Canadian Consensus for Biomarker Testing and Treatment of TRK Fusion Cancer in Pediatric Patients

 , , and
, , and 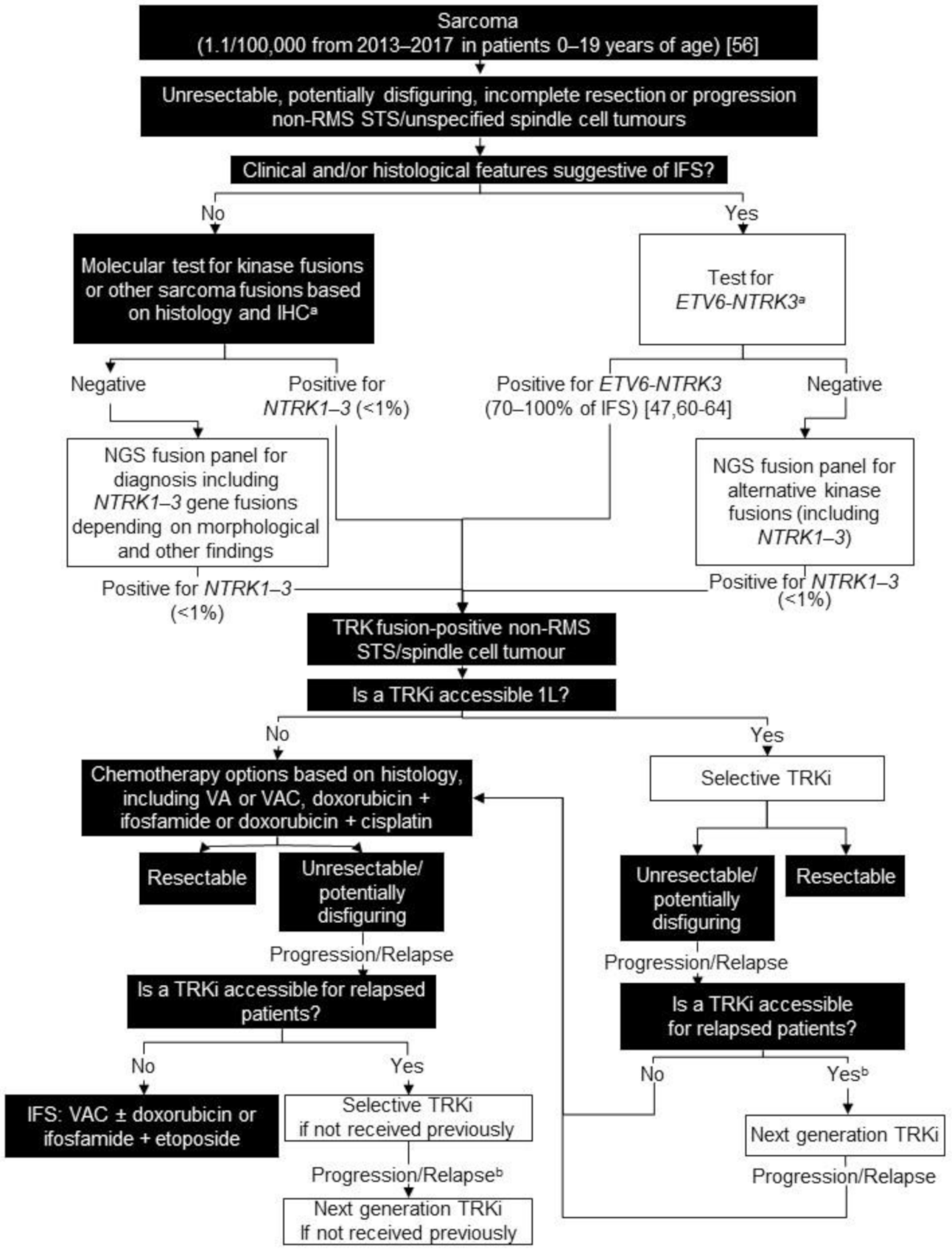{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Targeted Therapy for TRK Fusion Cancer
- Adult Phase I trial (NCT02122913),
- SCOUT (pediatric [≤21 years of age] Phase I/Phase II basket trial) (NCT02637687),
- NAVIGATE (adolescent/adult [≥12 years of age] Phase II basket trial) (NCT02576431).
- ALKA-372-001 (adult Phase I basket trial) (NCT02097810),
- STARTRK-1 (adult Phase I basket trial) (NCT02097810),
- STARTRK-2 (adult Phase II basket trial) (NCT02568267),
- STARTRK-NG (adolescent/pediatric [≤20 years of age] Phase I/II basket trial) (NCT02650401).
1.2. Development of Resistance to TRK Inhibitors
1.3. Regulatory and Funding Status of TRK Inhibitors in Canada, as of 2020
1.4. NTRK Gene Fusion Testing
1.4.1. Immunohistochemistry
1.4.2. Fluorescence In-Situ Hybridization (FISH)
1.4.3. Next-Generation Sequencing
1.5. Access to NTRK Gene Fusion Testing in Canada
2. Method to Achieve Consensus on TRK Fusion Cancer Algorithms
3. Non-Rhabdomyosarcoma (RMS) Soft Tissue Sarcoma (STS)/Unspecified Spindle Cell Tumours
3.1. Background
3.2. Testing Consensus
3.3. Treatment Consensus
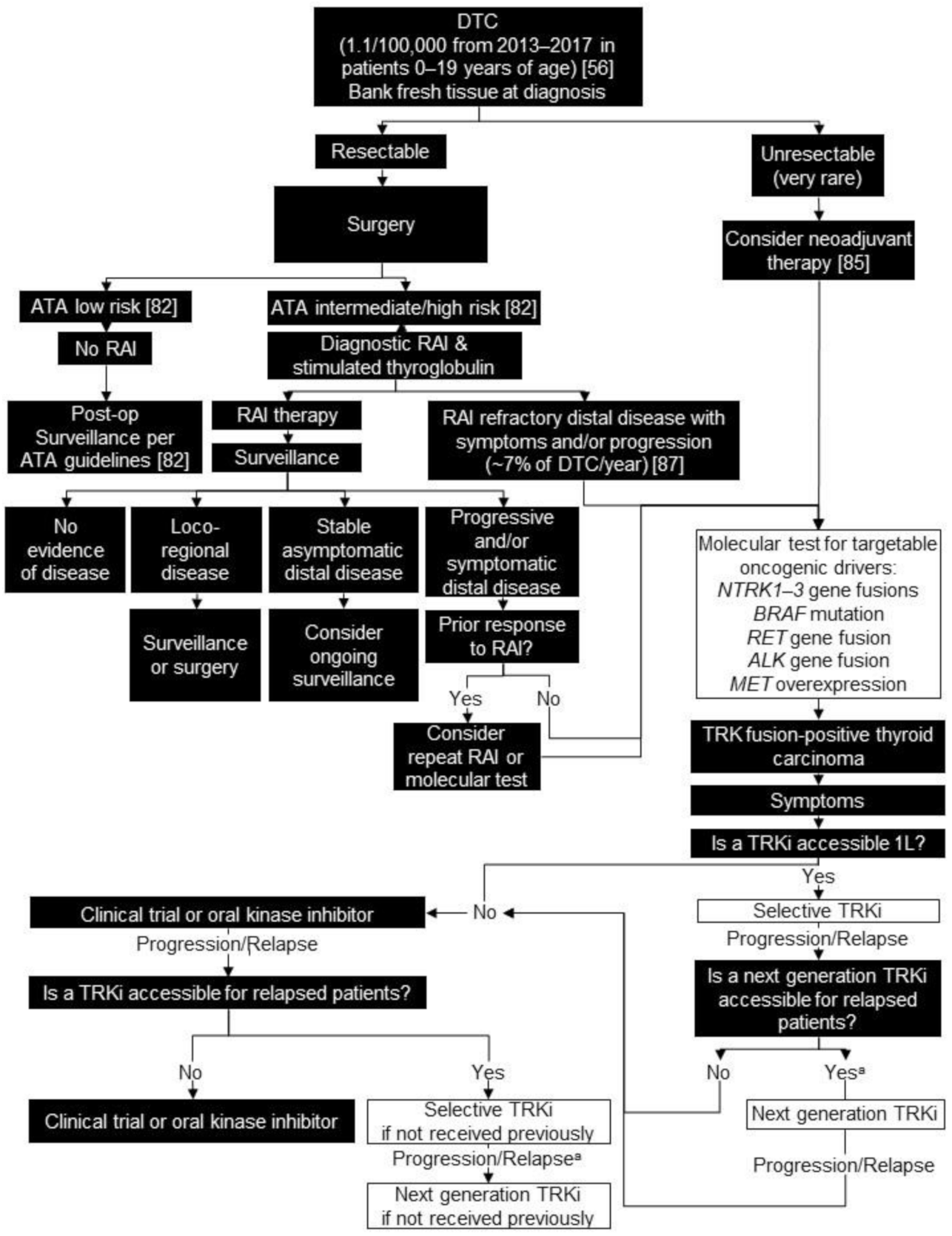
4. Differentiated Thyroid Carcinoma (DTC)
4.1. Background
4.2. Testing Consensus
4.3. Treatment Consensus
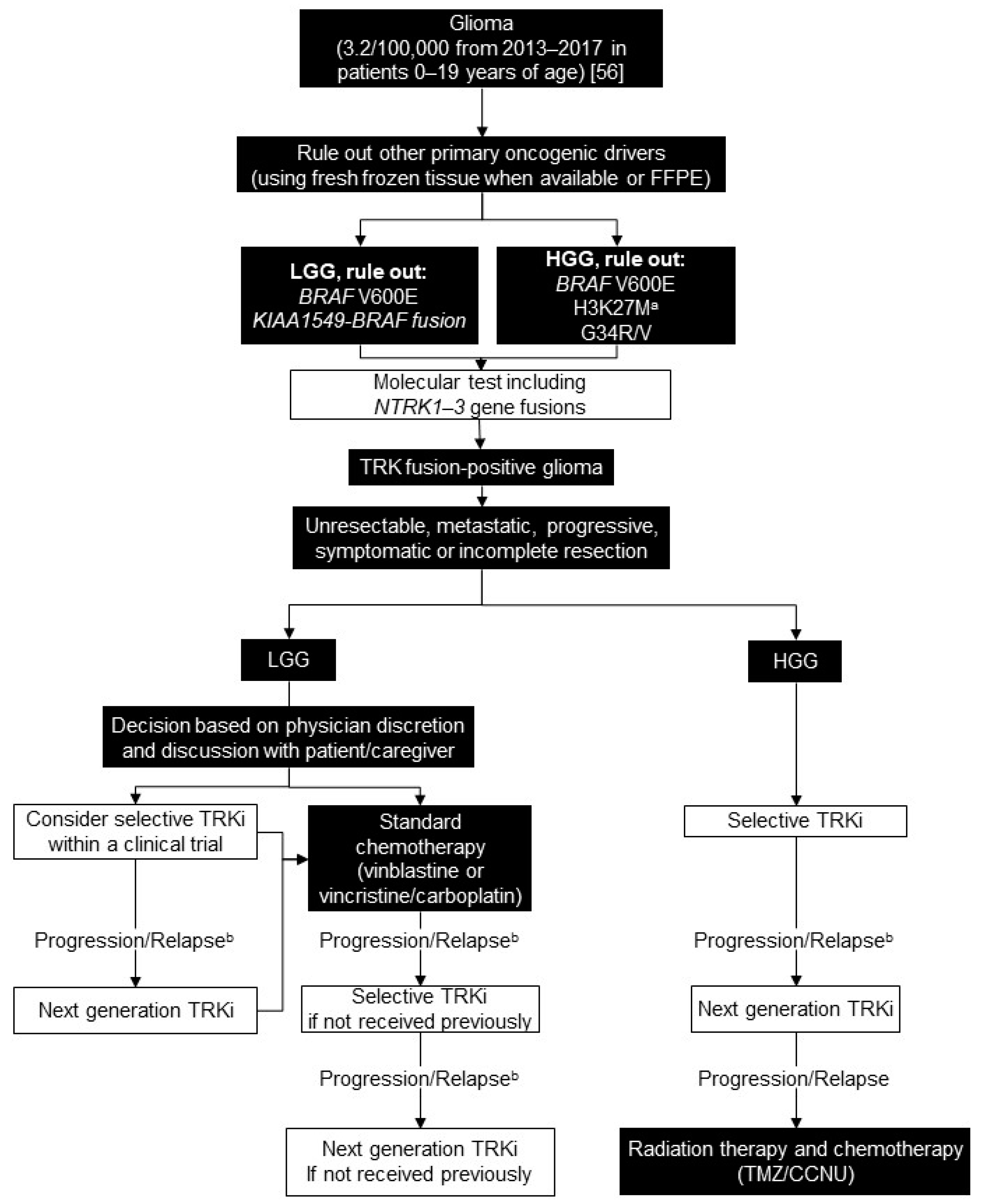
5. Glioma
5.1. Background
5.2. Testing Consensus
5.3. Treatment Consensus
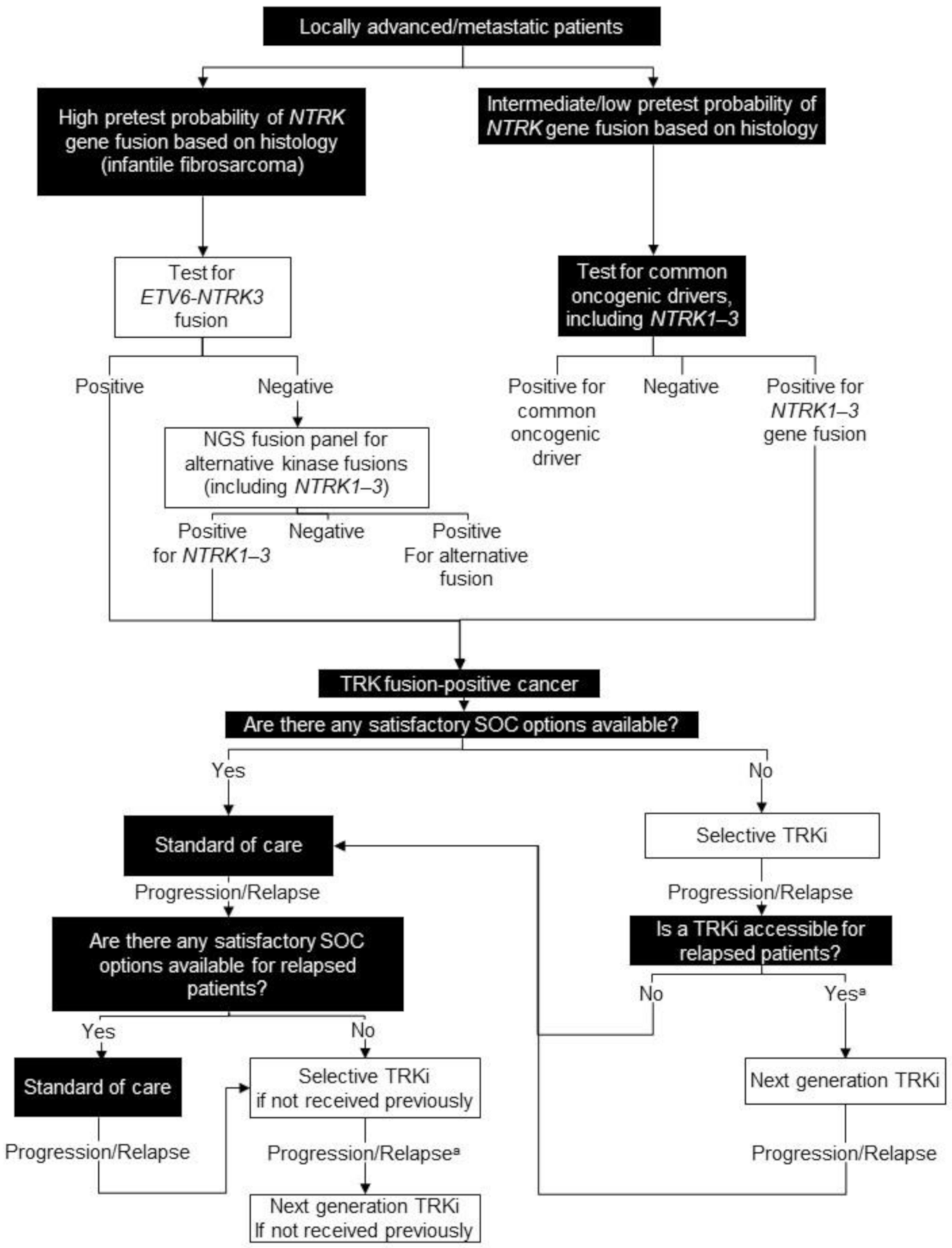
6. Tumour-Agnostic
6.1. Background
6.2. Testing Consensus
6.3. Treatment Consensus
7. Regulatory Landscape of TRK Inhibitor Therapy in Canada
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing Down an Old Oncogene in a New Era of Targeted Therapy. Cancer Discov. 2014, 5, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulciani, S.; Santos, E.; Lauver, A.V.; Long, L.K.; Aaronson, S.A.; Barbacid, M. Oncogenes in Solid Human Tumours. Nat. Cell Biol. 1982, 300, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Miranda, C.; Pierotti, M. Rearrangements of NTRK1 Gene in Papillary Thyroid Carcinoma. Mol. Cell. Endocrinol. 2010, 321, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Stephens, R.M.; Loeb, D.M.; Copeland, T.D.; Pawson, T.; Greene, L.A.; Kaplan, D.R. Trk Receptors Use Redundant Signal Transduction Pathways Involving SHC and PLC-γ1 to Mediate NGF Responses. Neuron 1994, 12, 691–705. [Google Scholar] [CrossRef]
- Holgado-Madruga, M.; Moscatello, D.K.; Emlet, D.R.; Dieterich, R.; Wong, A.J. Grb2-Associated Binder-1 Mediates Phosphatidylinositol 3-Kinase Activation and the Promotion of Cell Survival by Nerve Growth Factor. Proc. Natl. Acad. Sci. USA 1997, 94, 12419–12424. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Riccio, A.; Zhang, Y.; Ginty, D.D. Identification and Characterization of Novel Substrates of Trk Receptors in Developing Neurons. Neuron 1998, 21, 1017–1029. [Google Scholar] [CrossRef] [Green Version]
- U.S. National Library of Medicine. NTRK1 Neurotrophic Receptor Tyrosine Kinase 1 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/4914 (accessed on 1 April 2020).
- U.S. National Library of Medicine. NTRK2 Neurotrophic Receptor Tyrosine Kinase 2 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/4915 (accessed on 1 April 2020).
- U.S. National Library of Medicine. NTRK3 Neurotrophic Receptor Tyrosine Kinase 3 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/4916 (accessed on 1 April 2020).
- Latysheva, N.S.; Babu, M.M. Discovering and Understanding Oncogenic Gene Fusions through Data Intensive Computational Approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [Green Version]
- Lassen, U. How I treat NTRK Gene Fusion-Positive Cancers. ESMO Open 2019, 4, e000612. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.Y.; Goldman, D.A.; Hechtman, J.F.; Benayed, R.; Schram, A.M.; Cocco, E.; Shifman, S.; Gong, Y.; Kundra, R.; Solomon, J.P.; et al. TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin. Cancer Res. 2020, 26, 1624–1632. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.J.; Schram, A.M.; Hyman, D.M. Basket Studies: Redefining Clinical Trials in the Era of Genome-Driven Oncology. Annu. Rev. Med. 2018, 69, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Dubois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; Van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hong, D.S.; Bauer, T.; Lee, J.; Dowlati, A.; Brose, M.; Farago, A.; Taylor, M.; Shaw, A.; Montez, S.; Meric-Bernstam, F.; et al. Larotrectinib in Adult Patients with Solid Tumours: A Multi-Centre, Open-Label, Phase I Dose-Escalation Study. Ann. Oncol. 2019, 30, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.; Dubois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for Paediatric Solid Tumours Harbouring NTRK Gene Fusions: Phase 1 Results from a Multicentre, Open-Label, Phase 1/2 Study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Albert, C.M.; Pappo, A.; Geoerger, B.; Doz, F.F.; Nagasubramanian, N.R.; Bielack, S.; DuBois, S.G.M.D.; Schulte, J.; Shukla, N.; et al. Larotrectinib Demonstrates Durable Efficacy and Safety in an Expanded Dataset of Pediatric Patients with TRK Fusion Cancer. In Proceedings of the 52nd Congress of the International Society of Paediatric Oncology (SIOP), Virtual, 14–17 October 2020; p. 935. [Google Scholar]
- Perreault, S.; Doz, F.; Drilon, A.; Geoerger, B.; Boni, V.; Chisholm, J.; Dubois, S.G.; Grilley-Olson, E.J.; Hong, D.S.; Italiano, A.; et al. CTNI-67. Efficacy and Safety of Larotrectinib in Patients with Tropomyosin Receptor Kinase (TRK) Fusion Primary Central Nervous System (CNK) Tumors: An Expanded Dataset. Neuro-Oncology 2020, 22, ii58. [Google Scholar] [CrossRef]
- Joshi, S.K.; Davare, M.A.; Druker, B.J.; Tognon, C.E. Revisiting NTRKs as an Emerging Oncogene in Hematological Malignancies. Leukemia 2019, 33, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.; Pavlick, D.; Yoshimi, A.; Marcelus, C.; Chung, S.S.; Hechtman, J.F.; Benayed, R.; Cocco, E.; Durham, B.H.; Bitner, L.; et al. Oncogenic TRK Fusions Are Amenable to Inhibition in Hematologic Malignancies. J. Clin. Investig. 2018, 128, 3819–3825. [Google Scholar] [CrossRef]
- Desai, A.V.; Robinson, G.W.; Basu, E.M.; Foster, J.; Gauvain, K.; Sabnis, A.; Shusterman, S.; Macy, M.E.; Maese, L.; Yoon, J.; et al. Updated Entrectinib Data in Children and Adolescents with Recurrent or Refractory Solid Tumors, Including Primary CNS Tumors. J. Clin. Oncol. 2020, 38, 107. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent-Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A. TRK Inhibitors in TRK Fusion-Positive Cancers. Ann. Oncol. 2019, 30, viii23–viii30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, K.P.; Borazanci, E.; Shaw, A.T.; Katayama, R.; Shimizu, Y.; Zhu, V.W.; Sun, T.Y.; Wakelee, H.A.; Madison, R.; Schrock, A.B.; et al. US Phase 1 First-in-Human Study of Taletrectinib (DS-6051b/AB-106), a ROS1/TRK Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Health Canada. Summary Basis of Decision—Vitrakvi—Health Canada. Available online: https://hpr-rps.hres.ca/reg-content/summary-basis-decision-detailTwo.php?linkID=SBD00455 (accessed on 9 April 2020).
- Health Canada. Rozlytrek—Notice of Compliance with Conditions—Qualifying Notice. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/notice-compliance/conditions/rozlytrek-qualifying-notice.html (accessed on 9 April 2020).
- U.S. Food & Drug Administration. Rozlytrek Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf (accessed on 22 November 2020).
- Tsao, M.S.; Torlakovic, E.; Stockley, T.; Lo, B. CANTRK: A Canadian Multi-Centre NTRK Gene Fusion Testing Validation in Solid Tumors Project. In Proceedings of the Association for Molecular Pathology Annual Meeting, Virtual, 16–20 November 2020; ST07. [Google Scholar]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hechtman, J.F.; Benayed, R.; Hyman, D.M.; Drilon, A.; Zehir, A.; Frosina, D.; Arcila, M.E.; Dogan, S.; Klimstra, D.S.; Ladanyi, M.; et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am. J. Surg. Pathol. 2017, 41, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular Characterization of Cancers with NTRK Gene Fusions. Mod. Pathol. 2019, 32, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.J.; Zehir, A.; Sireci, A.N.; Aisner, D.L. Detection of Tumor NTRK Gene Fusions to Identify Patients Who May Benefit from Tyrosine Kinase (TRK) Inhibitor Therapy. J. Mol. Diagn. 2019, 21, 553–571. [Google Scholar] [CrossRef] [Green Version]
- Penault-Llorca, F.; Rudzinski, E.R.; Sepulveda, A.R. Testing Algorithm for Identification of Patients with TRK Fusion Cancer. J. Clin. Pathol. 2019, 72, 460–467. [Google Scholar] [CrossRef]
- Negri, T.; Tamborini, E.; Dagrada, G.P.; Greco, A.; Staurengo, S.; Guzzo, M.; Locati, L.D.; Carbone, A.; Pierotti, M.A.; Licitra, L.; et al. TRK-A, HER-2/neu, and KIT Expression/Activation Profiles in Salivary Gland Carcinoma. Transl. Oncol. 2008, 1, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Liu, L.; Cai, B.; He, Y.; Wan, X. Suppression of Anoikis by the Neurotrophic Receptor TrkB in Human Ovarian Cancer. Cancer Sci. 2008, 99, 543–552. [Google Scholar] [CrossRef]
- Wadhwa, S.; Nag, T.C.; Jindal, A.; Kushwaha, R.; Mahapatra, A.K.; Sarkar, C. Expression of the Neurotrophin Receptors Trk A and Trk B in Adult Human Astrocytoma and Glioblastoma. J. Biosci. 2003, 28, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Fletcher, C.D.M.; Hornick, J.L. Evaluation of Pan-TRK Immunohistochemistry in Infantile Fibrosarcoma, Lipofibromatosis-like Neural Tumour and Histological Mimics. Histopathology 2018, 73, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.; Bibeau, F.; Dietel, M.; Hechtman, J.; Troiani, T.; López-Rios, F.; Douillard, J.-Y.; et al. ESMO Recommendations on the Standard Methods to Detect NTRK Fusions in Daily Practice and Clinical Research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.P.; Linkov, I.; Rosado, A.; Mullaney, K.; Rosen, E.Y.; Frosina, D.; Jungbluth, A.A.; Zehir, A.; Benayed, R.; Drilon, A.; et al. NTRK Fusion Detection across Multiple Assays and 33,997 Cases: Diagnostic Implications and Pitfalls. Mod. Pathol. 2020, 33, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.-C.; Sung, Y.-S.; Argani, P.; Swanson, D.; Alaggio, R.; Tap, W.; Wexler, L.; Dickson, B.C.; Antonescu, C.R. NTRK3 Overexpression in Undifferentiated Sarcomas with YWHAE and BCOR Genetic Alterations. Mod. Pathol. 2020, 33, 1341–1349. [Google Scholar] [CrossRef]
- Solomon, J.P.; Hechtman, J.F. Detection of NTRK Fusions: Merits and Limitations of Current Diagnostic Platforms. Cancer Res. 2019, 79, 3163–3168. [Google Scholar] [CrossRef]
- Antonescu, C.R. Emerging Soft Tissue Tumors with Kinase Fusions: An Overview of the Recent Literature with an Emphasis on Diagnostic Criteria. Genes Chromosom. Cancer 2020, 59, 437–444. [Google Scholar] [CrossRef]
- Church, A.J.; Calicchio, M.L.; Nardi, V.; Skalova, A.; Pinto, A.; Dillon, D.A.; Gomez-Fernandez, C.R.; Manoj, N.; Haimes, J.D.; Stahl, A.J.; et al. Recurrent EML4–NTRK3 Fusions in Infantile Fibrosarcoma and Congenital Mesoblastic Nephroma Suggest a Revised Testing Strategy. Mod. Pathol. 2018, 31, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Knezevich, S.R.; Garnett, M.J.; Pysher, T.J.; Beckwith, J.B.; Grundy, P.E.; Sorensen, P.H. ETV6-NTRK3 Gene Fusions and Trisomy 11 Establish a Histogenetic Link between Mesoblastic Nephroma and Congenital Fibrosarcoma. Cancer Res. 1998, 58, 5046–5048. [Google Scholar]
- Wegert, J.; Vokuhl, C.; Collord, G.; Velasco-Herrera, M.D.C.; Farndon, S.J.; Guzzo, C.; Jorgensen, M.; Anderson, J.; Slater, O.; Duncan, C.; et al. Recurrent Intragenic Rearrangements of EGFR and BRAF in Soft Tissue Tumors of Infants. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.; Benayed, R.; Hechtman, J.; Ladanyi, M. Identifying Patients with NTRK Fusion Cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beadling, C.; Wald, A.I.; Warrick, A.; Neff, T.L.; Zhong, S.; Nikiforov, Y.E.; Corless, C.L.; Nikiforova, M.N. A Multiplexed Amplicon Approach for Detecting Gene Fusions by Next-Generation Sequencing. J. Mol. Diagn. 2016, 18, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikine, Y.; Kuo, F.; Lindeman, N.I. Clinical and Technical Aspects of Genomic Diagnostics for Precision Oncology. J. Clin. Oncol. 2017, 35, 929–933. [Google Scholar] [CrossRef]
- Geoerger, B.; Van Tilburg, C.M.; DuBois, S.G.; Albert, C.M.; Federman, N.; Nagasubramanian, R.; Doz, F.; Orbach, D.; Bielack, S.; Neerav, S.; et al. Larotrectinib Efficacy and Saffety in Paediatric Patients with TRK Fusion Cancer. In Proceedings of the 51st Congress of the International Society of Paediatric Oncology (SIOP), Lyon, France, 23–26 October 2019; FP114 SIOP19-0869. [Google Scholar]
- Drilon, A.E.; Dubois, S.G.; Farago, A.F.; Geoerger, B.; Grilley-Olson, J.E.; Hong, D.S.; Sohal, D.; Van Tilburg, C.M.; Ziegler, D.S.; Ku, N.; et al. Activity of Larotrectinib in TRK Fusion Cancer Patients with Brain Metastases or Primary Central Nervous System Tumors. J. Clin. Oncol. 2019, 37, 2006. [Google Scholar] [CrossRef]
- Smrke, A.; Wang, Y.; Simmons, C.E. Update on Systemic Therapy for Advanced Soft-Tissue Sarcoma. Curr. Oncol. 2020, 27, 25–33. [Google Scholar] [CrossRef]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric Rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute. Cancer Stat Facts: Childhood Brain and Other Nervous System Cancer (Ages 0–19). Available online: https://seer.cancer.gov/statfacts/html/childbrain.html (accessed on 28 August 2020).
- Enhanced Childhood Surveillance System. Cancer in Young People: A Report From the Enhanced Childhood Cancer Surveillance System. Available online: https://www.canada.ca/content/dam/hc-sc/documents/services/publications/science-research-data/cancer-young-people-canada-surveillance-2017-eng.pdf (accessed on 7 May 2020).
- Rudzinski, E.R.; Lockwood, C.M.; Stohr, B.A.; Vargas, S.O.; Sheridan, R.; Black, J.O.; Rajaram, V.; Laetsch, T.; Davis, J.L. Pan-Trk Immunohistochemistry Identifies NTRK Rearrangements in Pediatric Mesenchymal Tumors. Am. J. Surg. Pathol. 2018, 42, 927–935. [Google Scholar] [CrossRef]
- Agaram, N.P.; Zhang, L.; Sung, Y.-S.; Chen, C.-L.; Chung, C.T.; Antonescu, C.R.; Fletcher, C.D.M. Recurrent NTRK1 Gene Fusions Define a Novel Subset of Locally Aggressive Lipofibromatosis-like Neural Tumors. Am. J. Surg. Pathol. 2016, 40, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Knezevich, S.R.; McFadden, D.E.; Tao, W.; Lim, J.F.; Sorensen, P.H. A novel ETV6-NTRK3 Gene Fusion in Congenital Fibrosarcoma. Nat. Genet. 1998, 18, 184–187. [Google Scholar] [CrossRef]
- Bourgeois, J.M.; Knezevich, S.R.; Mathers, J.A.; Sorensen, P.H.B. Molecular Detection of the ETV6-NTRK3 Gene Fusion Differentiates Congenital Fibrosarcoma From Other Childhood Spindle Cell Tumors. Am. J. Surg. Pathol. 2000, 24, 937–946. [Google Scholar] [CrossRef]
- Rubin, B.P.; Chen, C.-J.; Morgan, T.W.; Xiao, S.; Grier, H.E.; Kozakewich, H.P.; Perez-Atayde, A.R.; Fletcher, J.A. Congenital Mesoblastic Nephroma t(12;15) is Associated withETV6-NTRK3 Gene Fusion. Am. J. Pathol. 1998, 153, 1451–1458. [Google Scholar] [CrossRef]
- Sheng, W.-Q.; Hisaoka, M.; Okamoto, S.; Tanaka, A.; Meis-Kindblom, J.M.; Kindblom, L.-G.; Ishida, T.; Nojima, T.; Hashimoto, H. Congenital-Infantile Fibrosarcoma. Am. J. Clin. Pathol. 2001, 115, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, M.L.; Ahn, P.; Perez-Atayde, A.R.; Gebhardt, M.C.; Shamberger, R.C.; Grier, H.E. Treatment of Infantile Fibrosarcoma With Chemotherapy and Surgery: Results from the Dana-Farber Cancer Institute and Children’s Hospital, Boston. J. Pediatr. Hematol. 2002, 24, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Orbach, D.; Rey, A.; Cecchetto, G.; Oberlin, O.; Casanova, M.; Thebaud, E.; Scopinaro, M.; Bisogno, G.; Carli, M.; Ferrari, A. Infantile Fibrosarcoma: Management Based on the European Experience. J. Clin. Oncol. 2010, 28, 318–323. [Google Scholar] [CrossRef]
- Orbach, D.; Brennan, B.; De Paoli, A.; Gallego, S.; Mudry, P.; Francotte, N.; Van Noesel, M.; Kelsey, A.; Alaggio, R.; Ranchère, D.; et al. Conservative Strategy in Infantile Fibrosarcoma Is Possible: The European Paediatric Soft Tissue Sarcoma Study Group Experience. Eur. J. Cancer 2016, 57, 1–9. [Google Scholar] [CrossRef]
- Linch, M.; Miah, A.B.; Thway, K.; Judson, I.; Benson, C. Systemic Treatment of Soft-Tissue Sarcoma—Gold Standard and Novel Therapies. Nat. Rev. Clin. Oncol. 2014, 11, 187–202. [Google Scholar] [CrossRef]
- Ferrari, A.; De Salvo, G.L.; Brennan, B.; Van Noesel, M.M.; De Paoli, A.; Casanova, M.; Francotte, N.; Kelsey, A.; Alaggio, R.; Oberlin, O.; et al. Synovial Sarcoma in Children and Adolescents: The European Pediatric Soft Tissue Sarcoma Study Group Prospective Trial (EpSSG NRSTS 2005). Ann. Oncol. 2015, 26, 567–572. [Google Scholar] [CrossRef]
- Demetri, G.D.; Albert, C.M.; Daniel, S.W.; Stefan, B.; Orbach, D.; DuBois, S.G.; Federman, N.; Geoerger, B.; Kummar, S.; Laetsch, T.W.; et al. Larotrectinib Efficacy and Safety in Patients with TRK Fusion Sarcomas. In Proceedings of the CTOS Annual Meetinng, Toyko, Japan, 5 November 2019. [Google Scholar]
- Wong, V.; Pavlick, D.; Brennan, T.; Yelensky, R.; Crawford, J.R.; Ross, J.S.; Miller, V.A.; Malicki, D.M.; Stephens, P.J.; Ali, S.; et al. Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib. J. Natl. Cancer Inst. 2016, 108, djv307. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.L.; Vargas, S.O.; Rudzinski, E.R.; Marti, J.M.L.; Janeway, K.; Forrest, S.; Winsnes, K.; Pinto, N.; Yang, S.E.; VanSandt, M.; et al. Recurrent RET Gene Fusions in Paediatric Spindle Mesenchymal Neoplasms. Histopathology 2020, 76, 1032–1041. [Google Scholar] [CrossRef]
- Xie, L.; Onysko, J.; Morrison, H. Childhood Cancer Incidence in Canada: Demographic and Geographic Variation of Temporal Trends (1992–2010). Health Promot. Chronic Dis. Prev. Can. 2018, 38, 79–115. [Google Scholar] [CrossRef] [Green Version]
- Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2019. Available online: https://www.cancer.ca/~/media/cancer.ca/CW/cancer%20information/cancer%20101/Canadian%20cancer%20statistics/Canadian-Cancer-Statistics-2019-EN.pdf?la=en (accessed on 7 May 2020).
- Hogan, A.R.; Zhuge, Y.; Perez, E.A.; Koniaris, L.G.; Lew, J.I.; Sola, J.E. Pediatric Thyroid Carcinoma: Incidence and Outcomes in 1753 Patients. J. Surg. Res. 2009, 156, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.J. Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol. Metab. Clin. N. Am. 2017, 46, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, J.D.; Sabbaghian, N.; Fahiminiya, S.; Chami, R.; Mete, O.; Acker, M.; Wu, M.K.; Shlien, A.; De Kock, L.; Foulkes, W.D. DICER1 Mutations Are Frequent in Adolescent-Onset Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 2009–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, V.A.; Rudzinski, E.R.; Hawkins, D.S. Thyroid Cancer in the Pediatric Population. Genes 2019, 10, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, M.L.; Vyas, M.; Horne, M.J.; Virk, R.K.; Morotti, R.; Liu, Z.; Tallini, G.; Nikiforova, M.N.; Christison-Lagay, E.R.; Udelsman, R.; et al. NTRK Fusion Oncogenes in Pediatric Papillary Thyroid Carcinoma in Northeast United States. Cancer 2016, 122, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Lazar, L.; Lebenthal, Y.; Steinmetz, A.; Yackobovitch-Gavan, M.; Phillip, M. Differentiated Thyroid Carcinoma in Pediatric Patients: Comparison of Presentation and Course between Pre-Pubertal Children and Adolescents. J. Pediatr. 2009, 154, 708–714. [Google Scholar] [CrossRef]
- Feinmesser, R.; Lubin, E.; Segal, Κ.; Noyek, A. Carcinoma of the Thyroid in Children—A Review. J. Pediatr. Endocrinol. Metab. 1997, 10, 561–568. [Google Scholar] [CrossRef]
- Hampson, S.; Stephens, D.; Wasserman, J.D. Young Age is Associated with Increased Rates of Residual and Recurrent Paediatric Differentiated Thyroid Carcinoma. Clin. Endocrinol. 2018, 89, 212–218. [Google Scholar] [CrossRef]
- Francis, G.L.; Waguespack, S.G.; Bauer, A.J.; Angelos, P.; Benvenga, S.; Cerutti, J.M.; Dinauer, C.A.; Hamilton, J.K.; Hay, I.D.; Luster, M.; et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015, 25, 716–759. [Google Scholar] [CrossRef] [Green Version]
- Fugazzola, L.; Elisei, R.; Fuhrer, D.; Jarzab, B.; Leboulleux, S.; Newbold, K.; Smit, J. 2019 European Thyroid Association Guidelines for the Treatment and Follow-Up of Advanced Radioiodine-Refractory Thyroid Cancer. Eur. Thyroid J. 2019, 8, 227–245. [Google Scholar] [CrossRef]
- Cabanillas, E.M.; McFadden, D.G.; Durante, C. Thyroid Cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Kazahaya, K.; Prickett, K.K.; Paulson, V.A.; Dahl, J.P.; Manning, S.C.; Rudzinski, E.R.; Rastatter, J.C.; Parikh, S.R.; Hawkins, D.S.; Brose, M.S.; et al. Targeted Oncogene Therapy before Surgery in Pediatric Patients with Advanced Invasive Thyroid Cancer at Initial Presentation: Is it Time for a Paradigm shift? JAMA Otolaryngol. Head Neck Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.; Drilon, A.; Farago, A.; Brose, M.; McDermott, R.; Sohal, D.; Oh, D.-Y.; Almubarak, M.; Bauman, J.; Chu, E.; et al. 1916P Larotrectinib Treatment of Advanced TRK Fusion Thyroid Cancer. Ann. Oncol. 2020, 31, S1086. [Google Scholar] [CrossRef]
- Schmidt, A.; Iglesias, L.; Klain, M.; Pitoia, F.; Schlumberger, M.J. Radioactive Iodine-Refractory Differentiated Thyroid Cancer: An Uncommon but Challenging Situation. Arch. Endocrinol. Metab. 2017, 61, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; De Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2015, 16, x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Linabery, A.M.; Ross, J.A. Trends in Childhood Cancer Incidence in the U.S. (1992–2004). Cancer 2007, 112, 416–432. [Google Scholar] [CrossRef]
- Whittle, I.R. The Dilemma of Low Grade Glioma. J. Neurol. Neurosurg. Psychiatry 2004, 75, ii31–ii36. [Google Scholar] [CrossRef] [Green Version]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric Low-Grade Glioma in the Era of Molecular Diagnostics. Acta Neuropathol. Commun. 2020, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ryall, S.; Zapotocky, M.; Fukuoka, K.; Nobre, L.; Stucklin, A.G.; Bennett, J.; Siddaway, R.; Li, C.; Pajovic, S.; Arnoldo, A.; et al. Integrated Molecular and Clinical Analysis of 1000 Pediatric Low-Grade Gliomas. Cancer Cell 2020, 37, 569–583.e5. [Google Scholar] [CrossRef]
- Jones, D.T.; the International Cancer Genome Consortium PedBrain Tumor Project; Hutter, B.; Jäger, N.; Korshunov, A.; Kool, M.; Warnatz, H.-J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; et al. Recurrent Somatic Alterations of FGFR1 and NTRK2 in Pilocytic Astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Lassaletta, Á.; Scheinemann, K.; Zelcer, S.; Hukin, J.; Wilson, B.A.; Jabado, N.; Carret, A.-S.; Lafay-Cousin, L.; Larouche, V.; Hawkins, C.; et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children with Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. J. Clin. Oncol. 2016, 34, 3537–3543. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.A.; Turner, C.D.; Poussaint, T.Y.; Scott, R.M.; Goumnerova, L.; Kieran, M.W. Chemotherapy for Progressive Low-Grade Gliomas in Children Older than Ten Years: The Dana-Farber Experience. Pediatr. Hematol. Oncol. 2003, 20, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Stucklin, A.S.G.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET Drive a Group of Infantile Hemispheric Gliomas. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Warren, K.E. Diffuse Intrinsic Pontine GliomA: Poised for Progress. Front. Oncol. 2012, 2, 205. [Google Scholar] [CrossRef] [Green Version]
- Broniscer, A.; Baker, S.J.; West, A.N.; Fraser, M.M.; Proko, E.; Kocak, M.; Dalton, J.; Zambetti, G.P.; Ellison, D.W.; Kun, L.E.; et al. Clinical and Molecular Characteristics of Malignant Transformation of Low-Grade Glioma in Children. J. Clin. Oncol. 2007, 25, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The Genomic Landscape of Diffuse Intrinsic Pontine Glioma and Pediatric Non-brainstem High-Grade Glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S.; et al. Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef] [Green Version]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.-F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M.; et al. Molecular and Clinicopathologic Features of Gliomas Harboring NTRK Fusions. Acta Neuropathol. Commun. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C.; et al. Pediatric High-Grade GliomA: Biologically and Clinically in Need of New Thinking. Neuro-Oncology 2016, 19, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Ullah, M.; De La Cruz, C.C.; Hunsaker, T.; Senn, C.; Wirz, T.; Wagner, B.; Draganov, D.; Vazvaei, F.; Donzelli, M.; et al. Entrectinib, a TRK/ROS1 Inhibitor with Anti-CNS Tumor Activity: Differentiation from other Inhibitors in its Class Due to Weak Interaction with P-Glycoprotein. Neuro-Oncology 2020, 22, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.L.; Lockwood, C.M.; Stohr, B.; Boecking, C.; Al-Ibraheemi, A.; Dubois, S.G.; Vargas, S.O.; Black, J.O.; Cox, M.C.; Luquette, M.; et al. Expanding the Spectrum of Pediatric NTRK-rearranged Mesenchymal Tumors. Am. J. Surg. Pathol. 2019, 43, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib inTRKFusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Ronsley, R.; Rassekh, S.R.; Shen, Y.; Lee, A.F.; Jantzen, C.; Halparin, J.; Albert, C.; Hawkins, D.M.; Amed, S.; Rothstein, R.; et al. Application of Genomics to Identify Therapeutic Targets in Recurrent Pediatric Papillary Thyroid Carcinoma. Mol. Case Stud. 2018, 4, a002568. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perreault, S.; Chami, R.; Deyell, R.J.; El Demellawy, D.; Ellezam, B.; Jabado, N.; Morgenstern, D.A.; Narendran, A.; Sorensen, P.H.B.; Wasserman, J.D.; et al. Canadian Consensus for Biomarker Testing and Treatment of TRK Fusion Cancer in Pediatric Patients. Curr. Oncol. 2021, 28, 346-366. https://doi.org/10.3390/curroncol28010038
Perreault S, Chami R, Deyell RJ, El Demellawy D, Ellezam B, Jabado N, Morgenstern DA, Narendran A, Sorensen PHB, Wasserman JD, et al. Canadian Consensus for Biomarker Testing and Treatment of TRK Fusion Cancer in Pediatric Patients. Current Oncology. 2021; 28(1):346-366. https://doi.org/10.3390/curroncol28010038
Chicago/Turabian StylePerreault, Sébastien, Rose Chami, Rebecca J. Deyell, Dina El Demellawy, Benjamin Ellezam, Nada Jabado, Daniel A. Morgenstern, Aru Narendran, Poul H. B. Sorensen, Jonathan D. Wasserman, and et al. 2021. "Canadian Consensus for Biomarker Testing and Treatment of TRK Fusion Cancer in Pediatric Patients" Current Oncology 28, no. 1: 346-366. https://doi.org/10.3390/curroncol28010038