
Abstract
Tweety-homolog 1 protein (Ttyh1) is abundantly expressed in neurons in the healthy brain, and its expression is induced under pathological conditions. In hippocampal neurons in vitro, Ttyh1 was implicated in the regulation of primary neuron morphology. However, the mechanisms that underlie transcriptional regulation of the Ttyh1 gene in neurons remain elusive. The present study sought to identify the promoter of the Ttyh1 gene and functionally characterize cis-regulatory elements that are potentially involved in the transcriptional regulation of Ttyh1 expression in rat dissociated hippocampal neurons in vitro. We cloned a 592 bp rat Ttyh1 promoter sequence and designed deletion constructs of the transcription factors specificity protein 1 (Sp1), E2F transcription factor 3 (E2f3), and achaete-scute homolog 1 (Ascl1) that were fused upstream of a luciferase reporter gene in pGL4.10[luc2]. The luciferase reporter gene assay showed the possible involvement of Ascl1, Sp1, and responsive cis-regulatory elements in Ttyh1 expression. These findings provide novel information about Ttyh1 gene regulation in neurons.
Similar content being viewed by others

Introduction
The Tweety-homolog1 (Ttyh1) gene encodes a transmembrane protein that putatively functions as a chloride channel [1,2,3,4]. TTYH1 is expressed in embryonic stem cells both during the early stages of brain development and in the adult brain [5]. Recently, TTYH1 has been discovered as one of the specific progenitor genes in human developing hypothalamus [6]. Ttyh1 is highly expressed in neurons in the healthy rat brain and dissociated hippocampal neurons, regardless of the age of the culture [7,8,9,10,11,12]. Ttyh1 was detected in neuropils, neuronal somata [8, 12], the presynaptic active zone of the rat brain [9], and invaginations of dendritic spines in vitro [12].
The importance of Ttyh1 in neural function has been underscored by its recently documented involvement in the regulation of neural morphology in vitro and aberrant neuronal structural plasticity in vivo [12]. Elevations of Ttyh1 expression were detected in the molecular layer of the dentate gyrus during epileptogenesis [11,12,13]. Our recent study revealed that Ttyh1 participates in shaping dendritic tree and dendritic spines morphology in hippocampal slices in vitro (Gorniak-Walas, submitted).
Ttyh1 mRNA is highly expressed in neurons in vitro and in vivo [7,8,9,10,11,12], but current knowledge of the transcriptional regulation of Ttyh1 in neurons is still lacking. Transcriptional regulation is a highly coordinated process and required for temporal- and tissue-specific gene expression. Therefore, the ability to identify promoter sequences and predict specific transcription factor binding sites is integral to unraveling the mechanism of Ttyh1 gene regulation.
Results
In silico Characterization of the Ttyh1 Gene Promoter
To gain insights into the transcription regulation of neuronal Ttyh1 expression, we analyzed the Ttyh1 promoter sequence in search of putative transcription factor binding sites. Using MatInspector software, we isolated a 630 bp fragment that contained the rat Ttyh1 promoter (Fig. 1). To identify the transcription start site (TSS) in the rat Ttyh1 promoter, the sequences of the mouse Ttyh1 promoter were retrieved from the Eukaryotic Promoter Database. Promoter sequences of rat Ttyh1 showed 93% sequence similarity to the mouse Ttyh1 promoter (Fig. 2). We found that the TSS was located 75 nt upstream of the ATG initiation codon (marked as + 1 in Figs. 1, 2).

The rat Ttyh1 promoter sequence obtained from MatInspector software. Putative binding sites for E2f3, Ascl1, and Sp1 are in boxes. The ATG initiation codon is in bold, and the TSS is highlighted in red (marked as + 1)

Amino acid alignment of the rat (R. norvegicus) and mouse (M. mulsculus) Ttyh1 gene promoter (NM_001106225 and NM_021324, respectively). Black and grey boxes indicate identical and similar nucleotides. Putative binding sites for E2f3, Ascl1, and Sp1 are indicated by arrows. The core sequences are in red boxes. The TSS and the ATG initiation codon are highlighted by blue and green boxes, respectively. The sequence alignment was carried out using T-Coffee, the alignment was formatted using BoxShade
Identification of Putative Binding Sites in the Rat Ttyh1 Gene Promoter
To identify potential cis-regulatory elements within the Ttyh1 promoter, we used MatInspector software. Although the Ttyh1 promoter included a number of cis-regulatory elements, we selected high-score potential binding sites for Sp1, Ascl1, and E2f3 upstream of TSS (−142 to −126 bp, 175 to −163 bp, and −376 to −360 bp, respectively; Figs. 1, 2, Table 1).
Functional Characterization of Regulatory Elements in the Rat Ttyh1 Gene Promoter
To interrogate the role of cis-regulatory elements in Ttyh1 expression, we cloned a 592 bp rat Ttyh1 promoter sequence (−541 to + 51 bp), designed deletion constructs for Sp1, E2f3, and Ascl1 that were fused to a luciferase reporter gene in pGL4.10[luc2], and performed a dual luciferase reporter assay in dissociated hippocampal neurons in vitro. This method allows identification of nucleotide sequences in the promoter region responsible for the regulation of gene expression [14].
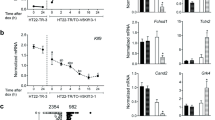
The coding sequences of E2f3, Ascl1, and SP1 were amplified by PCR and cloned into an N1-GFP plasmid. The expression of E2f3-GFP, Ascl1-GFP, and Sp1-GFP, was confirmed by Western blot (Fig. 3a, b). The transfection efficiency achieved using Lipofectamine2000 was lower for N1-EGFP plasmid contained the coding sequence of Sp1 (2361 bp) compared to plasmid contained the coding sequence of E2f3 (738 bp), and Ascl1 (702 bp). As the size of the plasmid can influence the transfection efficiency, transfection of hippocampal neurons with N1-Sp1-GFP (the total size of the plasmid: 7061 bp) could result in lower DNA uptake by the cells compared to N1-E2F3-GFP (the total size of the plasmid: 5438 bp) and N1-Ascl1-GFP (the total size of the plasmid: 5402 bp). Interestingly, we detected a 2.8-fold increase in Ttyh1 expression in E2f3-GFP-expressing neurons compared with control neurons (Fig. 3a, b).

Expression levels of Sp1-GFP, E2f3-GFP, Ascl1-GFP, and Ttyh1 proteins following transfection with respective vectors in hippocampal neurons in vitro. A Western blot analysis of the expression level of Sp1-GFP, E2f3-GFP, Ascl1-GFP, and Ttyh1 proteins. The arrows show the overexpressed Sp1-GFP, E2f3-GFP, and Ascl1-GFP, respectively. GAPDH was used as a loading control. B The intensity of Ttyh1 expression was normalized to GAPDH
Deletion constructs were devoid of predicted binding sites for E2f3 (−376 to −360 bp), Ascl1 (−175 to −163 bp), and Sp1 (−142 to −126 bp), that were linked upstream of the luciferase reporter gene in pGL4.10[luc2] (Fig. 4). The promoter deletion constructs were generated using site-directed mutagenesis by overlap extension based on polymerase chain reaction (PCR).

The luciferase constructs used in the experiments. Schematic representation of Ttyh1 promoter construct (pGL4.10-P), and the deletion constructs of the E2F3 (pGL4.10-E2f3), Ascl1 (pGL4.10-Ascl1), and Sp1 (pGL4.10-Sp1) used in the luciferase assay
Dissociated hippocampal neurons were transfected with the N1-GFP empty plasmid, or N1-GFP plasmid that contained coding sequences of E2f3, Ascl1, and Sp1 (N1-E2f3-GFP, N1-Ascl1-GFP, and N1-Sp1-GFP, respectively), and the pGL4.74[hRluc/TK] plasmid as an expression control. These plasmids were co-transfected with the pGL4.10[luc2] empty plasmid or pGL4.10[luc2] plasmid that encoded the Ttyh1 promoter (pGL4.10-P), and luciferase activity was monitored.
The analysis of promoter activity revealed that the Ttyh1 promoter (pGL4.10-P) exhibited high transcriptional activity, ensuring a strong level of luciferase gene expression.
This basal Ttyh1 promoter activity was 25-fold higher than the pGL4.10[luc2] empty plasmid and set to 100% for further comparisons (Fig. 5).

Transcriptional regulation of Ttyh1 promoter in hippocampal neurons in vitro. Luciferase reporter gene assays were used to determine Ttyh1 promoter activity in hippocampal neurons in vitro for the transcription factors E2f3 (A), Ascl1 (B), and Sp1 (C). The data are expressed as mean ± SEM. The data were collected from four biological replicates. Three technical replicates were performed for each sample. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test)
To investigate the functional role of E2f3, Ascl1, and Sp1 in the transcriptional regulation of Ttyh1 gene expression, we examined the effects of the overexpression of individual transcription factors on activity of the Ttyh1 gene promoter. As shown in Fig. 5a and c, we did not observe significant differences in promoter activity between E2f3-GFP- or Sp1-GFP-expressing neurons and control neurons that expressed GFP only. However, the Ttyh1 promoter exhibited 1.6-fold higher activity following Ascl1-GFP overexpression compared with basal Ttyh1 activity (Fig. 5b).
To further examine the functional role of predicted binding sites in Ttyh1 gene expression, we designed promoter deletion constructs for E2f3 (pGL4.10-E2f3), Ascl1 (pGL4.10-Ascl1), and Sp1 (pGL4.10-Sp1) predicted binding sites. As shown in Fig. 5, the promoter deletion constructs had basal activity that was similar to the full-length Ttyh1 promoter (pGL4.10-P). The activity of promoter deletion construct for Ascl1 (pGL4.10-Ascl1) was significantly lower in GFP-expressing neurons compared to the activity of the full-length Ttyh1 promoter (pGL4.10-P) in Ascl1-GFP-overexpressing neurons (p < 0.01). Interestingly, the deletion construct for Ascl1 (pGL4.10-Ascl1) significantly abolished Ttyh1 promoter activity in Ascl1-GFP-overexpressing neurons (p < 0.05) (Fig. 5b). The Ttyh1 promoter activity of deletion construct for Sp1 (pGL4.10-Sp1) was significantly decreased in Sp1-GFP-overexpressing neurons (p < 0.05) (Fig. 5c).
These data suggest that the Sp1 and Ascl1 transcription factors may be involved in the transcriptional regulation of Ttyh1 expression in neurons.
Discussion
TTYH1 is primary expressed in embryonic stem cells and continues to be expressed at later stages of brain development [5]. Ttyh1 expression is induced under pathological conditions, such as epilepsy [11,12,13], triple-negative breast cancer [15], glial tumors [16, 17], and in activated astrocytes in the epileptic brain [18]. To date, the mechanisms that underlie transcriptional regulation of the Ttyh1 gene remain elusive.
We generated a promoter of the rat Ttyh1 gene and identified cis-regulatory elements presumably involved in the transcriptional regulation of Ttyh1 in rat dissociated hippocampal neurons in vitro. We cloned a 592 bp rat Ttyh1 promoter sequence that was fused upstream of a luciferase reporter gene in pGL4.10[luc2] and designed promoter deletion constructs for Sp1 (−142 to −126 bp), Ascl1 (−175 to −163 bp), and E2f3 (−375 to −360 bp) predicted binding sites. Our findings provide evidence of the involvement of Sp1 and Ascl1 in Ttyh1 gene regulation in neurons.
Aachaete-scute like 1 (Ascl1) belongs to the basic-helix-loop-helix family and is expressed in the nervous system [19,20,21,22]. Ascl1 regulates gene expression during neurogenesis and controls cell-fate determination in neurons and glia [23,24,25,26]. Ascl1 has also been implicated in Parkinson’s disease and various cancers (e.g., glioma and neuroblastoma) [27, 28]. For example, high Ascl1 expression in glioma promotes neuronal differentiation and prevents tumor growth [29]. Ttyh1 regulates embryonic neural stem cell properties by positively regulating the Notch pathway [30, 31]. Evidence from human studies indicates that Ttyh1 is expressed in embryonic stem cells and neural structures and continues to be expressed at later stages of brain development [5]. These findings are consistent with our previous studies, in which Ttyh1 was expressed at high levels in rat hippocampal neurons in vitro, regardless of the age of the culture [12]. The present study showed a 1.6-fold increase in Ttyh1 promoter activity in Ascl1-GFP-overexpressing neurons compared with control neurons and significantly abolished activity of the deletion construct for Ascl1 (p < 0.05) compared with the full-length Ttyh1 promoter following Ascl1-GFP overexpression. Given the developmental function of Ascl1 in the central nervous system and high expression of Ttyh1 in embryonic stem cells and during brain development [5], Ascl1 appears to govern Ttyh1 gene expression during embryonic development.
Transcription factor Sp1 is widely expressed and regulates the expression of numerous genes that are involved in various processes, such as cell growth, metabolism, differentiation, and the immune response [32]. High SP1 expression was detected in the hippocampus after kainic acid administration in rats [33] and in the postmortem hippocampus in chronic schizophrenia patients [34]. Sp1 has been also implicated in tumorigenesis [35]. Previous studies implicated Ttyh1 in pathological conditions, such as epilepsy [11, 13] and glial tumor progression [16, 17]. In the present study, we observed a significant decrease in activity of the deletion construct for Sp1 compared with Ttyh1 promoter activity in Sp1-GFP-overexpressing neurons. Thus, Sp1 appears to be involved in Ttyh1 gene regulation in disease-related processes.
Transcription factor E2f3 plays an important role in regulating the cell cycle [36], and its dysregulation has been implicated in human cancers [37,38,39]. Previous studies indicated that miRNAs regulate the expression of E2f3 in various types of cancers. For example, E2f3 expression is posttranscriptionally regulated by miR-128 in glioma [40]. Under these conditions, miR-128 is downregulated, and E2F3 is highly expressed. Interestingly, miR-128 was significantly downregulated in the dentate gyrus and CA1 layer of the hippocampus in epileptic rats [41, 42]. An increase in Ttyh1 expression has been implicated in glial tumors [16, 17] and epilepsy [11, 13]. Therefore, we speculate that miR-128 may target E2f3 and regulate the expression of Ttyh1 under pathological conditions. Although the luciferase reporter gene assay in the present study did not reveal an impact of E2f3 on activity of the Ttyh1 promoter in hippocampal neurons in vitro, we observed a 2.8-fold increase in Ttyh1 expression in E2f3-GFP-expressing neurons compared with control neurons. Therefore, we cannot exclude the possibility that E2f3 interacts with another cis-regulatory sequence within the Ttyh1 promoter and drives the expression of Ttyh1 under pathological conditions.
The relevance of Ttyh1 to neuronal function is underscored by its high expression in the healthy brain and the induction of its expression in disease-related processes. The present study suggests that Ascl1 and Sp1 play a potential role in the modulation of Ttyh1 expression under physiological conditions or in brain pathology. The regulation of Ttyh1 expression appears to play a prominent role in proper brain development and function. Further studies should explore the precise mechanism of the interaction between Ascl1 and Sp1 and responsive cis-regulatory elements within the Ttyh1 promoter.
Materials and Methods
Sequence Analysis of the Ttyh1 Gene Promoter
To identify the promoter sequence of the rat Ttyh1 gene and check for potential binding sites, we used MatInspector software (MatInspector Release Professional 8.2, December 2014) [43]. For functional analysis, we selected transcription factor binding sites with a high degree of homology between species.
Cloning of the Rat Ttyh1 Gene Promoter
The rat genomic DNA was extracted from the tail using Genomic Mini (catalog no. 116-50, A&A Biotechnology, Gdynia, Poland). The rat Ttyh1 gene promoter was amplified from extracted genomic DNA using Phusion High-Fidelity DNA Polymerase (catalog no. F530L, Thermo Fisher Scientific, Waltham, MA, USA; forward primer, 5’-CAGTCTTCACAACGTGTGTAGGT-3’; reverse primer, 5’-TGCTGGTACTCTTGGTCGCG-3’). A 592 bp promoter sequence of the rat Ttyh1 gene upstream of the TSS was obtained by PCR using 5’-TGCAGCTAGCCAGTCTTCACAACGTGTGT-3’ (forward) and 5’-TATAAGCTTGACCGGGCCGGGAGC-3’ (reverse) primers that carried NheI and HindIII restriction sites, respectively. To design the Ttyh1 promoter construct (pGL4.10-K), the PCR product was cloned 5’-upstream of a luciferase reporter gene into pGL4.10[luc2] (catalog no. E6651, Promega, Madison, WI, USA) by restriction enzyme digestion using NheI (catalog no. R3131S, New England BioLabs, Ipswich MA, USA), HindIII (catalog no. R3104S, New England BioLabs, Ipswich MA, USA), and the Quick Ligation Kit (catalog no. M2200S, New England BioLabs, Ipswich MA, USA). Promoter deletion constructs that were devoid of the predicted binding site for Sp1 (−142 to −126 bp; pGL4.10-Sp1), E2f3 (−376 to −360 bp; pGL4.10-E2f3), and Ascl1 (−175 to −163 bp; pGL4.10-Ascl1) were designed using site-directed mutagenesis by overlap extension and PCR. Briefly, the promoter construct (pGL4.10-P) was amplified in two separate PCRs using F and R2 primers and F2 and R primers, with R2 and F2 overlapping primers (15 nt 3’-overhangs) upstream (R2) or downstream (F2) of the sequence that was deleted. The two PCR products, F-R2 and F2-R, were amplified in one reaction using forward and reverse primers. The mutated PCR product was introduced by restriction enzyme digestion (NheI, HindIII) and ligation (Quick Ligation Kit, catalog no. M2200S, New England BioLabs, Ipswich MA, USA). The following primers were used for the Ttyh1 promoter deletion constructs: F_Sp1 (5’-TGCAGCTAGCCAGTCTTCACAACGTGTGT-3’), R_Sp1 (5’-TATAAGCTTGACCGGGCCGGGAGC-3’), R2_Sp1 (5’-AATGGGCCCCTGGGCTCCCACCT-3’), F2_Sp1 (5’-AGGTGGGAGCCCAGGGGCCCAT-3’), F_E2f3 (5’-TATGCTAGCCAGTCTTCACAACGT-3’), R_E2f3 (5’-TATAAGCTTGACCGGGCCGG-3’), R2 (5’-ATTTGAGCAGCATTTAAAAATCCCTCATTT-3’), F2_E2f3 (5’-AAATGAGGGATTTTTAAATGCTGCTCAAAT-3’), F_Ascl1 (5’-TGCAGCTAGCCAGTCTTCACAACGTGTGT-3’), R_Ascl1 (5’-TATAAGCTTGACCGGGCCGGGAGC-3’), R2_Ascl1 (5’-CACCTGCTGCCTTGGTCTTTGCGAG-3’), and F2_Ascl1 (5’-CTCGCAAAGACCAAGGCAGCAGGTG-3’). The sequences of all of the constructs were verified by sequence analysis.
Cloning of Transcription Factor Coding Sequences
The N1-EGFP plasmids that contained the coding sequences of Sp1, E2f3, and Ascl1 were obtained by PCR using Phusion High-Fidelity DNA Polymerase (catalog no. F530S, Thermo Fisher Scientific, Waltham, MA, USA), a forward primer that carried the EcoRI restriction site, a reverse primer that carried the AgeI restriction site, and pCMV6-Sp1 (NM_012655, catalog no. RN213136, OriGene, Rockville, MA, USA), pCMV6-E2f3 (NM_001137626, catalog no. RN207430, OriGene Rockville, MA, USA), or pCMV-Ascl16 (NM_022384, catalog no. RN215503, OriGene Rockville, MA, USA) as a template. The PCR product was introduced into the N1-EGFP plasmid by restriction enzyme digestion (AgeI, catalog no. ER1462, Thermo Fisher Scientific, Waltham, MA, USA; EcoRI, catalog no. ER0271, Thermo Fisher Scientific, Waltham, MA, USA) and ligation (Quick Ligase, catalog no. M2200S, New England BioLabs, Ipswich MA, USA). The following primers were used to generate the expression constructs: F_Sp1 (5’-GCTATGAATTCGCCATGAGCGACCA-3’), R_Sp1 (5’-ACTAACCGGTGAGAAACCATTGCCAC-3’), F_E2f3 (5’-GCTATGAATTCGCCATGAGAAAGGG-3’), R_E2f3 (5’-AGCTACCGGTGAATTTTTCGAATATCTTG-3’), F_Ascl1 (5’-GATGAATTCATTATGGAGAGCTCT-3’), and R_Ascl1 (5’-AGCTACCGGTGAGAACCAGT-3’).
Dissociated Hippocampal Neurons In Vitro
Primary cultures of hippocampal neurons were prepared under sterile conditions from embryonic day 18 Wistar rat embryos (Animal House, Nencki Institute of Experimental Biology, Warsaw, Poland) according to a previously described procedure [12, 44]. All of the procedures were performed in accordance with the Animal Protection Act of Poland (Directive 2010/63/EU). No ethical approval is required under Polish law for tissue collection. Pregnant rats were anesthetized with 4% isoflurane, followed by swift decapitation with guillotine. The dam's abdomen area was rinsed with 70% EtOH and cut to expose the uterus and embryos. The fetuses were removed and placed in ice-cold Hank’s Balanced Salt Solution (catalog no. 14170-088, Thermo Fisher Scientific, Waltham, MA, USA). The fetuses were rapidly decapitated and brains were removed. Hippocampi were dissected from the brains and placed in ice-cold dissection medium. Hippocampi were incubated in HBSS with 0.25% trypsin (catalog no. 27250-0180, Thermo Fisher Scientific, Waltham, MA, USA) and 0.15 mg/ml deoxyribonuclease (catalog no. DN-25, Sigma-Aldrich, St. Louis, MO, USA) at 37 °C for 15 min. Dissociated hippocampal neurons were plated at a density of 5 × 104 cells per cm2 on poly-D-lysine-coated (50 µg/ml, catalog no. P7280, Sigma, St. Louis, MO, USA) multi-wells in G3 medium that contained Neurobasal (catalog no. 21103, Thermo Fisher Scientific, Waltham, MA, USA), B27 (catalog no. 17504, Thermo Fisher Scientific, Waltham, MA, USA), 0.5 mM GlutaMax (catalog no. 35050-038, Thermo Fisher Scientific, Waltham, MA, USA), 25 μM L-glutamate (catalog no. G1626, Sigma-Aldrich, St. Louis, MO, USA), and 10 mg/L gentamicin (catalog no. 15-750-037, Fisher Scientific, Waltham, MA, USA). After 5 days in culture, half of the medium was exchanged for G2 medium (G3 without L-glutamate). Hippocampal neurons were cultured at 37 °C in 95% O2 and 5% CO2. Cell cultures were inspected under the microscope during the culture. Neurons comprise > 30% of cells at 9th day in vitro (DIV).
Cell Transfection
Dissociated hippocampal neurons were transfected at 9 DIV with 1.3 μg pEGFP‐N1 (or N1-Sp1-GFP, N1‐E2f3-GFP, or N1‐Ascl1-GFP), 0.2 μg pGL4.10[luc2] that encoded the Ttyh1 gene promoter (or the appropriate pGL4.10[luc2] deletion construct), and 0.02 μg pGL4.74[hRluc/TK] using Lipofectamine2000 transfection reagent (catalog no. 11668027, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. As a positive control, cells were transfected with empty pEGFP‐N1, pGL4.10[luc2] that encoded the Ttyh1 gene promoter, and pGL4.74[hRluc/TK]. As a negative control, cells were transfected with empty pEGFP‐N1, N1-Sp1-GFP, pN1‐E2f3-GFP, or N1‐Ascl1-GFP, empty pGL4.10[luc2], and pGL4.74[hRluc/TK]. We did not observe transfection of other cell than neurons.
Cell Lysate Preparation
Cells were washed in 1 × phosphate-buffered saline and lysed in lysis buffer that contained 50 mM KCl, 50 mM PIPES, 10 mM EGTA, 2 mM MgCl2, 0.5% Triton X-100, 100 μM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol, supplemented with protease inhibitors (Roche). The cell lysates were clarified by centrifugation at 12,000 × g for 20 min at 4 °C. The protein concentration was determined using Protein Assay Dye Reagent (catalog no. 500-0006, BioRad, Hercules, CA, USA).
Western Blot
The cell lysates were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (catalog no. RPN303D, Cytiva, Marlborough, MA, USA). The membrane was blocked with 5% nonfat milk in TBST (0.5 M Tris, 0.9% NaCl, and 0.1% Tween 20, pH 8) for 1 h at room temperature, followed by overnight incubation at 4 °C in TBST that contained rabbit anti-GFP antibody (1:5000 dilution, catalog no. 598, MBL, Woburn, MA, USA), mouse anti-Ttyh1 antibody (1:1000 dilution, catalog no. WH0057348M4, Sigma-Aldrich, St. Louis, MO, USA), or mouse anti-GAPDH antibody (1:1000 dilution, catalog no. MAB374, Millipore, Burlington, MA, USA). After washing with TBST, the membranes were incubated with secondary antibody conjugated to horseradish peroxidase (1:5000 dilution, anti-rabbit horseradish peroxidase-linked antibody, catalog no. AP132P, Sigma-Aldrich, St. Louis, MO, USA; 1:5000 dilution, anti-mouse horseradish peroxidase-linked antibody, catalog no. ab6728, Abcam, Cambridge, MA, USA) in TBST for 2 h at room temperature. The membranes were washed with TBST and developed using Amersham ECL Western Blotting System (catalog no. RPN2108, Cytiva, Marlborough, MA, USA) according to the manufacturer’s instructions. Signal was registered using chemiluminescence western blotting using X-ray film with the automatic film processor. Densitometry was performed with Image Studio Lite Ver 5.2 software. The intensity of Ttyh1 expression was normalized to GAPDH.
Luciferase Assay
Luciferase activity was measured 24 h after transfection in a luminometer (TD-20/20) using the Dual Luciferase Reporter Assay System (catalog no. E1910, Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, cells were lysed with Passive Lysis Buffer, incubated at room temperature for 10 min on an orbital shaker, and centrifuged at 10,000 × g for 10 min. Luciferase Assay Reagent II and Stop&Glo reagent were sequentially added to the cell lysate to measure the activity of firefly (Photinus pyralis) and Renilla (Renilla reniformis), respectively. The activity of firefly was normalized to the activity of Renilla. Data were collected from four biological replicates. Three technical replicates were performed for each sample.
Statistical Analysis
Data were collected from four independent experiments. Three technical replicates were performed for each sample. The results are presented as mean ± standard error of the mean (SEM). The statistical analysis was performed using Prism 5.0 software (GraphPad, La Jolla, CA, USA). Datasets were tested using the two-tailed Student’s t-test. Values of p < 0.05 were considered statistically significant.
Data Availability
The manuscript has no associated data.
References
Bae Y, Kim A, Cho CH, Kim D, Jung HG, Kim SS, Yoo J, Park JY, Hwang EM (2019) TTYH1 and TTYH2 serve as LRRC8A-independent volume-regulated anion channels in cancer cells. Cells 8:6. https://doi.org/10.3390/cells8060562
Han YE, Kwon J, Won J, An H, Jang MW, Woo J, Lee JS, Park MG, Yoon BE, Lee SE, Hwang EM, Jung JY, Park H, Oh SJ, Lee CJ (2019) Tweety-homolog (Ttyh) family encodes the pore-forming subunits of the swelling-dependent volume-regulated anion channel (VRACswell) in the brain. Exp Neurobiol 28:183–215. https://doi.org/10.5607/en.2019.28.2.183
Suzuki M (2006) The Drosophila tweety family: molecular candidates for large-conductance Ca2+-activated Cl- channels. Exp Physiol 91:141–147. https://doi.org/10.1113/expphysiol.2005.031773
Suzuki M, Mizuno A (2004) A novel human Cl- channel family related to Drosophila flightless locus. J Biol Chem 279:22461–22468. https://doi.org/10.1074/jbc.M313813200
Kleinman CL et al (2014) Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46:39–44. https://doi.org/10.1038/ng.2849
Zhou X, Zhong SJ, Peng HH, Liu J, Ding WY, Sun L, Ma Q, Liu ZY, Chen RG, Wu Q, Wang XQ (2020) Cellular and molecular properties of neural progenitors in the developing mammalian hypothalamus. Nat Commun 11:4063. https://doi.org/10.1038/s41467-020-17890-2
Kumada T, Yamanaka Y, Kitano A, Shibata M, Awaya T, Kato T, Okawa K, Abe T, Oshima N, Nakahata T, Heike T (2010) Ttyh1, a Ca2+-binding protein localized to the endoplasmic reticulum, is required for early embryonic development. Dev Dyn 239:2233–2245. https://doi.org/10.1002/dvdy.22348
Matthews CA, Shaw JE, Hooper JA, Young IG, Crouch MF, Campbell HD (2007) Expression and evolution of the mammalian brain gene Ttyh1. J Neurochem 100:693–707. https://doi.org/10.1111/j.1471-4159.2006.04237.x
Morciano M, Beckhaus T, Karas M, Zimmermann H, Volknandt W (2009) The proteome of the presynaptic active zone: from docked synaptic vesicles to adhesion molecules and maxi-channels. J Neurochem 108:662–675. https://doi.org/10.1111/j.1471-4159.2008.05824.x
Nothwang HG, Koehl A, Friauf E (2006) Comparative gene expression analysis reveals a characteristic molecular profile of the superior olivary complex. Anat Rec A Discov Mol Cell Evol Biol 288A:409–423. https://doi.org/10.1002/ar.a.20301
Stefaniuk M, Lukasiuk K (2010) Cloning of expressed sequence tags (ESTs) representing putative epileptogenesis-related genes and the localization of their expression in the normal brain. Neurosci Lett 482:230–234. https://doi.org/10.1016/j.neulet.2010.07.045
Stefaniuk M, Swiech L, Dzwonek J, Lukasiuk K (2010) Expression of Ttyh1, a member of the Tweety family in neurons in vitro and in vivo and its potential role in brain pathology. J Neurochem 115:1183–1194. https://doi.org/10.1111/j.1471-4159.2010.07023.x
Lukasiuk K, Kontula L, Pitkanen A (2003) cDNA profiling of epileptogenesis in the rat brain. Eur J Neurosci 17:271–279. https://doi.org/10.1046/j.1460-9568.2003.02461.x
Surgucheva I, Surguchov A (2008) Gamma-synuclein: Cell-type-specific promoter activity and binding to transcription factors. J Mol Neurosci 35:267–271. https://doi.org/10.1007/s12031-008-9074-6
Zhong GS, Lou WY, Shen QY, Yu K, Zheng YJ (2020) Identification of key genes as potential biomarkers for triple-negative breast cancer using integrating genomics analysis. Mol Med Rep 21:557–566. https://doi.org/10.3892/mmr.2019.10867
Feng YP, Hurst J, Almeida-De-Macedo M, Chen X, Li L, Ransom N, Wurtele ES (2012) Massive human co-expression network and its medical applications. Chem Biodivers 9:868–887. https://doi.org/10.1002/cbdv.201100355
Jung E et al (2017) Tweety-homolog 1 drives brain colonization of gliomas. J Neurosci 37:6837–6850. https://doi.org/10.1523/JNEUROSCI.3532-16.2017
Wiernasz E, Kaliszewska A, Brutkowski W, Bednarczyk J, Gorniak M, Kaza B, Lukasiuk K (2014) Ttyh1 protein is expressed in glia in vitro and shows elevated expression in ectivated astrocytes following status epilepticus. Neurochem Res 39:2516–2526. https://doi.org/10.1007/s11064-014-1455-3
Britz O, Mattar P, Nguyen L, Langevin LM, Zimmer C, Alam S, Guillemot F, Schuurmans C (2006) A role for proneural genes in the maturation of cortical progenitor cells. Cereb Cortex 16:I138–I151. https://doi.org/10.1093/cercor/bhj168
Chouchane M, Costa MR (2019) Instructing neuronal identity during CNS development and astroglial-lineage reprogramming: roles of NEUROG2 and ASCL1. Brain Res 1705:66–74. https://doi.org/10.1016/j.brainres.2018.02.045
Dennis DJ, Han SS, Schuurmans C (2019) bHLH transcription factors in neural development, disease, and reprogramming. Brain Res 1705:48–65. https://doi.org/10.1016/j.brainres.2018.03.013
Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL (1993) Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75:463–476. https://doi.org/10.1016/0092-8674(93)90381-y
Dennis DJ, Wilkinson G, Li S, Dixit R, Adnani L, Balakrishnan A, Han S, Kovach C, Gruenig N, Kurrasch DM, Dyck RH, Schuurmans C (2017) Neurog2 and Ascl1 together regulate a postmitotic derepression circuit to govern laminar fate specification in the murine neocortex. Proc Natl Acad Sci U S A 114:4934–4943. https://doi.org/10.1073/pnas.1701495114
Fode C, Ma QF, Casarosa S, Ang SL, Anderson DJ, Guillemot F (2000) A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes Dev 14:67–80. https://doi.org/10.1101/gad.14.1.67
Kim HJ, McMillan E, Han FB, Svendsen CN (2009) Regionally specified human neural progenitor cells derived from the mesencephalon and forebrain undergo increased neurogenesis following overexpression of ASCL1. Stem Cells 27:390–398. https://doi.org/10.1634/stemcells.2007-1047
Nieto M, Schuurmans C, Britz O, Guillemot F (2001) Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron 29:401–413. https://doi.org/10.1016/S0896-6273(01)00214-8
Ide M, Yamada K, Toyota T, Iwayama Y, Ishitsuka Y, Minabe Y, Nakamura K, Hattori N, Asada T, Mizuno Y, Mori N, Yoshikawa T (2005) Genetic association analyses of PHOX2B and ASCL1 in neuropsychiatric disorders: evidence for association of ASCL1 with Parkinson’s disease. Hum Genet 117:520–527. https://doi.org/10.1007/s00439-005-1342-8
Phillips HS, Kharbanda S, Chen RH, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173. https://doi.org/10.1016/j.ccr.2006.02.019
Park NI et al (2017) ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells. Cell Stem Cell 21:411–411. https://doi.org/10.1016/j.stem.2017.06.004
Kim J, Han D, Byun SH, Kwon M, Cho JY, Pleasure SJ, Yoon K (2018) Ttyh1 regulates embryonic neural stem cell properties by enhancing the Notch signaling pathway. EMRO Rep 19:11. https://doi.org/10.15252/embr.201745472
Wu HN, Cao XL, Fang Z, Zhang YF, Han WJ, Yue KY, Cao Y, Zheng MH, Wang LL, Han H (2019) Deficiency of Ttyh1 downstream to Notch signaling results in precocious differentiation of neural stem cells. Biochem Biophys Res Commun 514:842–847. https://doi.org/10.1016/j.bbrc.2019.04.181
Wierstra I (2008) Sp1: emerging roles - beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 372:1–13. https://doi.org/10.1016/j.bbrc.2008.03.074
Feng ZH, Chang RCC, Bing GY, Hudson P, Tiao NY, Jin L, Hong JS (1999) Long-term increase of Sp-1 transcription factors in the hippocampus after kainic acid treatment. Brain Res Mol Brain Res 69:144–148. https://doi.org/10.1016/s0169-328x(99)00099-6
Pinacho R, Villalmanzo N, Roca M, Iniesta R, Monje A, Haro JM, Meana JJ, Ferrer I, Gill G, Ramos B (2013) Analysis of Sp transcription factors in the postmortem brain of chronic schizophrenia: a pilot study of relationship to negative symptoms. J Psychiatr Res 47:926–934. https://doi.org/10.1016/j.jpsychires.2013.03.004
Black AR, Black JD, Azizkhan-Clifford J (2001) Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol 188:143–160. https://doi.org/10.1002/jcp.1111
Leone G, DeGregori J, Yan Z, Jakoi L, Ishida S, Williams RS, Nevins JR (1998) E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev 12:2120–2130. https://doi.org/10.1101/gad.12.14.2120
Li XP, Li H, Zhang R, Liu J (2015) MicroRNA-449a inhibits proliferation and induces apoptosis by directly repressing E2F3 in gastric cancer. Cell Physiol Biochem 35:2033–2042. https://doi.org/10.1159/000374010
Ren XS, Yin MH, Zhang X, Wang Z, Feng SP, Wang GX, Luo YJ, Liang PZ, Yang XQ, He JX, Zhang BL (2014) Tumor-suppressive microRNA-449a induces growth arrest and senescence by targeting E2F3 in human lung cancer cells. Cancer Lett 344:195–203. https://doi.org/10.1016/j.canlet.2013.10.031
Tang GD, Wu J, Xiao GL, Huo L (2015) miR-203 sensitizes glioma cells to temozolomide and inhibits glioma cell invasion by targeting E2F3. Mol Med Rep 11:2838–2844. https://doi.org/10.3892/mmr.2014.3101
Cui JG, Zhao Y, Sethi P, Li YY, Mahta A, Culicchia F, Lukiw WJ (2010) Micro-RNA-128 (miRNA-128) down-regulation in glioblastoma targets ARP5 (ANGPTL6), Bmi-1 and E2F–3a, key regulators of brain cell proliferation. J Neurooncol 98:297–304. https://doi.org/10.1007/s11060-009-0077-0
Bot AM, Debski KJ, Lukasiuk K (2013) Alterations in miRNA levels in the dentate gyrus in epileptic rats. PLoS One. https://doi.org/10.1371/journal.pone.0076051
Gorter JA, Iyer A, White I, Colzi A, van Vliet EA, Sisodiya S, Aronica E (2014) Hippocampal subregion-specific microRNA expression during epileptogenesis in experimental temporal lobe epilepsy. Neurobiol Dis 62:508–520. https://doi.org/10.1016/j.nbd.2013.10.026
Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T (2005) Matlnspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942. https://doi.org/10.1093/bioinformatics/bti473
Araujo IM, Carreira BP, Pereira T, Santos PF, Soulet D, Inacio A, Bahr BA, Carvalho AP, Ambrosio AF, Carvalho CM (2007) Changes in calcium dynamics following the reversal of the sodium-calcium exchanger have a key role in AMPA receptor-mediated neurodegeneration via calpain activation in hippocampal neurons. Cell Death Differ 14:1635–1646. https://doi.org/10.1038/sj.cdd.4402171
Acknowledgements
The authors wish to thank Dr. Kinga Szydlowska (Laboratory of Epileptogenesis, Nencki Institute of Experimental Biology) for the support with the imaging studies.
Funding
This work was supported by the National Science Centre, Poland grant (2015/19/N/NZ3/03268 to MGW and 2011/03/B/NZ4/00302 to KL).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection were performed by Malgorzata Gorniak-Walas. Testing the purity of the cell cultures was performed by KN. Data analysis and interpretation was performed by Malgorzata Gorniak-Walas and Katarzyna Lukasiuk. The manuscript was written by MGW and KL.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest. This manuscript has not been published and is not under consideration for publication elsewhere.
Ethical Approval
All of the procedures were performed in accordance with the Animal Protection Act of Poland (Directive 2010/63/EU). No ethical approval is required under Polish law for tissue collection.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gorniak-Walas, M., Nizinska, K. & Lukasiuk, K. Cloning and Functional Analysis of Rat Tweety-Homolog 1 Gene Promoter. Neurochem Res 46, 2463–2472 (2021). https://doi.org/10.1007/s11064-021-03374-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03374-2