new targets for therapeutic intervention in metabolic disease and complications such as CKD.
Inflammation and metabolic disease
A growing consensus suggests that inflammation is an important driver of type 1 diabetes mellitus and type 2 diabetes mellitus (T2DM) and associated vascular complications5. Inflammation in pancreatic islet β-cells results in cell depletion and loss of function6,7. Findings from experimental models and observational studies in humans demonstrate a key role for macrophages in islet β-cell inflammation in obesity and T2DM, driven largely by synergistic responses to IFNγ, TNF and IL-1β8. Islet autoimmunity might also contribute to the functional decline of β-cells during the course of T2DM9.
Studies over the past few decades have provided insights into the cellular and physiological mechanisms by which obesity-associated inflammation contributes to insulin resistance and glucose intolerance. Adipose tissues are metabolically active endocrine organs that secrete a range of adipokines, cytokines and chemokines to regulate energy homeostasis. However, in the setting of excessive calorie intake, the capacity to store fat in white adipose tissue can be exceeded, leading to ectopic lipid accumulation in non-adipose tissues, such as skeletal muscle, liver, kidneys and pancreas, contributing to an inflammatory response in these tissues10. Increased pro-inflammatory signalling has been observed in the three classic target tissues of insulin — adipose tissue, liver and muscle — as well as in the central nervous system and gastrointestinal tract11,12.
Adipose inflammation
Macrophages can adopt a spectrum of phenotypes. At the most simplistic level, classically activated, pro-inflammatory M1 macrophages are thought to promote tissue injury, whereas anti-inflammatory M2 macrophages may contribute to a reparative phenotype and the resolution of injury. In the setting of obesity, crosstalk between adipocytes and activated M1 macrophages maintains a chronic state of inflammation in adipose tissue. Activated macrophages release pro-inflammatory chemokines, including C-C motif chemokine 2 (CCL2, also known as monocyte chemoattractant protein 1 (MCP1)), TNF and IL-6, and recruit monocytes from the circulation into the site of adipose inflammation13,14. Pro-inflammatory lipids are also present within adipose tissue, including leukotriene B4 (LTB4), which is derived from arachidonic acid and enhances macrophage chemotaxis, pro-inflammatory cytokine production and insulin resistance15,16. Of note, the LTB4 receptor LTB4R1/BLT1 has been proposed as a therapeutic target in the context of obesity-associated adipose inflammation17. An important role of a particular family of lipids — ceramides — as drivers of insulin resistance has also been proposed, since inhibition of ceramide biosynthesis attenuated insulin resistance, hepatic steatosis and β-cell apoptosis in mice on an obesogenic diet18. Several other immune cell populations are also implicated in obesity-induced inflammation, including pro-inflammatory neutrophils, CD4+ T-helper (Th1) cells and CD8+ cytotoxic T cells, which are known to accumulate and proliferate in obese adipose tissue, thereby contributing to local and systemic inflammation and metabolic dysfunction of adipose tissue19,20,21.
Distinct immune cell populations, including eosinophils, reparative M2 macrophages, type 2 innate lymphoid cells, invariant natural killer cells, B cells and regulatory T cells, act to counteract these pro-inflammatory effects. These cells reside in adipose tissue under normal conditions but are reduced in obese adipose tissue22,23,24. In addition to anti-inflammatory immune cells, adipose tissue can secrete a variety of anti-inflammatory adipokines. Adiponectin, for example, is produced primarily by white adipose tissue and acts as an insulin sensitizer. Circulating levels of adiponectin are reduced in obesity and insulin resistance25. Moreover, adiponectin knockout mice show high levels of TNF in adipose tissue and in the circulation, indicating that adiponectin exerts anti-inflammatory effects26. These anti-inflammatory effects are mediated not only by suppression of TNF but also through induction of anti-inflammatory genes in human leukocytes, including genes encoding IL-10 and the interleukin 1 receptor antagonist, IL-1RA27. The role of immunometabolism as a driver of disease has been well described elsewhere11,28.
The adipose–renal axis
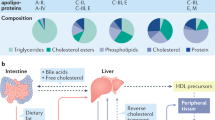
The interactions of adipose tissue with the kidney via the adipose–renal axis is important for normal kidney function as well as the response of the kidney to metabolic insults (Fig. 1). In the setting of metabolic disease, adipose hypertrophy, accumulation of M1 macrophages and the resulting insulin resistance drive adipokine dysfunction and dyslipidaemia. Elevated plasma triglycerides, fatty acids and LDL cholesterol can induce kidney dysfunction by promoting lipotoxicity, insulin resistance and pro-inflammatory pathways — an entity referred to as obesity-related glomerulopathy (ORG). In the kidneys, the ectopic accumulation of lipid occurs in all major cell types, leading to structural and functional changes in mesangial cells, podocytes and proximal tubule cells29,30. Lipid uptake is mediated in part by CD36 — a multifunctional scavenger receptor for long-chain fatty acids, oxidized lipids and advanced glycation end-products31. Several signalling pathways — including Toll-like receptor, NLRP3 inflammasome and TGFβ signalling pathways — act downstream of CD36 and have been implicated in the accumulation of lipids within the kidney. As a result, CD36 is considered an attractive therapeutic target for the prevention of kidney inflammation and fibrosis31. Adipose–renal crosstalk is also thought to be mediated by adipose tissue-derived paracrine and endocrine signals, including adipokines, cytokines and metabolites, which further impair kidney cell function. Key signals amongst these include adipose tissue-derived leptin, TNF and angiotensin II, which promote oxidative stress, inflammation and fibrotic scarring, ultimately contributing to glomerular and tubular damage32,33. Further details about adipose–renal crosstalk can be found elsewhere29,34.

In the context of excess calorie intake, the body’s capacity to store fat in adipose tissue is exceeded and multiorgan ectopic lipid accumulation ensues. This situation is characterized by adipocyte hypertrophy, insulin resistance, dysregulation of inflammatory cytokines and adipokines, and the recruitment of macrophages and other immune cells to the inflamed adipose tissue. Adipose tissue-derived leptin, tumour necrosis factor (TNF), IL-6, IL-1β, interferon-γ (IFNγ), and C-C motif chemokine 2 (CCL2/MCP1) stimulate pro-inflammatory signalling pathways in multiple organs, including the kidneys. Renal lipotoxicity occurs in all major kidney cell populations, including mesangial cells, podocytes and proximal tubule epithelial cells. Lipid accumulation within the kidney is mediated, in part, via the scavenger receptor CD36, leading to activation of multiple inflammatory pathways, including those mediated via Toll-like receptors, NLRP3 inflammasome and TGFβ signalling. The overall burden of lipotoxic effects caused by the ectopic accumulation of lipids and adipokines in the kidney results in oxidative stress, activation of the renin–angiotensin aldosterone system (RAAS), inflammation and fibrosis, ultimately leading to glomerular and tubular compartment damage. FFA, free fatty acid; iNKT, invariant natural killer; LTB4, leukotriene B4.
Diabetic kidney disease
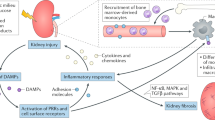
An estimated 30–40% of individuals with type 1 or type 2 diabetes develop diabetic kidney disease (DKD). Established risk factors for DKD include hyperglycaemia, hypertension, obesity and smoking35. The major pathological changes observed in DKD arise as a result of metabolic and haemodynamic stress and include: glomerular endothelial cell injury, which leads to endothelial dysfunction; mesangial matrix expansion, which drives increased intraglomerular pressure; podocyte foot process effacement, which contributes to impaired glomerular filtration; and proximal tubule damage36,37. Within the glomerulus, podocytes are considered to be the major insulin-sensitive cell population, with glucose uptake mediated via glucose transporters 1 and 4 (GLUT1 and GLUT 4, respectively); reduced insulin responsiveness of podocytes may contribute to DKD pathogenesis38,39. Beyond injury of resident kidney cell populations, infiltrating macrophages also contribute to the progression of DKD40,41. The local generation of chemokines, such as CCL2, promotes monocyte infiltration whereas upregulation of adhesion molecules facilitates the adhesion of these monocytes to endothelia. The early stages of DKD are characterized by glomerular basement membrane thickening and elevated levels of pro-inflammatory and pro-fibrotic factors (Fig. 2). Among the key pro-fibrotic factors, TGFβ1 drives extracellular matrix accumulation and fibrosis within the kidney42, whereas other members of the TGFβ superfamily, including bone morphogenetic protein agonists and antagonists, act to enhance or modulate the responses to TGFβ1 (refs43,44). Another pro-fibrotic cytokine, connective tissue growth factor (CTGF; also known as CCN2) is activated in glomerular and tubular epithelial cells by high glucose, TGFβ1 and advanced glycation end-products in experimental models of DKD as well as in kidney biopsy tissue from patients with DKD45,46.

a | As a consequence of diabetes, prolonged hyperglycaemia, dyslipidaemia and hypertension drive multiorgan damage. In the kidney, for example, pro-inflammatory signalling contributes to glomerular and tubular damage and altered kidney morphology and function. The production of growth factors (for example, transforming growth factor-β1, platelet-derived growth factor and connective tissue growth factor), as well as pro-inflammatory mediators (for example, IL-1β, IL-6, monocyte chemoattractant protein-1, nuclear factor κB and tumour necrosis factor) by infiltrating leukocytes and resident kidney cells lead to kidney damage via mesangial cell activation and proliferation, tubule epithelial cell damage and disruption of the tubular basement membrane, glomerular endothelial dysfunction, and podocyte effacement and loss. b | Key aspects of this process that might be modulated by specialized pro-resolving lipid mediators include leukocyte recruitment and activation152,181, dedifferentiation of epithelial cells within the kidney tubule126,151,181, glomerular endothelial cell and myofibroblast activation128,180,191,196, podocyte foot process effacement192, M1 to M2 macrophage phenotype shifts41,180,197 and mesangial cell activation and matrix accumulation116,118,184,198,199. AGE-RAGE, advanced glycation end-products–receptor for advanced glycation end-products; ECM, extracellular matrix; LX, lipoxin; MaR, maresin; PD, protectin; Rv, resolvin; sLX, synthetic lipoxin.
Inflammation in diabetic kidney disease
Inflammation is a feature of most kidney pathologies, and can lead to glomerulosclerosis, tubular atrophy, vascular injury and fibrosis. The principal source of pro-inflammatory mediators that drive kidney injury in the context of diabetes and obesity are infiltrating macrophages and resident kidney cells47. Several large-scale studies have demonstrated that DKD is associated with high circulating levels of pro-inflammatory cytokines, including C-reactive protein (CRP), IL-6, intercellular adhesion molecule-1 (ICAM-1), plasminogen activator inhibitor 1 and the soluble TNF receptors sTNFR-1 and sTNFR-2 (refs48,49). As a result, there is considerable interest in defining inflammatory biomarkers with prognostic value in DKD50,51. As in DKD, levels of circulating CRP, TNF and IL-6 are also elevated in ORG, whereas as mentioned above, concentrations of the anti-inflammatory adipokine adiponectin are reduced in obesity52,53. Oxidative stress is also frequently observed in DKD and ORG, driving the production of reactive oxygen species, which further exacerbates the inflammatory response. As discussed below, these findings indicate that use of anti-inflammatory and antioxidative agents may be beneficial in the treatment of DKD and ORG.
Anti-inflammatory therapy
Transcriptional analyses of human kidney biopsy tissue have been instructive in defining specific inflammatory pathways associated with DKD progression and in identifying potential therapeutic targets54,55. For example, the identification of JAK–STAT signalling55 led to initiation of a phase II trial, which showed that the JAK1,2 kinase inhibitor baricitinib decreases albuminuria in patients with T2DM at high risk of DKD56. A phase II study showed that combining a TGFβ1-specific neutralizing monoclonal antibody with conventional renin–angiotensin system (RAS) blockade does not confer any additional protective effect over that provided by RAS blockade alone in patients with DKD57.
Researchers have also targeted CCL2-mediated CCR2 activation, which promotes leukocyte infiltration and activates inflammatory signalling cascades58. Deletion of Ccl2 in mice with streptozotocin-induced diabetes attenuated macrophage accumulation and kidney damage59. Similarly, the use of a CCR2 antagonist attenuated mesangial matrix deposition and macrophage-driven glomerulosclerosis in a transgenic iNOS-Tg mouse model of diabetic glomerulosclerosis60. In patients with T2DM and kidney disease, a randomized phase II trial demonstrated the therapeutic potential of the CCR2 antagonist CCX140-B, with significant reductions in albuminuria when given in addition to standard care61. In 2017, the phase III CANTOS trial demonstrated that the human monoclonal anti-IL-1β antibody, canakinumab, reduced the rate of major adverse cardiovascular events among patients with CKD at high risk of atherosclerosis. In that trial, the observed cardiovascular benefits of canakinumab occurred in the absence of any clear benefit or harm in terms of renal events at follow-up62,63.
Of note, therapies used in the treatment of kidney disease and other associated pathologies probably exert anti-inflammatory effects. These include conventional agents, such as RAS blockers64,65, but also newer classes of glucose-lowering drugs, such as incretin-based agents (for example, glucagon-like peptide 1 (GLP-1) agonists and dipeptidyl peptidase-4 inhibitors) and sodium–glucose co-transporter 2 (SGLT2) inhibitors66. In addition to their effects on pancreatic β-cells, growing evidence suggests that some GLP-1 receptor agonists such as liraglutide exert direct effects on distinct kidney cell populations, including epithelial cells, mesangial cells and podocytes67. The actions of GLP-1 agonists extend beyond glucose lowering, with anti-inflammatory effects reported through the inhibition of NF-κB and vascular adhesion molecules (for example, CCL2/MCP1, ICAM1 and VCAM1)68. SGLT2 inhibitors prevent reabsorption of glucose and sodium from the glomerular filtrate and have demonstrated consistent and robust effects on renal protection69,70. Although the precise mechanisms of their renoprotective effects remain unclear, the haemodynamic and metabolic benefits of SGLT2 inhibition may reduce intraglomerular pressure and vascular wall tension, as well as suppress signalling by inflammatory and/or fibrotic components in the proximal tubule71,72.
In addition to pharmaceutical agents, bariatric surgery resolves various complications of diabetes, including kidney disease73,74,75. Bariatric surgery reduces circulating levels of pro-inflammatory molecules while stimulating anti-inflammatory adiponectin, which may contribute to the overall renoprotective effect of this intervention73,75,76.
Challenges of targeting inflammation
Chronic inflammation is a major driver of many common and serious diseases. To date, research has largely focused on the pro-inflammatory mediators that exacerbate such conditions, leading to the development of anti-inflammatory strategies, as described above. However, concerns remain regarding the possibility that direct targeting of inflammatory molecules might adversely affect their role in maintaining host defence. This concern might be of particular relevance in the context of diabetes, given the elevated risk of severe viral and bacterial infection associated with this entity77,78. This risk was exemplified in the CANTOS study, in which canakinumab treatment was associated with significantly more deaths attributable to infection, suggesting that anti-inflammatory strategies might compromise patient immune responses62,63. Of note, diabetes is recognized as a major risk factor for adverse outcomes following severe acute respiratory distress syndrome coronavirus 3 (SARS-CoV-2) infection and beyond antiviral drugs, great interest exists in the potential utility of anti-inflammatory strategies to manage the ‘cytokine storm’ associated with coronavirus disease 19 (COVID-19). In this context, the corticosteroid dexamethasone and anti-IL-6 receptor antibody, tocilizumab, have been investigated as potential life-saving therapies for patients with COVID-19 who are critically ill79,80. However, while anti-inflammatory approaches hold promise as adjuvant therapies for diseases such as diabetes and COVID-19, a need exists to consider alternative therapeutic approaches that modulate inflammation without suppressing innate immune responses and thereby compromising susceptibility to infection. In this context it is important to consider the processes and mediators that underpin self-limiting inflammation in an effort to mimic the resolution of this process and suppress tissue remodelling and fibrosis. As described below, the discovery of different classes of endogenous bioactive lipids, including SPMs, and fatty acid esters of hydroxy fatty acids (FAHFAs) that modulate inflammation has led to exploration of their utility in experimental models of acute and chronic inflammatory disease. Evidence also suggests that SPMs exert antibacterial and antiviral effects in the absence of immunosuppressive properties, suggesting that SPM-based therapies may be an alternative strategy to anti-inflammatory agents in the management of inflammatory metabolic disorders, such as diabetes and DKD, and potentially in the context of SARS-CoV-2 infection81.
Specialized pro-resolving lipid mediators
The precise temporal regulation of the onset, magnitude and duration of an inflammatory response reflects responses to multiple signals. In the context of self-limiting inflammation, it is now appreciated that the ‘braking switch’ for inflammation does not merely reflect a dissipation of inflammatory signals and chemokine gradients but is dynamically regulated by cytokines, peptides and lipid mediators that have distinct roles in promoting the resolution of inflammation (Fig. 3). Of particular note, promoting the resolution of inflammation is not physiologically equivalent to an anti-inflammatory response. The resolution of inflammation reflects several distinct processes that are mediated by SPMs, including the cessation of neutrophil diapedesis and the promotion of apoptotic neutrophil efferocytosis at the inflammatory focus to prevent the release of neutrophil histotoxic contents82,83,84,85. Evidence also indicates that SPMs have an important roles in the anti-inflammatory and immunomodulatory properties of stem cells, including the modulation of stem cell proliferation, migration and wound healing capacity86.

During the early initiation phase of inflammation, local production of pro-inflammatory cytokines (for example, TNF, IL-1β and IL-6) and eicosanoids promotes the recruitment of neutrophils to the site of tissue injury. Here, cyclooxygenase (COX) and lipoxygenase (LO) enzymes catalyse the conversion of the ω-6 polyunsaturated fatty acid (PUFA) arachidonic acid into a series of pro-inflammatory mediators, including prostaglandins (PGs), thromboxanes (TXs), and leukotrienes (LTs). Subsequently, a ‘class switch’ in lipid mediator biosynthesis occurs via a process involving the prostaglandins generated in the initiation phase of inflammation, leading to the synthesis of specialized pro-resolving lipid mediators (SPMs) and the resolution phase of inflammation. The ω-6 PUFA arachidonic acid produces lipoxins (LXs) and aspirin-triggered lipoxins (15-epi-LXs). The ω-3 PUFAs, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are precursors of a series of lipid mediators including resolvins (RVs), protectins (PD) and maresins (MaRs). 18S-E-series (18S-RvE1 and 18S-RvE2) resolvins and MaR conjugates in tissue regeneration (MCTR) have also been discovered, which are derived from the Rv and MaR biosynthesis pathways, respectively. Aspirin acetylation of COX-2 leads to distinct SPM biosynthetic pathways from arachidonic acid, EPA and DHA. These aspirin-triggered (AT) SPMs show the same biological actions as the native mediators.
Biosynthesis of specialized pro-resolving lipid mediators
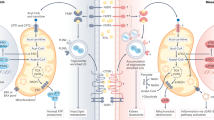
SPMs are derived from the metabolism of ω-3 and ω-6 polyunsaturated fatty acids (PUFAs)82 (Fig. 3; Fig. 4). The local biosynthesis of SPMs is a transcellular biosynthetic process, involving enzyme and receptor contributions from resident cells and recruited leukocytes at the site of injury. SPMs include lipoxins, which are generated from the ω-6 PUFA arachidonic acid, and resolvins, protectins and maresins (MaRs), which are derived from the ω-3 PUFAs eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The characterization of SPM biosynthesis has typically relied on analyses of exudates from mouse models of self-limiting inflammation. However, the production of SPMs in the resolution phase of inflammation in humans has been demonstrated using a skin blister model. That study also demonstrated that administration of exogenous SPMs accelerates the resolution of inflammation87.

The formation of specialized pro-resolving lipid mediators (SPMs) is achieved via transcellular biosynthesis. This highly coordinated process requires contributions from enzymes (for example, lipoxygenase (LO)-resident and cyclooxygenase 2 (COX-2)-resident endothelial cells, epithelial cells and polymorphonuclear cells (PMN) and platelets at the site of inflammation. SPMs can be derived from arachidonic acid (AA) (part a), eicosapentaenoic acid (EPA) (part b) and docosahexaenoic acid (DHA) (part c). 18S-RvE, 18S E-series resolvins; ASA, acetylsalicylic acid (aspirin); AT-PD1, aspirin-triggered protectin D1; AT-RvD, aspirin-triggered D-series resolvins; ATLs, aspirin-triggered lipoxins; LXA4, lipoxin A4; LXB4, lipoxin B4; RvD, D-series resolvins; RvE, E-series resolvins; MaR, maresins; PD1, protectin D1.
ω-6 PUFA-derived lipoxins
Several lipoxygenase enzymes, including 5-LO, 12-LO and 15-LO, synthesize endogenous lipoxins. 5S,6R,15S-trihydroxy-7,9,13-trans-11-cis-eicosatetraenoic acid (LXA4) and its positional isomer 5S,14R,15S-trihydroxy-6,10,12-trans-8-cis-eicosatetraenoic acid (LXB4) are the principal lipoxin species formed in mammals from the ω-6 PUFA arachidonic acid88,89,90,91. Several routes of biosynthesis have been described that require distinct lipoxygenase enzymes and transcellular metabolism. For example, sequential lipoxygenation of arachidonic acid by 15-LO in epithelial cells and monocytes, and 5-LO in neutrophils leads to lipoxin production90,91. Alternatively, lipoxin biosynthesis can occur in platelets through interactions with leukocytes whereby platelet-derived 12-LO stimulates the conversion of leukocyte-derived leukotriene A4 to LXA4 and LXB4 (ref.92). SPMs, including lipoxins, can also be produced in response to low-dose aspirin. For example, acetylation of cyclooxygenase 2 (COX-2) by aspirin triggers the production of 15-epi-lipoxin A4 (aspirin-triggered LXA4, also known as ATL)93,94.
ω-3 PUFA-derived resolvins, maresins and protectins
The ω-3 PUFAs, EPA and DHA, generate distinct classes of resolvins, MaRs and protectins through transcellular interactions between infiltrating leukocytes and tissue-resident endothelial or epithelial cells at the site of inflammation (Fig. 4). D-series resolvins (RvDs) are synthesized from DHA-derived 17S-hydroperoxy-DHA via 15-LO, which is then converted by 5-LO to RvD1–RvD4 (ref.84). Aspirin-triggered D-series resolvins (AT-RvD) can also be produced through aspirin-acetylated COX-2-mediated conversion of DHA to 17R-hydroxy-DHA, which is then transformed via neutrophil-derived 5-LO to form AT-RvD1-4 epimers84. EPA generates E-series resolvins (RvE) through transcellular mechanisms, with two stereoselective biosynthesis pathways described. For example, in vascular endothelial cells, aspirin-acetylated COX-2 converts EPA to 18R-hydroxy-EPA (18R-HEPE), which is then released and converted to RvE1 and RvE2 by 5-LO in neutrophils95. By contrast, conversion of EPA to 18S-hydroxy-EPA (18S-HEPE) by aspirin-acetylated COX-2, leads to 5-LO-mediated biosynthesis of 18S-RvE1 and 18S-RvE2 (ref.96). Interestingly, comparison of RvE1 and 18S-RvE1 epimers in this series indicated that 18S-RvE1 displays an enhanced receptor affinity and potency, but undergoes more rapid metabolic inactivation compared with RvE1 (ref.96). Alternatively, EPA can be converted to resolvins (RvE1 and RvE2) via aspirin-independent pathways by cytochrome P450 and 5-LO97.
MaRs are generated by the sequential conversion of DHA by 12-LO to a 14-hydroxyperoxide intermediate that undergoes epoxidation to form 13S,14S-epoxy-MaR. This epoxide precursor is enzymatically hydrolysed to form MaR1 (refs98,99). A novel family of SPMs — termed MaR conjugates in tissue regeneration — are also derived from the MaR biosynthesis pathway100.
The protectin family of SPMs are thought to be produced through the actions of 15-LO or aspirin-acetylated COX-2 on DHA, producing the D1 protectins PD1 and NPD1 and the aspirin-triggered protectin AT-PD1, respectively101. In the past few years, several additional anti-inflammatory SPMs have been identified, including ω-3 docosapentaenoic acid (DPA)-derived SPMs (for example, the protectin n-3 DPA series, resolvin n-3 DPA series and the MaR n-3 DPA series)102,103,104,105,106.
Bioactions of SPMs
SPMs typically exert their responses through actions on G protein-coupled receptors107,108. The lipoxins were the first SPMs identified and thus most is known about the molecular mechanisms underpinning their bioactions. LXA4 and 15-epi-LXA4 bind to the LXA4 receptor N-formyl peptide receptor 2 (ALX/FPR2), a member of the formyl peptide receptor family. This receptor is uncommonly promiscuous as it binds both lipids and peptides (for example, it binds LXA4 and RvD1 as well as annexin A1-derived peptides and serum amyloid A), and ALX/FPR2 agonists can elicit both pro-resolving effects (for example, in the case of LXA4 and annexin A1-derived peptides) or pro-inflammatory effects (for example, in the case of serum amyloid A)109,110. The structural and molecular mechanisms that underlie these diverse functional responses include biased agonism-induced receptor homodimerization and heterodimerization, recruitment of β-arrestin and receptor internalization111,112,113 (Fig. 5). The proposed role of ALX/FPR2 as a master regulator of effective resolution in inflammation is highlighted by the finding that persistent inflammation is observed in mice with deletion of functional ALX/FPR2 (ref.114). In a study of patients with severe trauma, the presence of a single nucleotide polymorphism in the promoter region of ALX/FPR2 was associated with decreased gene and protein expression of ALX/FPR2 and enhanced susceptibility to sepsis115, suggesting a failure to resolve inflammation coupled with compromised immunomodulatory responses.

Both pro-inflammatory and anti-inflammatory (pro-resolving) signals are mediated by the G protein-coupled receptor ALX/FPR2 (referred to as FPR2 in the figure). Endogenous ligands at this receptor include ω-3 and ω-6 polyunsaturated fatty acid-derived anti-inflammatory lipids (lipoxin A4 (LXA4), 15-epi-lipoxin A4 (ATL) and resolvin D1 (RvD1). Protein ligands with anti-inflammatory effects (for example, annexin A1 (AnxA1); annexin A1-derived peptide Ac2-26) and pro-inflammatory effects (for example, serum amyloid A (SAA)) have also been described. Ligand–receptor interactions are believed to occur at distinct receptor pockets, with different affinities. ALX/FPR2 homodimers and ALX/FPR2–FPR1 heterodimers eliciting pro-resolving and pro-inflammatory effects have been described. ALX/FPR2 receptor internalization is essential for LXA4-stimulated and AnxA1-stimulated phagocytosis. Crosstalk has also been reported between ALX/FPR2 and several receptor kinases implicated in organ fibrosis, including transforming growth factor-β1 receptor (TGFβR1), platelet-derived growth factor receptor (PDGFR) and epidermal growth factor receptor (EGFR).
Receptor crosstalk between ALX/FPR2 and serine or tyrosine receptor kinases for growth factors such as TGFβ, PDGF, EGF and VEGF has also been described, and may contribute to the alleviation of organ fibrosis by lipoxins116,117,118. The D-series resolvin RvD1 is an agonist of ALX/FPR2 and G protein-coupled receptor 32 (DRV1/GPR32), whereas RvD2 binds G protein-coupled receptor 18 (DRV2/GPR18)109,119,120. The E-series resolvin RvE1 binds and activates chemerin receptor 23 (ERV1/ChemR23)95. RvE1 and RvE2 also act as competitive antagonists of LTB4 and its receptor, LTB4R1/BLT1 (refs121,122).
ALX/FPR2 was initially found to be expressed by leukocytes, but was subsequently shown to be expressed by cells of diverse lineages throughout the body123,124. ALX/FPR2 is expressed throughout the kidneys, with expression reported in fibroblasts, myofibroblasts, mesangial cells, proximal tubule epithelia, macrophages and endothelial cells116,125,126,127,128. Several studies have sought to investigate the role of ALX/FPR2 in a range of diseases. For example, upregulation of ALX/FPR2 has been reported in atherosclerotic lesions129,130 compared with levels in healthy control tissue; conversely, ALX/FPR2 expression is decreased in human abdominal aortic aneurysm lesions compared with levels in healthy controls131.
Although our understanding of the downstream signalling events following receptor activation by SPMs remains incomplete, several pathways have been described. These include SPM-mediated attenuation of ERK and NF-κB activation, as well as inhibition of p38 mitogen-activated protein kinase95,132. Several novel micro-RNA mechanisms have also been identified that may in part explain the bioactions of SPMs, including LXA4-mediated upregulation of let-7, and mir-126-5p126,133,134, and RvD1-mediated regulation of miR-21, miR-146b, miR-219 and miR-155 (refs135,136). Of note, let-7 target networks are dysregulated in renal biopsy samples from patients with DKD, and let-7 mimetics have been proposed as a strategy to target pro-fibrotic factors (such as collagen and TGFβ receptor 1) and pro-inflammatory factors (for example, IL-6, IL-1β, TNF and NF-κB) that are upregulated in DKD126,129,133,137. SPMs stimulate monocyte recruitment and promote non-phlogistic (that is, non-inflammatory) phagocytosis of apoptotic neutrophils at an inflammatory site138 (Fig. 6). SPMs also act to limit neutrophil recruitment139, and may exert direct effects on lymphocyte populations, by enhancing B cell differentiation and activation140, natural killer cell-mediated clearance of neutrophils and eosinophils141 and activation of regulatory T cells142. In addition to their effects on recruited immune cells, SPMs also exert protective effects on local tissue-resident cell populations, including kidney mesangial, podocyte and tubule epithelial cells, as well as vascular smooth muscle cells and endothelial cells, as discussed below143 (Fig. 2b).

Specialized pro-resolving lipid mediators (SPMs) are biosynthesized at the site of tissue injury via interactions between resident endothelial and/or epithelial cells and infiltrating polymorphonuclear cells (PMN). SPMs exert their actions via engaging with specific pro-resolving G protein-coupled receptors expressed on resident tissue cells and PMNs. Pro-resolving SPM bioactions include attenuation of endothelial cell activation, blocking of neutrophil extravasation, promoting non-phlogistic monocyte recruitment, promoting an (M2) resolving macrophage phenotype, and stimulating neutrophil apoptosis and macrophage efferocytosis. SPMs are produced by, and act on, stem cells, and evidence suggests that SPMs mediate the immunomodulatory and anti-inflammatory effects of these cells. PGs, prostaglandins; LTs, leukotrienes.
Resolution pharmacology
Resolution pharmacology is a therapeutic paradigm that seeks to make targeted use of endogenous pro-resolving mediators to treat acute and chronic inflammation. For example, supplementation of SPM precursors has been investigated as a therapeutic approach in acute and chronic conditions that have an inflammatory component. This approach is based on a growing body of evidence which shows that circulating levels of SPM are depleted in patients with inflammatory and immune disorders such as severe asthma and cystic fibrosis, suggesting that a resolution deficit contributes to these pathologies144. Interestingly, transgenic mice with overexpression of arachidonate 5-lipoxygenase-activating protein (ALOX5AP) to enhance LXA4 generation by adipose tissue were demonstrated to be protected against diet-induced obesity through browning of white adipose tissue and prevention of insulin resistance, hepatic steatosis and inflammation145. Similarly, the exogenous administration of RvD1 in obese, diabetic, db/db mice lead to improvements in glucose tolerance and adipose insulin sensitivity, which was associated with increased adiponectin and reduced IL-6 production within adipose tissue146.
Challenges remain in the exploitation of the therapeutic potential of SPMs. Many of the endogenously generated mediators are unsuitable as drugs as they are chemically and or biologically unstable and their synthesis is challenging. To circumvent this challenge, synthetic SPMs and agonists of SPM receptors have been developed, some of which have shown potential in preclinical models of acute inflammation, and cardiac and kidney disease. LXA4 and ATL mimetics show enhanced pharmacokinetic properties and efficacy compared with endogenous lipoxins in vivo147,148,149. For example, LXA4 mimics have demonstrated an ability to promote macrophage-mediated neutrophil phagocytosis, and attenuate kidney ischaemia–reperfusion injury (IRI) and obesity-induced adipose inflammation41,110,150,151,152. The replacement of the triene core of LXA4 with a benzene ring has led to the generation of several aromatic mimetics153 that are protective in experimental models of diabetes-associated kidney disease and atherosclerosis, with evidence that these mimetics can halt and even reverse established disease129,154. Building on these findings, several synthetic imidazole and oxazole-containing lipoxin mimics have demonstrated potent effects in mouse models of peritonitis and arthritis147,155. Other synthetic agonists of ALX/FPR2 have shown efficacy in myocardial IRI156,157, and in preventing the development of experimental heart failure158. Beyond lipoxins, great interest exists in the generation of synthetic resolvins. An RvE1 analogue, 19-(p-fluorophenoxy)-RvE1, which is designed to resist rapid metabolic inactivation, reduced inflammation in an in vivo model of zymosan-induced peritonitis159,160,161. Similarly, a metabolically stable analogue of aspirin-triggered RvD1, 17R-hydroxy-19-p-fluorophenoxy-resolvin D1 methyl ester, demonstrated efficacy in experimental models of lung injury by modulating the inflammatory milieu161.
Several clinical trials have offered further insight into the potential of SPM therapy. In a randomized controlled trial of SPMs in infants with eczema, administration of AT-LXA4 and a synthetic mimic of LXB4, 15R/S-methyl-LXB4, significantly reduced eczema severity162. Similar protective findings have been reported for an inhaled LXA4 mimic, 5S,6R-LXA4 methyl ester, in childhood asthma, suggesting that synthetic SPMs might represent a promising therapeutic strategy for asthma163. Synthetic mimetics of the resolvin RvE1, the MaR1 and the protectin NPD1 are also being explored as therapies in dry eye syndrome, neurodegeneration and hearing loss164.
SPMs in metabolic and kidney disease
As is the case with many common and complex diseases, defects in the resolution phase of inflammation may underlie the pathophysiology of diabetes mellitus and associated complications. Thus, interest exists in understanding the role of both pro-inflammatory and anti-inflammatory factors in diabetic complications, and whether strategies that promote the resolution of inflammation might alleviate these complications.Approaches that have involved dietary supplementation with fish oils that are enriched in ω-3 and ω-6 PUFAs and their metabolites have yielded conflicting results in individuals with DKD165,166,167. Concerns also exist regarding the purity of such supplements and the potential risk of increasing LDL levels168. Clinical responses to high-purity SPM precursors, such as EPA administered either alone or together with DHA have been more encouraging. For example, the REDUCE-IT trial demonstrated that administration of a highly purified EPA ethyl ester reduced the risk of cardiovascular disease in patients with established cardiovascular disease or risk factors such as diabetes169,170.
Several in vivo and in vitro studies have also suggested that SPMs may confer direct protective effects on pancreatic islet β-cells. Oral supplementation of arachidonic acid enhanced plasma LXA4 levels and insulin sensitivity, and attenuated pancreatic tissue NF-κB activation in a rat model of T2DM171. In support of this finding, although administration of a second-generation synthetic EPA-ester — epeleuton — did not reduce liver stiffness and alanine aminotransferase activity in a phase IIa study in patients with non-alcoholic fatty liver disease, secondary analyses revealed intriguing metabolic effects. Specifically, the study investigators observed decreased levels of plasma triglycerides, vLDL and total cholesterol together with decreases in plasma glucose, HbA1c, insulin resistance and markers of endothelial activation172. These findings are further supported by experimental evidence that EPA-derived RvE1 decreases hyperinsulinaemia and hyperglycaemia in obese mice173.
Impaired wound healing is a well-described clinical manifestation of both type 1 and type 2 diabetes, and stimulating the resolution of inflammation with SPMs may represent a strategy to promote wound healing in this context174,175. In response to a skin wound, non-diabetic Balb/C mice produce the DHA-derived mediator 14S,21R-dihydroxy-DHA (14S,21R-diHDHA)176. However, obese, diabetic mice demonstrate reduced levels of 14S,21R-diHDHA at the wound site, whereas treatment of the wounds with this DHA-derived SPM stimulates wound closure177,178, suggesting that synthesis of this lipid mediator may be required to stimulate effective wound repair.
A 2017 study found lower circulating levels of ATL in patients with DKD than in those with non-diabetic kidney disease, which were restored by 12 months of low-dose aspirin treatment179. These findings indicate that deficits in the production of SPMs may underlie DKD and that restoration of these lipid mediators could represent a potential therapeutic approach. In line with this proposal, we have demonstrated renoprotective effects of an ATL analogue in models of ischaemic injury151,152. Moreover, in a unilateral ureteric obstruction model of kidney tubulointerstitial fibrosis and inflammation, administration of LXA4 and a synthetic benzo-LXA4 analogue attenuated kidney injury and macrophage infiltration180. Specifically, the lipoxins prevented the deposition of collagen and TGFβ1-induced fibroblast activation and proliferation. Protective effects of LXA4 and benzo-LXA4 have also been demonstrated in a model of ORG in association with a decrease in glomerular expansion and oxidative stress41. Although lipoxin administration did not restore obesity-mediated impairment of glucose tolerance, it reduced serum levels of alanine aminotransferase and hepatic triglycerides and promoted an M2-macrophage (CD206+) phenotype in adipose tissue. These lipoxin-mediated protective effects were observed in both wild-type and adiponectin-knockout mice, suggesting that they are not mediated via adiponectin signalling. We have also demonstrated renoprotective effects of LXA4 and benzo-LXA4 in ApoE-knockout mice with streptozotocin-induced diabetes154. At a cellular level, lipoxins exert their pro-resolving effects on distinct kidney cell types, including mesangial cells, kidney fibroblasts and tubule epithelial cells, and attenuate the effects of several pro-fibrotic cytokines, including PDGF, TGFβ1 and CTGF116,118,126 (Fig. 2b).
Other SPM classes have also demonstrated renoprotective effects. In a model of kidney IRI, administration of exogenous D-series resolvins and the protectin PD1 attenuated leukocyte infiltration, ultimately preserving kidney function181. Interestingly, dietary ω-3 PUFA increases endogenous kidney levels of protectin D1 and 17-hydroxy-DHA, the latter being a metabolic marker for D-series resolvins182. A 2019 study also demonstrated that MaR1 is renoprotective in a mouse model of IRI, attenuating inflammation and oxidative stress by inhibiting the TLR4–MAPK–NF-κB pathway and activating the Nrf2 pathway183. In vitro studies demonstrated the ability of MaR1 to inhibit high glucose-induced fibrotic responses in mesangial cells, suggesting that MaR1 may also function as an antifibrotic molecule in this setting184. Together, these data suggest that SPMs may have additional cellular sites of action in the kidney beyond their effects on leukocyte trafficking that contribute to their pro-resolving activity. However, further investigation in this area is warranted (Fig. 2; Table 1).
Branched fatty acid esters of hydroxy fatty acids
In addition to SPMs, another class of endogenous lipids — branched FAHFAs — have also been found to have beneficial anti-inflammatory and antidiabetic effects185. Production of FAHFAs occurs at sites of organ lipogenesis, such as adipose, kidney and liver tissues, where dietary fatty acids (for example, palmitic, palmitoleic, stearic, oleic and linoleic acids, and DHA) are esterified to form hydroxy fatty acids185,186. FAHFAs such as 13-DHAHLA, 9-DHAHLA (formed by DHA esterification to 9- and 13-hydroxyoctadecadienoic acid (HLA)), and 14-DHAHDHA (formed by DHA esterification to 14-hydroxy-DHA (HDHA)) are synthesized by the adipocytes of obese mice and of patients with diabetes in response to dietary intervention with ω-3 PUFAs. These FAHFAs then act to reduce macrophage activation and enhance phagocytosis186. Another FAHFA family member, branched esters of palmitic acid hydroxystearic acid (PAHSA), has also been shown to exert antidiabetic and anti-inflammatory effects185,187. Specifically, treatment of diet-induced obese mice with 5-PAHSA or 9-PAHSA isomers enhanced insulin-stimulated glucose uptake in adipose tissue via increased secretion of GLP-1 and insulin185. That study also showed that PAHSA concentrations are significantly lower in insulin-resistant individuals than in individuals who are insulin-sensitive. Mechanistic insights indicate that 5-PAHSA is regulated by adipose triglyceride lipase and stimulates lipogenesis188. Interestingly, investigation of the in vivo metabolic benefits of two other classes of FAHFAs — 9-PAHPA and 9-OAHPA — in healthy mice demonstrated their ability to increased insulin sensitivity without modifying glucose tolerance189. However, the potential metabolic benefits of this family of FAHFAs were offset by evidence of hepatic steatosis and fibrosis. Thus, it is evident that additional studies are required to define the precise mechanisms of action of the FAHFAs and their potential role in DKD.
Conclusions
Despite major advances in our understanding of the pathophysiology of kidney disease in diabetes and obesity and the emergence of promising new therapies, a great need remains for additional interventions. A large body of experimental evidence now suggests that endogenous pro-resolving lipids have exciting potential as adjuvant therapeutics in diabetes and its complications, and may have benefits over conventional targeting of pro-inflammatory pathways that may have detrimental effects on immune function and tissue repair. From a therapeutic perspective, one major limitation of approaches to harnessing endogenous SPMs is their relative instability — a concern that could be circumvented through the design of synthetic mimics. The evidence that lipoxins can attenuate the development of experimental DKD and reverse established kidney and macrovascular disease demonstrates proof-of-principle from the perspective of pharmacodynamic efficacy. Advances in the modular synthesis of compounds with enhanced pharmacokinetic properties hold further promise for the application of these agents in diabetic complications and is the focus of current investigations as adjuncts to conventional therapies. Our understanding of genetic profiles within SPM biosynthesis pathways is poorly understood, and further investigations are warranted to ensure that personalized administration of SPMs is considered. Furthermore, the extent to which existing therapies modulate inflammation in diabetes and obesity remains unclear, and indeed whether levels of pro-resolving lipids are changed in response to existing therapies. A future challenge will be to determine whether a deficit exists in the production of these lipid classes in patients with diabetes. As our understanding of the endogenous pathways that produce different classes of pro-resolving lipids improves, it is anticipated that novel therapeutic strategies may be developed to target these pathways in order to alleviate kidney disease in individuals with diabetes and obesity.
References
Saeedi, P. et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 157, 107843 (2019).
Gregg, E. W., Hora, I. & Benoit, S. R. Resurgence in diabetes-related complications. 321, 1867–1868 (2019).
Ejerblad, E. et al. Obesity and risk for chronic renal failure. J. Am. Soc. Nephrol. 17, 1695–1702 (2006).
Fullerton, J. N. & Gilroy, D. W. Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551–567 (2016).
Skyler, J. S. et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 66, 241–255 (2017).
Usmani-Brown, S. et al. β cell responses to inflammation. Mol. Metab. 27S, S104–S113 (2019).
Imai, Y., Dobrian, A. D., Morris, M. A. & Nadler, J. L. Islet inflammation: a unifying target for diabetes treatment? Trends Endocrinol Metab. 24, 351–360 (2013).
Ying, W., Fu, W., Lee, Y. S. & Olefsky, J. M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 16, 81–90 (2020).
Brooks-Worrell, B. M., Boyko, E. J. & Palmer, J. P. Impact of islet autoimmunity on the progressive β-cell functional decline in type 2 diabetes. Diabetes Care 37, 3286–3293 (2014).
Samuel, V. T. & Shulman, G. I. Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871 (2012).
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017).
da Silva Rosa, S. C., Nayak, N., Caymo, A. M. & Gordon, J. W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 8, e14607 (2020).
Wellen, K. E. & Hotamisligil, G. S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Invest. 112, 1785–1788 (2003).
Kamei, N. et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 281, 26602–26614 (2006).
Li, P. et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 21, 239–247 (2015).
Ying, W. et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J. Clin. Invest. 127, 1019–1030 (2017).
Spite, M. et al. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J. Immunol. 187, 1942–1949 (2011).
Chaurasia, B. et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365, 386–392 (2019).
Wu, H. et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 115, 1029–1038 (2007).
Nishimura, S. et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 15, 914–920 (2009).
Talukdar, S. et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 18, 1407–1412 (2012).
Wang, Q. & Wu, H. T cells in adipose tissue: critical players in immunometabolism. Front. Immunol. 9, 2509 (2018).
Nishimura, S. et al. Adipose natural regulatory B cells negatively control adipose tissue inflammation. Cell Metab. 18, 759–766 (2013).
Molofsky, A. B. et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 210, 535–549 (2013).
Arita, Y. et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 257, 79–83 (1999).
Maeda, N. et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med. 8, 731–737 (2002).
Wolf, A. M., Wolf, D., Rumpold, H., Enrich, B. & Tilg, H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem. Biophys. Res. Commun. 323, 630–635 (2004).
Lee, Y. S., Wollam, J. & Olefsky, J. M. An integrated view of immunometabolism. Cell 172, 22–40 (2018).
de Vries, A. P. et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2, 417–426 (2014).
Gai, Z. et al. Lipid accumulation and chronic kidney disease. Nutrients 11, 722 (2019).
Yang, X. et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 13, 769–781 (2017).
Li, C. et al. Intrarenal renin-angiotensin system mediates fatty acid-induced ER stress in the kidney. Am. J. Physiol. Ren. Physiol. 310, F351–F363 (2016).
Sieber, J. et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am. J. Physiol. Ren. Physiol. 299, F821–F829 (2010).
Zhu, Q. & Scherer, P. E. Immunologic and endocrine functions of adipose tissue: implications for kidney disease. Nat. Rev. Nephrol. 14, 105–120 (2018).
Alicic, R. Z., Rooney, M. T. & Tuttle, K. R. Diabetic kidney disease: challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 12, 2032–2045 (2017).
Russell, N. D. & Cooper, M. E. 50 years forward: mechanisms of hyperglycaemia-driven diabetic complications. Diabetologia 58, 1708–1714 (2015).
Reidy, K., Kang, H. M., Hostetter, T. & Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Invest. 124, 2333–2340 (2014).
Coward, R. J. et al. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes 56, 1127–1135 (2007).
Welsh, G. I. et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 12, 329–340 (2010).
Tang, P. M., Nikolic-Paterson, D. J. & Lan, H. Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 15, 144–158 (2019).
Borgeson, E. et al. Lipoxin A4 attenuates obesity-induced adipose inflammation and associated liver and kidney disease. Cell Metab. 22, 125–137 (2015).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Zeisberg, M. et al. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968 (2003).
Brazil, D. P., Church, R. H., Surae, S., Godson, C. & Martin, F. BMP signalling: agony and antagony in the family. Trends Cell Biol. 25, 249–264 (2015).
Ito, Y. et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 53, 853–861 (1998).
Murphy, M. et al. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J. Biol. Chem. 274, 5830–5834 (1999).
Mathew, A. V., Okada, S. & Sharma, K. Obesity related kidney disease. Curr. Diabetes Rev. 7, 41–49 (2011).
Groop, P. H. et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes 58, 1651–1658 (2009).
Lopes-Virella, M. F. et al. Baseline markers of inflammation are associated with progression to macroalbuminuria in type 1 diabetic subjects. Diabetes Care 36, 2317–2323 (2013).
Niewczas, M. A. et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 25, 805–813 (2019).
Scurt, F. G. et al. Systemic inflammation precedes microalbuminuria in diabetes. Kidney Int. Rep. 4, 1373–1386 (2019).
Visser, M., Bouter, L. M., McQuillan, G. M., Wener, M. H. & Harris, T. B. Elevated C-reactive protein levels in overweight and obese adults. JAMA 282, 2131–2135 (1999).
Park, H. S., Park, J. Y. & Yu, R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-α and IL-6. Diabetes Res. Clin. Pract. 69, 29–35 (2005).
Woroniecka, K. I. et al. Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369 (2011).
Berthier, C. C. et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 58, 469–477 (2009).
Tuttle, K. R. et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a phase 2 randomized controlled clinical trial. Nephrol. Dial. Transpl. 33, 1950–1959 (2018).
Voelker, J. et al. Anti-TGF-β1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 28, 953–962 (2017).
Giunti, S., Barutta, F., Perin, P. C. & Gruden, G. Targeting the MCP-1/CCR2 system in diabetic kidney disease. Curr. Vasc. Pharmacol. 8, 849–860 (2010).
Chow, F. Y. et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 69, 73–80 (2006).
Kanamori, H. et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 360, 772–777 (2007).
de Zeeuw, D. et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. 3, 687–696 (2015).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Verma, S., Mathew, V. & Farkouh, M. E. Targeting inflammation in the prevention and treatment of type 2 diabetes: insights from CANTOS. J. Am. Coll. Cardiol. 71, 2402–2404 (2018).
Merino, A. et al. Losartan prevents the development of the pro-inflammatory monocytes CD14+CD16+ in haemodialysis patients. Nephrol. Dial. Transpl. 27, 2907–2912 (2012).
Gamboa, J. L. et al. Comparative effects of angiotensin-converting enzyme inhibition and angiotensin-receptor blockade on inflammation during hemodialysis. J. Am. Soc. Nephrol. 23, 334–342 (2012).
Kahn, S. E., Cooper, M. E. & Del Prato, S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383, 1068–1083 (2014).
Kawanami, D. & Takashi, Y. GLP-1 receptor agonists in diabetic kidney disease: from clinical outcomes to mechanisms. Front. Pharmacol. 11, 967 (2020).
Alicic, R. Z., Cox, E. J., Neumiller, J. J. & Tuttle, K. R. Incretin drugs in diabetic kidney disease: biological mechanisms and clinical evidence. Nat. Rev. Nephrol. 17, 227–244 (2021).
Neal, B. et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 377, 644–657 (2017).
Perkovic, V. et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 380, 2295–2306 (2019).
Sasson, A. N. & Cherney, D. Z. Renal hyperfiltration related to diabetes mellitus and obesity in human disease. World J. Diabetes 3, 1–6 (2012).
Heerspink, H. J. L. et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 62, 1154–1166 (2019).
Neff, K. J. et al. The effect of bariatric surgery on renal function and disease: a focus on outcomes and inflammation. Nephrol. Dial. Transpl. 28 (Suppl 4) iv73–iv82 (2013).
Brolin, R. E. Bariatric surgery and long-term control of morbid obesity. JAMA 288, 2793–2796 (2002).
Fenske, W. K. et al. Effect of bariatric surgery-induced weight loss on renal and systemic inflammation and blood pressure: a 12-month prospective study. Surg. Obes. Relat. Dis. 9, 559–568 (2013).
Canney, A. L. et al. Improvements in diabetic albuminuria and podocyte differentiation following Roux-en-Y gastric bypass surgery. Diab. Vasc. Dis. Res. 17, 1479164119879039 (2020).
Knapp, S. Diabetes and infection: is there a link? – A mini-review. Gerontology 59, 99–104 (2013).
Carey, I. M. et al. Risk of infection in type 1 and type 2 diabetes compared with the general population: a matched cohort study. Diabetes Care 41, 513–521 (2018).
Mahase, E. Covid-19: Low dose steroid cuts death in ventilated patients by one third, trial finds. BMJ 369, m2422 (2020).
Salama, C. et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 384, 20–30 (2021).
Pal, A., Gowdy, K. M., Oestreich, K. J., Beck, M. & Shaikh, S. R. Obesity-driven deficiencies of specialized pro-resolving mediators may drive adverse outcomes during SARS-CoV-2 infection. Front. Immunol. 11, 1997 (2020).
Serhan, C. N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014).
Serhan, C. N. et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332 (2007).
Serhan, C. N. et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 (2002).
Serhan, C. N. & Petasis, N. A. Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943 (2011).
Romano, M., Patruno, S., Pomilio, A. & Recchiuti, A. Proresolving lipid mediators and receptors in stem cell biology: concise review. Stem Cell Transl. Med. 8, 992–998 (2019).
Motwani, M. P. et al. Novel translational model of resolving inflammation triggered by UV-killed E. coli. J. Pathol. Clin. Res. 2, 154–165 (2016).
Serhan, C. N., Hamberg, M. & Samuelsson, B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl Acad. Sci. USA 81, 5335–5339 (1984).
Serhan, C. N. On the relationship between leukotriene and lipoxin production by human neutrophils: evidence for differential metabolism of 15-HETE and 5-HETE. Biochim. Biophys. Acta 1004, 158–168 (1989).
Serhan, C. N., Hamberg, M., Samuelsson, B., Morris, J. & Wishka, D. G. On the stereochemistry and biosynthesis of lipoxin B. Proc. Natl Acad. Sci. USA 83, 1983–1987 (1986).
Serhan, C. N. et al. Lipoxin A. Stereochemistry and biosynthesis. J. Biol. Chem. 261, 16340–16345 (1986).
Serhan, C. N. & Sheppard, K. A. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Invest. 85, 772–780 (1990).
Claria, J., Lee, M. H. & Serhan, C. N. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol. Med. 2, 583–596 (1996).
Claria, J. & Serhan, C. N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl Acad. Sci. USA 92, 9475–9479 (1995).
Arita, M. et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722 (2005).
Oh, S. F., Pillai, P. S., Recchiuti, A., Yang, R. & Serhan, C. N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Invest. 121, 569–581 (2011).
Arita, M., Clish, C. B. & Serhan, C. N. The contributions of aspirin and microbial oxygenase to the biosynthesis of anti-inflammatory resolvins: novel oxygenase products from omega-3 polyunsaturated fatty acids. Biochem. Biophys. Res. Commun. 338, 149–157 (2005).
Dalli, J. et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 27, 2573–2583 (2013).
Abdulnour, R. E. et al. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc. Natl Acad. Sci. USA 111, 16526–16531 (2014).
Dalli, J., Chiang, N. & Serhan, C. N. Identification of 14-series sulfido-conjugated mediators that promote resolution of infection and organ protection. Proc. Natl Acad. Sci. USA 111, E4753–E4761 (2014).
Hansen, T. V., Vik, A. & Serhan, C. N. The protectin family of specialized pro-resolving mediators: potent immunoresolvents enabling innovative approaches to target obesity and diabetes. Front. Pharmacol. 9, 1582 (2018).
Serhan, C. N., Chiang, N. & Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Aspects Med. 64, 1–17 (2018).
Vik, A., Dalli, J. & Hansen, T. V. Recent advances in the chemistry and biology of anti-inflammatory and specialized pro-resolving mediators biosynthesized from n-3 docosapentaenoic acid. Bioorg. Med. Chem. Lett. 27, 2259–2266 (2017).
Hansen, T. V., Dalli, J. & Serhan, C. N. The novel lipid mediator PD1n-3 DPA: an overview of the structural elucidation, synthesis, biosynthesis and bioactions. Prostaglandins Other Lipid Mediat. 133, 103–110 (2017).
Dalli, J., Colas, R. A. & Serhan, C. N. Novel n-3 immunoresolvents: structures and actions. Sci. Rep. 3, 1940 (2013).
Drouin, G., Rioux, V. & Legrand, P. The n-3 docosapentaenoic acid (DPA): A new player in the n-3 long chain polyunsaturated fatty acid family. Biochimie 159, 36–48 (2019).
Pirault, J. & Back, M. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front. Pharmacol. 9, 1273 (2018).
Sansbury, B. E. & Spite, M. Resolution of acute inflammation and the role of resolvins in immunity, thrombosis, and vascular biology. Circ. Res. 119, 113–130 (2016).
Krishnamoorthy, S. et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl Acad. Sci. USA 107, 1660–1665 (2010).
Maderna, P. et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 24, 4240–4249 (2010).
Zhuang, Y. et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nat. Commun. 11, 885 (2020).
Cooray, S. N. et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl Acad. Sci. USA 110, 18232–18237 (2013).
Gobbetti, T. et al. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc. Natl Acad. Sci. USA 111, 18685–18690 (2014).
Dufton, N. et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 184, 2611–2619 (2010).
Zhang, H. et al. The common promoter polymorphism rs11666254 downregulates FPR2/ALX expression and increases risk of sepsis in patients with severe trauma. Crit. Care 21, 171 (2017).
McMahon, B. et al. Lipoxin, leukotriene, and PDGF receptors cross-talk to regulate mesangial cell proliferation. FASEB J. 16, 1817–1819 (2002).
Mitchell, D. et al. Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol. 164, 937–946 (2004).
Rodgers, K., McMahon, B., Mitchell, D., Sadlier, D. & Godson, C. Lipoxin A4 modifies platelet-derived growth factor-induced pro-fibrotic gene expression in human renal mesangial cells. Am. J. Pathol. 167, 683–694 (2005).
Norling, L. V., Dalli, J., Flower, R. J., Serhan, C. N. & Perretti, M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler. Thromb. Vasc. Biol. 32, 1970–1978 (2012).
Chiang, N., Dalli, J., Colas, R. A. & Serhan, C. N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 212, 1203–1217 (2015).
Oh, S. F. et al. Resolvin E2 formation and impact in inflammation resolution. J. Immunol. 188, 4527–4534 (2012).
Arita, M. et al. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917 (2007).
Becker, E. L. et al. Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 292, 129–135 (1998).
Migeotte, I., Communi, D. & Parmentier, M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor. Rev. 17, 501–519 (2006).
Neymeyer, H. et al. Activation of annexin A1 signalling in renal fibroblasts exerts antifibrotic effects. Acta Physiol. 215, 144–158 (2015).
Brennan, E. P. et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J. Am. Soc. Nephrol. 24, 627–637 (2013).
Mitchell, D. et al. The lipoxin A4 receptor is coupled to SHP-2 activation: implications for regulation of receptor tyrosine kinases. J. Biol. Chem. 282, 15606–15618 (2007).
Baker, N., O’Meara, S. J., Scannell, M., Maderna, P. & Godson, C. Lipoxin A4: anti-inflammatory and anti-angiogenic impact on endothelial cells. J. Immunol. 182, 3819–3826 (2009).
Brennan, E. P. et al. Lipoxins protect against inflammation in diabetes-associated atherosclerosis. Diabetes 67, 2657–2667 (2018).
Petri, M. H. et al. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 105, 65–74 (2015).
Petri, M. H. et al. Resolution of inflammation through the lipoxin and ALX/FPR2 receptor pathway protects against abdominal aortic aneurysms. JACC Basic Transl. Sci. 3, 719–727 (2018).
Fredman, G. et al. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl Acad. Sci. USA 111, 14530–14535 (2014).
Brennan, E. et al. Protective effect of let-7 miRNA family in regulating inflammation in diabetes-associated atherosclerosis. Diabetes 66, 2266–2277 (2017).
Codagnone, M. et al. Lipoxin A4 stimulates endothelial miR-126-5p expression and its transfer via microvesicles. FASEB J. 31, 1856–1866 (2017).
Recchiuti, A., Krishnamoorthy, S., Fredman, G., Chiang, N. & Serhan, C. N. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits. FASEB J. 25, 544–560 (2011).
Rajasagi, N. K., Bhela, S., Varanasi, S. K. & Rouse, B. T. Frontline Science: Aspirin-triggered resolvin D1 controls herpes simplex virus-induced corneal immunopathology. J. Leukoc. Biol. 102, 1159–1171 (2017).
Wang, B. et al. Transforming growth factor-β1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 85, 352–361 (2014).
Schwab, J. M., Chiang, N., Arita, M. & Serhan, C. N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874 (2007).
Campbell, E. L. et al. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 21, 3162–3170 (2007).
Ramon, S., Gao, F., Serhan, C. N. & Phipps, R. P. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J. Immunol. 189, 1036–1042 (2012).
Barnig, C. et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 5, 174ra126 (2013).
Krishnamoorthy, N. et al. Cutting edge: maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J. Immunol. 194, 863–867 (2015).
Borgeson, E. & Godson, C. Molecular circuits of resolution in renal disease. Sci. World J. 10, 1370–1385 (2010).
Philippe, R. & Urbach, V. Specialized pro-resolving lipid mediators in cystic fibrosis. Int. J. Mol. Sci. 19, 2865 (2018).
Elias, I. et al. ALOX5AP overexpression in adipose tissue leads to LXA4 production and protection against diet-induced obesity and insulin resistance. Diabetes 65, 2139–2150 (2016).
Hellmann, J., Tang, Y., Kosuri, M., Bhatnagar, A. & Spite, M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 25, 2399–2407 (2011).
de Gaetano, M. et al. Asymmetric synthesis and biological evaluation of imidazole- and oxazole-containing synthetic lipoxin A4 mimetics (sLXms). Eur. J. Med. Chem. 162, 80–108 (2019).
Guilford, W. J. & Parkinson, J. F. Second-generation beta-oxidation resistant 3-oxa-lipoxin A4 analogs. Prostaglandins Leukot. Essent. Fatty Acids 73, 245–250 (2005).
Maciuszek, M., Cacace, A., Brennan, E., Godson, C. & Chapman, T. M. Recent advances in the design and development of formyl peptide receptor 2 (FPR2/ALX) agonists as pro-resolving agents with diverse therapeutic potential. Eur. J. Med. Chem. 213, 113167 (2021).
O’Sullivan, T. P. et al. Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. J. Med. Chem. 50, 5894–5902 (2007).
Kieran, N. E. et al. Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. 64, 480–492 (2003).
Leonard, M. O. et al. 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J. Am. Soc. Nephrol. 13, 1657–1662 (2002).
Petasis, N. A. et al. Design and synthesis of benzo-lipoxin A4 analogs with enhanced stability and potent anti-inflammatory properties. Bioorg. Med.Chem. Lett. 18, 1382–1387 (2008).
Brennan, E. P. et al. Lipoxins regulate the early growth response-1 network and reverse diabetic kidney disease. J. Am. Soc. Nephrol. 29, 1437–1448 (2018).
Galvao, I. et al. Therapeutic potential of the FPR2/ALX agonist AT-01-KG in the resolution of articular inflammation. Pharmacol. Res. 165, 10544 (2021).
Fu, T. et al. Therapeutic potential of lipoxin A4 in chronic inflammation: focus on cardiometabolic disease. ACS Pharmacol. Transl. Sci. 3, 43–55 (2020).
Qin, C. X. et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 8, 14232 (2017).
Asahina, Y. et al. Discovery of BMS-986235/LAR-1219: a potent formyl peptide receptor 2 (FPR2) selective agonist for the prevention of heart failure. J. Med. Chem. 63, 9003–9019 (2020).
Arita, M. et al. Metabolic inactivation of resolvin E1 and stabilization of its anti-inflammatory actions. J. Biol. Chem. 281, 22847–22854 (2006).
Hong, S. et al. Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol. 180, 3512–3519 (2008).
Tang, H. et al. Protective actions of aspirin-triggered (17R) resolvin D1 and its analogue, 17R-hydroxy-19-para-fluorophenoxy-resolvin D1 methyl ester, in C5a-dependent IgG immune complex-induced inflammation and lung injury. J. Immunol. 193, 3769–3778 (2014).
Wu, S. H., Chen, X. Q., Liu, B., Wu, H. J. & Dong, L. Efficacy and safety of 15(R/S)-methyl-lipoxin A(4) in topical treatment of infantile eczema. Br. J. Dermatol. 168, 172–178 (2013).
Kong, X., Wu, S. H., Zhang, L. & Chen, X. Q. Pilot application of lipoxin A4 analog and lipoxin A4 receptor agonist in asthmatic children with acute episodes. Exp. Ther. Med. 14, 2284–2290 (2017).
Basil, M. C. & Levy, B. D. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16, 51–67 (2016).
Chewcharat, A., Chewcharat, P., Rutirapong, A. & Papatheodorou, S. The effects of omega-3 fatty acids on diabetic nephropathy: a meta-analysis of randomized controlled trials. PLoS ONE 15, e0228315 (2020).
de Boer, I. H. et al. Effect of vitamin D and omega-3 fatty acid supplementation on kidney function in patients with type 2 diabetes: a randomized clinical trial. JAMA 322, 1899–1909 (2019).
Han, E. et al. Effects of omega-3 fatty acid supplementation on diabetic nephropathy progression in patients with diabetes and hypertriglyceridemia. PLoS ONE 11, e0154683 (2016).
Bradberry, J. C. & Hilleman, D. E. Overview of omega-3 fatty acid therapies. P T. 38, 681–691 (2013).
Bhatt, D. L. et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N. Engl. J. Med. 380, 11–22 (2019).
Peterson, B. E. et al. Reduction in revascularization with icosapent ethyl: insights from REDUCE-IT revascularization analyses. Circulation 143, 33–44 (2021).
Gundala, N. K. V., Naidu, V. G. M. & Das, U. N. Amelioration of streptozotocin-induced type 2 diabetes mellitus in Wistar rats by arachidonic acid. Biochem. Biophys. Res. Commun. 496, 105–113 (2018).
Climax, J. et al. Effects of epeleuton, a novel synthetic second-generation n-3 fatty acid, on non-alcoholic fatty liver disease, triglycerides, glycemic control, and cardiometabolic and inflammatory markers. J. Am. Heart Assoc. 9, e016334 (2020).
Pal, A. et al. Resolvin E1 derived from eicosapentaenoic acid prevents hyperinsulinemia and hyperglycemia in a host genetic manner. FASEB J. 34, 10640–10656 (2020).
Hellmann, J., Tang, Y. & Spite, M. Proresolving lipid mediators and diabetic wound healing. Curr. Opin. Endocrinol. Diabetes Obes. 19, 104–108 (2012).
Tang, Y. et al. Proresolution therapy for the treatment of delayed healing of diabetic wounds. Diabetes 62, 618–627 (2013).
Lu, Y., Tian, H. & Hong, S. Novel 14,21-dihydroxy-docosahexaenoic acids: structures, formation pathways, and enhancement of wound healing. J. Lipid Res. 51, 923–932 (2010).
Tian, H., Lu, Y., Shah, S. P. & Hong, S. 14S,21R-dihydroxydocosahexaenoic acid remedies impaired healing and mesenchymal stem cell functions in diabetic wounds. J. Biol. Chem. 286, 4443–4453 (2011).
Tian, H., Lu, Y., Shah, S. P. & Hong, S. Autacoid 14S,21R-dihydroxy-docosahexaenoic acid counteracts diabetic impairment of macrophage prohealing functions. Am. J. Pathol. 179, 1780–1791 (2011).
Goicoechea, M. et al. Low dose aspirin increases 15-epi-lipoxin A4 levels in diabetic chronic kidney disease patients. Prostaglandins Leukot. Essent. Fatty Acids 125, 8–13 (2017).
Borgeson, E. et al. Lipoxin A(4) and benzo-lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 25, 2967–2979 (2011).
Duffield, J. S. et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 177, 5902–5911 (2006).
Hassan, I. R. & Gronert, K. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J. Immunol. 182, 3223–3232 (2009).
Qiu, Y., Wu, Y., Zhao, H., Sun, H. & Gao, S. Maresin 1 mitigates renal ischemia/reperfusion injury in mice via inhibition of the TLR4/MAPK/NF-κB pathways and activation of the Nrf2 pathway. Drug Design Dev. Ther. 13, 739–745 (2019).
Tang, S. et al. Maresin 1 mitigates high glucose-induced mouse glomerular mesangial cell injury by inhibiting inflammation and fibrosis. Mediators Inflamm. 2017, 2438247 (2017).
Yore, M. M. et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 159, 318–332 (2014).
Kuda, O. et al. Docosahexaenoic acid-derived fatty acid esters of hydroxy fatty Acids (FAHFAs) with anti-inflammatory properties. Diabetes 65, 2580–2590 (2016).
Lee, J. et al. Branched fatty acid esters of hydroxy fatty acids (FAHFAs) protect against colitis by regulating gut innate and adaptive immune responses. J. Biol. Chem. 291, 22207–22217 (2016).
Paluchova, V. et al. Lipokine 5-PAHSA is regulated by adipose triglyceride lipase and primes adipocytes for de novo lipogenesis in mice. Diabetes 69, 300–312 (2020).
Benlebna, M. et al. Long-term high intake of 9-PAHPA or 9-OAHPA increases basal metabolism and insulin sensitivity but disrupts liver homeostasis in healthy mice. J. Nutr. Biochem. 79, 108361 (2020).
Guo, Y. P., Jiang, H. K., Jiang, H., Tian, H. Y. & Li, L. Lipoxin A4 may attenuate the progression of obesity-related glomerulopathy by inhibiting NF-κB and ERK/p38 MAPK-dependent inflammation. Life Sci. 198, 112–118 (2018).
Qu, X. et al. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 228, 506–519 (2012).
Zhang, X. et al. Resolvin D1 protects podocytes in adriamycin-induced nephropathy through modulation of 14-3-3β acetylation. PLoS ONE 8, e67471 (2013).
Luan, H. et al. Resolvin D1 protects against ischemia/reperfusion-induced acute kidney injury by increasing Treg percentages via the ALX/FPR2 pathway. Front. Physiol. 11, 285 (2020).
Chen, J. et al. Aspirin-triggered resolvin D1 down-regulates inflammatory responses and protects against endotoxin-induced acute kidney injury. Toxicol. Appl. Pharmacol. 277, 118–123 (2014).
Sun, S. et al. Maresin 1 mitigates sepsis-associated acute kidney injury in mice via inhibition of the NF-κB/STAT3/MAPK pathways. Front. Pharmacol. 10, 1323 (2019).
Sun, Y. B., Qu, X., Li, X., Nikolic-Paterson, D. J. & Li, J. Endothelial dysfunction exacerbates renal interstitial fibrosis through enhancing fibroblast Smad3 linker phosphorylation in the mouse obstructed kidney. PLoS ONE 8, e84063 (2013).
Mitchell, S. et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 13, 2497–2507 (2002).
Wu, S. H. et al. Lipoxin A4 inhibits connective tissue growth factor-induced production of chemokines in rat mesangial cells. Kidney Int. 69, 248–256 (2006).
Wu, S. H., Wu, X. H., Liao, P. Y. & Dong, L. Signal transduction involved in protective effects of 15(R/S)-methyl-lipoxin A(4) on mesangioproliferative nephritis in rats. Prostaglandins Leukot. Essent. Fatty Acids 76, 173–180 (2007).
Acknowledgements
Work in the authors’ laboratories is supported by Science Foundation Ireland awards (SFI 15/IA/3152, SFI/US/B3130), a Strategic Research Award from JDRF (2-SRA-2017-507-S-B) and The National Health and Medical Research Council (NHMRC) of Australia. E.B. is supported by a University College Dublin Ad Astra Fellowship.
Author information
Authors and Affiliations
Contributions
E.B. and C.G. researched data for the article. All authors contributed to writing, reviewing and editing the article before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Nephrology thanks B. Conway and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Sterile inflammation
-
An inflammatory response arising in the absence of a pathogen (for example, toxin exposure, mechanical tissue injury and ischaemia).
- Long-chain fatty acids
-
Non-esterified straight-chain fatty acids containing 12 or more carbon atoms, with important roles as components of membranes and metabolic energy sources.
- Advanced glycation end-products
-
Proteins or lipids that have become glycated as a result of exposure to sugars.
- Positional isomer
-
Positional isomers have the same carbon backbone and functional groups, but differ in the positioning of functional groups on the backbone.
- Transcellular metabolism
-
In the context of eicosanoid biosynthesis, a pathway whereby one cell type synthesizes an intermediate that is further metabolized by a different cell.
- Triene core
-
An unsaturated hydrocarbon containing three double bonds between carbon atoms.
Rights and permissions
About this article

Cite this article
Brennan, E., Kantharidis, P., Cooper, M.E. et al. Pro-resolving lipid mediators: regulators of inflammation, metabolism and kidney function. Nat Rev Nephrol 17, 725–739 (2021). https://doi.org/10.1038/s41581-021-00454-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-021-00454-y
This article is cited by
-
Klotho exerts protection in chronic kidney disease associated with regulating inflammatory response and lipid metabolism
Cell & Bioscience (2024)
-
Specialized pro-resolving mediators in vascular inflammation and atherosclerotic cardiovascular disease
Nature Reviews Cardiology (2024)
-
Predictors of Kidney Function Outcomes and Their Relation to SGLT2 Inhibitor Dapagliflozin in Patients with Type 2 Diabetes Mellitus Who Had Chronic Heart Failure
Advances in Therapy (2024)
-
Interface between Resolvins and Efferocytosis in Health and Disease
Cell Biochemistry and Biophysics (2024)
-
HMGN1 down-regulation in the diabetic kidney attenuates tubular cells injury and protects against renal inflammation via suppressing MCP-1 and KIM-1 expression through TLR4
Journal of Endocrinological Investigation (2024)
