
Abstract
Along with intrinsic evolution, adaptation to selective pressure in new environments might have resulted in the circulatory SARS-CoV-2 strains in response to the geoenvironmental conditions of a country and the demographic profile of its population. With this target, the current study traced the evolutionary route and mutational frequency of 198 Bangladesh-originated SARS-CoV-2 genomic sequences available in the GISAID platform over a period of 13 weeks as of 14 July 2020. The analyses were performed using MEGA X, Swiss Model Repository, Virus Pathogen Resource and Jalview visualization. Our analysis identified that majority of the circulating strains strikingly differ from both the reference genome and the first sequenced genome from Bangladesh. Mutations in nonspecific proteins (NSP2-3, NSP-12(RdRp), NSP-13(Helicase)), S-Spike, ORF3a, and N-Nucleocapsid protein were common in the circulating strains with varying degrees and the most unique mutations (UM) were found in NSP3 (UM-18). But no or limited changes were observed in NSP9, NSP11, Envelope protein (E) and accessory factors (NSP7a, ORF 6, ORF7b) suggesting the possible conserved functions of those proteins in SARS-CoV-2 propagation. However, along with D614G mutation, more than 20 different mutations in the Spike protein were detected basically in the S2 domain. Besides, mutations in SR-rich region of N protein and P323L in RDRP were also present. However, the mutation accumulation showed a significant association (p = 0.003) with sex and age of the COVID-19-positive cases. So, identification of these mutational accumulation patterns may greatly facilitate vaccine development deciphering the age and the sex-dependent differential susceptibility to COVID-19.
Similar content being viewed by others


Introduction
In the past two decades, Coronaviruses mainly of the β-coronavirus family Coronaviridae and the subfamily Coronavirinae have been a major subject of deeper investigations due to their emergence, re-emergence and associated public health impact [1, 2]. Among the seven coronaviruses (229E, OC43, NL63, HKU1, SARS-CoV (Severe Acute Respiratory Syndrome Coronavirus), MERS-CoV (Middle East respiratory syndrome Coronavirus) and SARS-CoV-2 responsible for coronavirus disease 2019 (COVID-19)) causing human infections, the newly emerged single-stranded RNA beta-coronavirus SARS-CoV-2 has been wreaking havoc around the world since its emergence in mid-December 2019 in the Chinese city of Wuhan [1,2,3] and was first reported from Bangladesh on March 8, 2020. Currently, the COVID-19 disease has 1.9% of case fatality rate in Bangladesh, which is significantly lower than a lot of countries as Mexico, China, Italy, Spain, Canada, etc. [4].
This 29903-kb enveloped virus consists of a 5′‐untranslated region (5′‐UTR), spike (S), envelope (E), matrix (M), nucleocapsid (N) gene and 3′‐UTR4, among which E, M, S and N proteins are involved in protecting the genome by forming the structure of the virus [5]. On the other hand, among the 16 nonstructural proteins (NSPs), four NSPs (NSP12, NSP13, NSP14, NSP16) are involved in synthesizing and processing the viral RNA [5], while the remaining proteins are crucial cofactors facilitating the function of viral enzymes [6]. So, the current circulating strain might have evolved through the ongoing evolutionary process of mutations in these genes since its emergence [7]. Errors made by RdRp despite having proofreading activity [8] along with a direct response to selective pressure on the viral genome and homologous recombination may lead to mutational accumulation in the SARS-CoV-2 genome [9], while according to recent studies on mutation analysis, no recombination events were reported [10] and the sequence diversity of SARS-CoV-2 so far is very low [11]. On the contrary, the receptor-binding domain (RBD) in the S protein is the most variable genomic part in the beta-coronavirus group [12, 13], and some sites of S protein might be subjected to positive selection [14]. However, despite these variabilities in the SARS-CoV-2 genome, one key question remains as to whether these mutations have any functional impact on the pathogenicity of SARS-CoV-2. The previous experiences with MERS-CoV and SARS-CoV [15], the close relatives of SARS-CoV-2 [12, 13], showed that a single mutation might be significant enough to confer resistance to neutralizing antibodies against those viruses. Meanwhile, during the rampant spread of SARS-CoV-2 around the world, it has undergone multiple antigenic drifts including several mutations compromising the containment and diagnostics strategies along with the effectivity of repurposed drugs [16], which suggested that the virus will be active and spreading for a year or more before vaccines are available [12, 13]. Besides, based on amino acid changes of the genomes, 3 major clades (S, G, and V) were proposed in many more studies [12, 13, 17]. Another study by Tai et al. suggested that amino acid variations in the genome are associated with the stability of RBD/ACE2 structure [18]. Also, the primer–template mismatches might affect the stability and the functionality of polymerase [19]. On the other hand, a study by Su et al. revealed that the deletion of 382 nucleotides towards the 3’ end of the viral genome may have an impact on the viral phenotype [20]. Thus, these mutational analyses justify the potential of mutations in affecting the viral infectivity and adaptability to the new environment as well as explaining the differential rates of infection and mortality worldwide conducive to controlling the pandemic. Meanwhile, the data avalanche, especially the complete genome sequences in Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org/) has resulted in an unprecedented expeditious effort towards understanding the implications of genome diversity [21, 22] in pathogenicity, drug repositioning [12, 13] or developing diagnostic and preventive strategies [23]. Concurrently with the global sequence data, legionary complete genome sequences have been submitted from Bangladesh in GISAID since the first submission on 14 July, 2020 [24]. So, the current study was designed to investigate the genomic diversity of SARS-CoV-2 strains isolated from the country as well as analyzing the temporal profile of the mutational accumulations in the genome. Ultimately, this study will give an insight into the circulating strains of the country to devise a more effective containment strategy and efficient treatment regimen along with adding values to the global understanding of SARS-CoV-2 genome evolution and molecular basis of its pathogenicity, infectivity and drug/vaccine targets.
Materials and methods
Retrieval of SARS-CoV-2 genome sequences from the database
A total of 226 complete genome sequences of SARS-CoV-2 isolated from Bangladesh were retrieved from the GISAID virus database (https://www.gisaid.org/, last access 14 July 2020) along with the collection date and the patient history (Supplementary Material SM1 & Supplementary Table ST1). Alignment of the retrieved genome sequences of the SARS-CoV-2 strains was executed using online based Virus Pathogen Resource (https://www.viprbrc.org/) database and MEGA X tool [25] to remove ambiguous and low-quality sequences. Later, the MSA file was opened with Jalview visualization software to eliminate the redundancy of the studied sequences [26]. Finally, the complete viral genomes sequenced from both male and female patients, reported from Bangladesh were analyzed using the reference genome (NC_045512.2).
Determination of mutational accumulation and frequencies
The nucleotide positions with corresponding amino acid of each protein were identified using two databases: Swiss Model Repository (https://swissmodel.expasy.org/repository/) and the GISAID (https://www.gisaid.org/). An initial analysis was performed to identify the phylogenetic clusters using an analysis tool named Virus Pathogen Database and Analysis Resource (ViPR) and then all the retrieved sequences from GISAID were provided as input in the aforementioned database to collect mutational information in comparison with the reference genome (NC_045512). Moreover, GISAID was explored for the determination of mutational accumulation and frequency (MF) in the circulating genome for a total of 13 weeks. MF was calculated using the following formula:
Ancestral history analysis of SARS-CoV-2 sequences
To infer the evolutionary relationships among the examined sequences, the sequences were aligned with relevant reference sequences retrieved from NCBI database using the neighbor-joining approach [27]. The Molecular Evolutionary Genetics Analysis across Computing Platforms (MEGA X) [25] software was used to construct a phylogenetic tree applying the neighbor-joining method [28] and evolutionary distances were computed using the Kimura–Nei method [29]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches.
Statistical analysis
Statistical analysis was performed using SPSS software for Windows, version 20.0 (SPSS Inc., Chicago, IL, USA). The association of mutational frequency with age, and sex were computed using the χ2 tests. The output from the analysis was considered to be significant at p ≤ 0.05.
Results
Genome analysis reveals SARS-CoV-2 ancestral biology
In our study, initially a total no. of 226 complete SARS-CoV-2 genome sequences isolated from the Bangladeshi patients submitted between 18 April and 14 July, 2020 were taken into account for further analysis. Interestingly, the majority of the collected genomes were clade B and/or L type (SM1 & ST1). From the initially selected 226 genomes, redundant sequences were removed using Jalview visualization software and sequences containing legionary characters (N, R, X, and Y) and sequences without complete patient history were excluded from the study. After completing all of the above screening processes, finally 198 unique SARS-CoV-2 genome sequences were selected for further mutational analyses and the metadata of all the studied 198 sequences are summarized in ST1. The phylogenetic tree of 198 complete sequences reveals that the circulating strains in Bangladesh are different from the reference sequence (NC_045512 (mark as blue)). The analysis also shows that circulating strains’ lineage is divided into several sub-lineages. Phylogenetic analysis also segregated the closely related strain into cluster A to Z with threshold value 0.00005. Cluster A contains six sequences which all are collected in June 2020 (EPI_ISL_483627 (12-06-2020), EPI_ISL_483627 (16-06-2020), EPI_ISL_483635 (17-06-2020), EPI_ISL_4836289 (27-06-2020), EPI_ISL_483689 (26-06-2020), and EPI_ISL_4836214 (14-02-2020). Sequences isolated from April to May, 2020 were grouped into clusters B, W, X, Z). However, sequences collected in June rather than others months were more distant from the reference sequence (Fig. 1). Besides, the majority of the sequences were also more distant from the 1st Bangladeshi reported sequence (mark as red) (Fig. 1).

Phylogenetic tree of the studied whole genome sequences of SARS-CoV-2 Bangladesh outbreak. The optimal tree with the sum of branch length = 0.01209279 is shown. The tip of branches corresponds to the accession numbers with country originated, sources, released year and week of sequences. The taxon colored with red, green, pink and yellow for denoting April, May, June and July month, respectively. Closely related genome sequences with minimum branch deviation (cut of 0.00005) were represented in clusters (cluster A to Z). Reference sequence (NC_045512) form Wuhan, China, and 1st declared sequences form Bangladesh were marked as blue and red, respectively. There were a total of 29,011 positions in the final dataset. The tree reveals the history of the common ancestry of all 198 SARS-CoV-2 genome sequences from Bangladesh outbreak. The lines of a tree represent evolutionary lineages. Sequences were grouped by the taxon and shown as red, green, pink and yellow mark colors for April, May, June and July, respectively
Accumulation of SARS-CoV-2 mutation
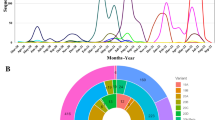
In our analysis, a total number of 13 weeks were considered for the calculation mutation accumulation (Supplementary Table ST2). Because of the presence of one genome (EPI_ISL_468077) during the 1st week, it was considered with week 2 and declared these 2 weeks as 1st week (W1). For the determination of the mutational frequency, the genome of SARS-CoV-2 was split into five regions (Fig. 2). Our analysis reveals that during the initial 5 weeks, circulating viral genomes accumulate fewer mutations with low MF except genomic region 2. However, the mutational frequencies augment after 35 days and continued it until week 9. Interestingly, after 9 weeks, MF persisted similar or even marginally down in 10th and 11th weeks (Fig. 3). After week 11, all the regions except R4 seems to be increasing sharply again. Overall, R5 accumulates the highest MF over the time period (13 weeks) followed by R4 and R2 (Fig. 3) and regions 2 (R2) and R3 appears to be more conserved. The structural proteins such as N and S demonstrate the highest mutation rates (p = 0.001) over the time period (13 weeks) (Fig. 4). Interestingly, NSP9, NSP11 proteins did not accumulate any mutation over the time period. Unique mutations (UM) were also calculated and summarized in Table 1. In the analysis, R1 (p = 0.002) followed by R4 (p = 0.004) and R5 (p = 0.003) were observed to have high frequencies of unique mutations (Fig. 5) and in comparison with other weeks, week 9 to 11 showed the highest UMs frequency. Moreover, after the initial 4 weeks, the frequency seems to have significantly increased (p = 0.002) drastically. Notably, the rate of MF had been also observed until week 9 (p = 0.003) at an uneven rate. After week 11, UM frequency drops drastically. Most interestingly, after week 12 unique mutation in the region 1 and 5 fell to approximately zero but the mutations in the other three regions significantly varied (p = 0.015) (Fig. 2). In terms of proteins, NSP8, NSP9, NSP11, 2’-O-ribose methyltransferase, Matrix, ORF7b had no unique mutations. Moreover, several mutations were found that persisted, (I120F, T412I, L37F, P323L, G204R, R203K, and D614G) for more than 6 weeks (Table 1 and Fig. 5).

Mapping of SARS-CoV-2 genome regions and proteins. The SARS-CoV-2 genome was divided into five regions and the location of each protein in the different regions is schematically presented

Mutational frequency of five genomic segments of SARS-CoV-2. Mutational frequency was calculated by the ratio of the number of total protein mutations and the number of genome sequences in each week. The SARS-CoV-2 genome was divided into five regions, which are represented as R1–R5. Here, red arrows indicate the significant MF variation over time in the various genomic parts of the predominantly circulating SARS-CoV-2 in Bangladesh

Week-wise comparative amino acid mutational frequency of SARS-CoV-2 proteins. Mutational frequency was calculated by the ratio of the number of total amino acid mutations and the number of genome sequences in each week. W1–W13 represent different weeks

Unique protein mutation of five genomic regions of SARS-CoV-2. A. Region-wise unique mutation distributions in the studied genomes. Unique mutations are calculated by removing the redundant mutations, which occur in more than 1 week. The SARS-CoV-2 genome was divided into five regions, which are represented as R1–R5. B. Unique mutational frequency of five genomic segments of SARS-CoV-2. Mutational frequency was calculated by the ratio of the number of total protein mutations and the number of genome sequences in each week
Variability of the spike (S) protein
In our analysis, more than 20 nonsynonymous mutation sites were identified in the S protein, in which 13 (S13I, Q14H, P26L, H49Y, G75V, T75I, S95F, T95I, V127F, D138H, N211Y, Y248H, and S255F) were observed in the N-terminal domain (NTD) (Fig. 6). The fusion peptide region, S’ including heptad repeats HR1 and HR2 regions contains 3 mutated regions (G769V, A783S, T791I), 2 (D936Y, S939Y), 1 (K1191N), respectively.

Mapping of mutations in different domains of spike protein. S1 and S2 are subdomains, N-terminal domain (NTD), C-terminal domain (CTD), receptor binding domain (RBD), fusion peptides (FP), heptad repeats (HR1, HR2) regions, while S includes heptad repeats (HR1, HR2) regions. Changes in amino acid (AA) sequence from the reference genome are shown by the arrow
Sex and age-based mutational accumulation analysis
The sex-based UM analysis revealed that men (70) harbored more frequency than women (36) (Table 2). Among the five segregated regions, R1 (p = 0.018) and R4 (p = 0.002) accumulated more mutations in men than the other regions. This is also true in case of women, but with a much lower frequency (p < 0.05). Protein NSP10, Helicase, 3’ to 5’ exonuclease, ORF6, ORF7a had no mutation in case of women. NSP2, NSP3 RdRp, ORF8 and N proteins possessed 2–6 unique amino acid mutations per protein in the male-originated virus (Table 1). Moreover, region 3 had the lowest unique mutation frequency in viral sequences retrieved from female patients. This aforementioned analysis data suggest differences in COVID-19 infection based on the sex of the infected individual. On the other hand, all age groups (4) accumulated the highest number of mutations in the virus genome R5 (p < 0.05), while the age group of 47–67 years harbors the highest number of mutational accumulation (p = 0.004) followed by the group of 26–46 years (p < 0.05) (Fig. 7). However, the age group 67 to 95 years had the highest mutational frequency in R5 (p = 0.001), while a gradual increase of the mutational frequency was observed in case of age group 26–46 (p < 0.05) and 47 to 67 (p < 0.05) in all regions except R2. Most interestingly, age group 0 to 25 has approximately mutational accumulation rates similar to the age group 26 to 46 years (Fig. 7).

Age-based mutational frequency of five genomic segments of SARS-CoV-2. Mutational frequency was calculated by the ratio of the number of total protein mutations and the number of genome sequences in each week. The SARS-CoV-2 genome was divided into five regions, which are represented as R1–R5. All the studied patients were segregated into 4 age groups (0 to 25 years-blue color, 26 to 46 years-green color, 47–67 years-red color, 68 to 95 years-violet color)
Discussion
Considering the lack of definitive drug and vaccine against COVID-19, studying SARS-CoV-2 genomes is of great importance to elucidate the molecular basis of pathogenesis and evolution for explaining differences in region-specific mortality rates and individual-dependent susceptibility to SARS-CoV-2. Analysis of 198 high-quality complete genome of SARS-CoV-2 from Bangladesh revealed that the circulating strains are of many sub-lineages harboring the same ancestry as Wuhan virus, although their direct evolution from the reference Wuhan virus was not found. Two previous reports by Parvez et al. [30] and Hasan et al. [31] were consistent with our findings that the contributory strains in the SARS-CoV-2 outbreak in Bangladesh might be arising from the different regions of the world other than China. Besides, that majority of the Bangladeshi isolates were found to fall within the clade B belonging to L type (Supplementary Table ST2). While these types were estimated to be more aggressive and capable of rapid transmission, human intervention had been reported to decrease the relative frequency of the L type [15]. A similar type of A type isolates was also reported circulating into the European countries by Forster et al. [32], while a recent study reported the emergence of European and North American mutant variants in Southeast Asia including Bangladesh [24]. However, mutational frequency analysis of the SARS-CoV-2 whole genomes has shown fluctuations of mutational frequency over time, which can be associated with the increase or decrease rate of infections among the population of Bangladesh [33]. Among the 5 regions of SARS-CoV-2 genomes divided to determine the region of mutational hotspots, regions (R) 1, 4, 5 showed a greater tendency to accumulate mutations (p < 0.05) compared to region 2 and 3. On the other hand, the temporal profile of mutational analysis showed elevated mutation rate in 7th to 10th week and the mutation rate was increasing over time. This finding was also consistent with the study carried out in USA [5]. Regions 2 and 3 of higher conservancy harbored NSP4, NSP 5, NSP6, NSP 7, NSP8, NSP 9, NSP10, and NSP11, while the conservative nature of these proteins was also reported previously [5, 34]. Besides, NSP9 and NSP11 had no record to accumulate any amino acid substitutions over a period of 11 weeks in USA, which was evident in our study in case of NSP9. However, another report by Liang et al. [35] revealed only 2 mutations in nonstructural protein 11. So, our analysis along with others literature conclude that these two nonstructural proteins (NSP9 and NSP11) could serve as potential targets for the diagnostics, treatment or vaccine development of SARS-CoV-2. However, Nucleocapsid and Spike protein harbor maximum number of mutations, which contradicts the finding of the study carried out by Kaushal et al. [5] reporting higher mutation in the region of ORF8 and helicase. However, the mutational frequencies in these regions may positively facilitate the virus to adapt to not only external interactions with host cells, but also internal interactions within the host cells [19]. While the mutation site of N protein does not elicit much antibody response, region 603–634 of the S protein of SARS has been shown to be a major immunodominant epitope in S protein [36]. So, changes in this epitope by mutation could alter the sensitivity of the IgG/IgM tests conducted. These changes are actually due to the positive selection pressure in SARS-CoV-2 [37, 38]. However, despite the high frequency of mutations in the spike protein, the notable escape mutations making the virus capable of escaping the neutralizing antibodies or the mutations affecting ACE2 binding were absent [39]. But considering the effects of mutations in Spike protein on the folding of the RBD thus modulating the viral infection via interacting either with ACE2 receptor or neutralizing antibodies, mutations found in these samples should be analyzed further for their effect on the folding of RBD [40]. Additionally ORF8, ORF7a and ORF7b showed mutations in many SARS-CoV-2 isolates, which might result in significant adaptation of coronavirus from human-to-human transmission as well as in contributing to the viral pathogenesis in the host by inhibiting bone marrow stromal antigen 2 (BST-2), which restricts the release of coronaviruses from affected cells [41, 42]. In this study, signature nonsynonymous mutations leading to amino acid changes of P323L in the RdRp was found (Supplementary Table ST2), which is involved in the replication of the viral genome.
Moreover, D614G in the spike glycoprotein is also predominant in Bangladesh-originated SARS-CoV-2 genome, which should be of urgent concern considering the dominance of this mutation globally since early February in Europe [34]. Notably, the D614G mutation is close to the furin recognition site for cleavage of the spike protein, which plays an important role in virus entry. So, mutations in S protein including D614G need to be evaluated carefully, as S protein is essential for the entry of the virus in the host cell by binding to the ACE2 receptor leading to the escape from antibody inhibition allowing infected and recovered patients to become infected again [43] and these mutations may have resulted in the evolution of a new subtype with more transmissible ability [44]. Interestingly, one clinical study regarding this specific mutation did not report significant increase of disease severity associated with this mutation [34]. Notably, few other previous studies although suggest the involvement of the diseases severity with the specific mutation (D614, P323L) in the SARS-CoV-2 genome [45]. Most interestingly, in two studies by Shishir et al. and Garvin et al. suggested the involvement of the higher mortality rate in Bangladesh due to the mutation in NSP2, NSP13, and spike protein in the circulating SARS-COV-2 genome in our country, which is in accordance of our study [46, 47].
Several unique mutations in NSP3 followed by S, ORF3a, NSP2, RdRp, helicase, E and N protein were observed in this study. All of the UM-containing regions are very crucial in the virus genome because of their contributions in SARS-CoV-2 virulence as well as pathogenicity [48]. Reports from other countries including Italy show male-to-female ratio being 3:1 in Italy. The rate of accumulating mutations was found to be higher in males than female patients. Interestingly, the infection and mortality rates were also disproportionately higher in males than females of Bangladesh. In terms of mortality, it was 79.24% for males and 20.76% for females [46]. Similar phenomena have also been [49]. The mortality rate was also reported high in males compared to females from China showing 2.4 times higher mortality in males [50], New York State of USA (42% females vs. 58% males [51]. However, age-stratified mortality rate was also evident [51], while the mutational accumulation in this study also showed an age-stratified pattern. The highest number of mutation accumulation was observed in age group of 47–67 years, followed by group of 26 to 46 years, which might explain the infection rate in Bangladesh [33]. On the other hand, the temporal analysis of mutation accumulation also showed mutations (P323L, I120F, D614G, R203K, G204R) that persist over a longer period of time. Such persistent mutations were also found to be circulating in other parts of the worlds, which reveal similar mutation-accumulating behavior of the genomes across the world [2, 52, 53]. Moreover, RdRp, E, and N genes are the target genes for designing primers and probes in RT-PCR-based SARS-CoV-2 diagnosis [54], owing to their high sequence conservation. Although the effect of primer-template mismatches on laboratory diagnostics of SARS-CoV-2 is not clear totally. So any kind of changes in the RDRP, E and N reasons might play a vital role in the diagnosis of the SARS-CoV-2 [54]. Unfortunately, in our studied genome we have found majority of the genomic variation in the RdRp and N region which might be more alarming. The study by Khan et al. [55] reported 7 sets of genomic diagnostic assay out of 27 assay contain mismatches or mutation which is also supported by our studied results where 5 sets of genomic diagnostic assay contain the following mismatches (Table 3) with having 100% in some cases which is very much alarming for the diagnostic procedure in Bangladesh.
However, our analysis along with literature reviews suggests that the mutational accumulations in the SARS-CoV-2 genome depend on country and continent, while most of the vaccine attempts and diagnostic kits are based on the genome sequence of the original viral isolate from Wuhan. So, the region-specific mutations in the SARS-CoV-2 genome may make these vaccines ineffective [56]. Therefore, continuous monitoring of mutation accumulation and the consequences of these mutations on receptor binding affinity, genome replication and propagation ability, pathogenicity as well as host–pathogen interaction need to be evaluated. On the other hand, RdRp, E, and N genes should be considered as the target genes for designing primers and probes in RT-PCR-based SARS-CoV-2 diagnosis, owing to their high sequence conservation.
Data availability
Complete genome sequences or SARS-CoV-2 are available in GISAID dataset along with reference sequences in the NCBI dataset. The accession number and sequences of the studied genomes are available in Supplementary table 1 and Supplementary file 1.
Abbreviations
- COVID-19:
-
Coronavirus disease 2019
- ACE2:
-
Angiotensin converting enzyme 2
- MSA:
-
Multiple sequence alignment
- SARS:
-
Severe acute respiratory syndrome
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SNP:
-
Single nucleotide polymorphisms
- UM:
-
Unique mutations
- WHO:
-
The World Health Organization
- GISAID:
-
Global Initiative on Sharing All Influenza Data
References
Mim MA, Rakhi NN, Saha O, Rahaman MM (2020) Recommendation of fecal specimen for routine molecular detection of SARS-CoV-2 and for COVID-19 discharge criteria. Pathog Global Health 114:168
Saha O, Rakhi NN, Towhid ST, Rahaman MM (2020) Reactivation of severe acute respiratory coronavirus-2 (SARS-CoV-2): hoax or hurdle? Int J Healthcare Manage. https://doi.org/10.1080/20479700.2020.1782660
Rahaman MM, Saha O, Rakhi NN, Chowdhury MMK, Sammonds P, Kamal AM (2020) Overlapping of locust swarms with COVID-19 pandemic: a cascading disaster for Africa. Pathog Global Health 114:285
Fani M, Teimoori A, Ghafari S (2020) Comparison of the COVID-2019 (SARS-CoV-2) pathogenesis with SARS-CoV and MERS-CoV infections. Future Virol. https://doi.org/10.2217/fvl-2020-0050
Kaushal N, Gupta Y, Goyal M, Khaiboullina SF, Baranwal M, Verma SC (2020) Mutational frequencies of SARS-CoV-2 genome during the beginning months of the outbreak in USA. Pathogens 9(7):565
Da Silva SJR, da Silva CTA, Mendes RPG, Pena L (2020) Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J Med Virol 92:1427
Dawood AA (2020) Mutated COVID-19, may foretells mankind in a great risk in the future. New Microbes New Infect 35:100673
Sevajol M, Subissi L, Decroly E, Canard B, Imbert I (2014) Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res 194:90–99
Hon CC, Lam TY, Shi ZL, Drummond AJ, Yip CW, Zeng F, Leung FCC (2008) Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J Virol 82(4):1819–1826
Yu WB (2020) Decoding evolution and transmissions of novel pneumonia coronavirus (SARS-CoV-2) using the whole genomic data Comparative analyses of the chloroplast genome in carnivorous plants View project. Zool Res 41:247
Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG, Razeq J (2020) Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell 181(5):990–996
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579:265–269
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 6:14. https://doi.org/10.1038/s41421-020-0153-3
Lv L, Li G, Chen J, Liang X, Li Y (2020) Comparative genomic analysis revealed specific mutation pattern between human coronavirus SARS-CoV-2 and Bat-SARSr-CoV RaTG13. Front Microbiol 11:584717
Tang XC, Agnihothram SS, Jiao Y, Stanhope J, Graham RL, Peterson EC, Baric RS (2014) Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc Natl Acad Sci USA 111(19):E2018–E2026
Coppee F, Lechien JR, Decleves AE, Tafforeau L, Saussez S (2020) Severe acute respiratory syndrome coronavirus 2: virus mutations in specific European populations. New Microbes New Infect 36:100696
Saha O, Hossain MS, Rahaman MM (2020) Genomic exploration light on multiple origin with potential parsimony-informative sites of the severe acute respiratory syndrome coronavirus 2 in Bangladesh. Gene Rep 21:100951
Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, Du L (2020) Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 17(6):613–620
Kim JS, Jang JH, Kim JM, Chung YS, Yoo CK, Han MG (2020) Genome-wide identification and characterization of point mutations in the SARS-CoV-2 genome. Osong Public Health Res Perspect 11(3):101
Su YC, Anderson DE, Young BE, Linster M, Zhu F, Jayakumar J, Chia WN (2020) Discovery and genomic characterization of a 382-nucleotide deletion in ORF7b and ORF8 during the early evolution of SARS-CoV-2. MBio 11(4):e01610
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452. https://doi.org/10.1038/s41591-020-0820-9
Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L et al (2020) Genomic diversity of SARS-CoV-2 in coronavirus disease 2019 patients. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa203
Zhao S, Chen H (2020) Modeling the epidemic dynamics and control of COVID-19 outbreak in China. Quant Biol 8:11–19. https://doi.org/10.1007/s40484-020-0199-0
Islam MM, Rakhi NN, Islam OK, Saha O, Rahaman MM (2020) Challenges to be considered to evaluate the COVID-19 preparedness and outcome in Bangladesh. Int J Healthcare Manage 13:263
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Rahman MS, Hoque MN, Islam MR, Akter S, Rubayet-Ul-Alam ASM, Siddique MA, Hossain MA (2020) Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2 etiologic agent of global pandemic COVID-19: an in silico approach. PeerJ 8:e9572
Saha P, Banerjee AK, Tripathi PP, Srivastava AK, Ray U (2020) A virus that has gone viral: amino acid mutation in S protein of Indian isolate of Coronavirus COVID-19 might impact receptor binding, and thus, infectivity. Biosci Rep 40:BSR20201312
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Parvez MSA, Rahman MM, Morshed MN, Rahman D, Anwar S, Hosen MJ (2020) Genetic analysis of SARS-CoV-2 isolates collected from Bangladesh: insights into the origin, mutation spectrum, and possible pathomechanism. Comput Biol Chem 90:107413
Hossain MS, Hami I, Sawrav MSS, Rabbi MF, Saha O, Bahadur NM, Rahaman MM (2020) Drug repurposing for prevention and treatment of COVID-19: a clinical landscape. Discoveries 8(4):e121
Forster P, Forster L, Renfrew C, Forster M (2020) Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci USA 117(17):9241. https://doi.org/10.1073/pnas.2004999117
Saha O, Rakhi NN, Sultana A, Rahman MM, Rahaman MM (2020) SARS-CoV-2 and COVID-19: a threat to Global Health. Discov Rep 3:e13
Korber B, Fischer W, Gnanakaran SG, Yoon H, Theiler J, Abfalterer W, Partridge DG (2020) Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 25:2000045
Liang Q, Li J, Guo M, Tian X, Liu C, Wang X, Yang X, Wu P, Xiao Z, Qu Y (2020) Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. bioRxiv 395:311
He Y, Zhou Y, Wu H, Luo B, Chen J et al (2004) Identification of immunodominant sites on the Spike protein of severe acute respiratory syndrome (SARS) coronavirus: Implication for developing SARS diagnostics and vaccines. J Immunol 173:4050–4057. https://doi.org/10.4049/jimmunol.173.6.4050
Velazquez-Salinas L, Zarate S, Eberl S, Gladue DP, Novella I, Borca MV (2020) Positive selection of ORF3a and ORF8 genes drives the evolution of SARS-CoV-2 during the 2020 COVID-19 pandemic. bioRxiv 4:vey035
Benvenuto D, Giovanetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M (2020) The 2019-new coronavirus epidemic: evidence for virus evolution. J Med Virol 92:455–459
Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN et al (2021) Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29(1):44–57
Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS et al (2020) Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182(5):1295–1310
Decaro N, Lorusso A (2020) Novel human coronavirus (SARS-CoV-2): a lesson from animal coronaviruses. Vet Microbiol 244:108693
Taylor JK, Coleman CM, Postel S, Sisk JM, Bernbaum JG, Venkataraman T, Frieman MB (2015) Severe acute respiratory syndrome coronavirus ORF7a inhibits bone 291 marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation 292 interference. J Virol 89(23):11820–11833
Huang AT, Garcia-Carreras B, Hitchings MD, Yang B, Katzelnick LC, Rattigan SM et al (2020) A systematic review of antibody-mediated immunity to coronaviruses: kinetics, correlates of protection, and association with severity. Nat Commun 11(1):1–16
Maitra A, Sarkar MC, Raheja H et al (2020) Mutations in SARS-CoV-2 viral RNA identified in Eastern India: possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J Biosci 45(1):76. https://doi.org/10.1007/s12038-020-00046-1
Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD et al (2020) SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 11(1):1–9
Shishir TA, Naser IB, Faruque SM (2021) In silico comparative genomics of SARS-CoV-2 to determine the source and diversity of the pathogen in Bangladesh. PLoS ONE 16(1):e0245584
Garvin MR, Prates ET, Pavicic M, Jones P, Amos BK, Geiger A et al (2020) Potentially adaptive SARS-CoV-2 mutations discovered with novel spatiotemporal and explainable AI models. Genome Biol 21(1):1–26
Consortium CSME (2004) Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303:1666–1669
Chakravarty D, Nair SS, Hammouda N, Ratnani P, Gharib Y, Wagaskar V, Tewari AK (2020) Sex differences in SARS-CoV-2 infection rates and the potential link to prostate cancer. Commun Biol 3(1):1–12
Jian-Min Jin PB et al (2020) Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health 8:152. https://doi.org/10.3389/fpubh.2020.00152
Guilmoto CZ (2020) COVID-19 death rates by age and sex and the resulting mortality vulnerability of countries and regions in the world. medRxiv. https://doi.org/10.1101/2020.05.17.20097410
Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen KY (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9:221–236
Ou J, Zhou Z, Dai R, Zhang J, Lan W, Zhao S, Wu J, Seto D, Cui L, Zhang G (2020) Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. BioRxiv. https://doi.org/10.1101/2020.03.15.991844
Khailany RA, Safdar M, Ozaslan M (2020) Genomic characterization of a novel SARS-CoV-2. Gene Rep 1006:82
Khan KA, Cheung P (2020) Presence of mismatches between diagnostic PCR assays and coronavirus SARS-CoV-2 genome. R Soc Open Sci 7(6):200636
Towhid ST, Rakhi NN, Arefin AS, Saha O, Mamun S, Moniruzzaman M, Rahaman MM (2020) COVID-19 and the cardiovascular system: how the first post-modern pandemic ‘weakened’ our hearts. Discov Rep 3:e15
Acknowledgements
We acknowledge the Ministry of Science and Technology, Bangladesh for Special Allocation Project 2019-2020 to M.M.R and Bangladesh Bureau of Education Information and Statistics (BANBEIS) for research Grant No. LS2019935 to M.M.R. The authors would like to acknowledge Bangabandhu Science & Technology Fellowship Trust for supporting OtO.S. with PhD fellowship.
Author information
Authors and Affiliations
Contributions
OS, RNS, and II carried out the studies (data collection and data analysis). OS drafted the manuscript. OS and MMR developed the hypothesis, supervised the whole work and MMR, MSH, II, NNR and RNS critically reviewed the drafted manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
The authors confirm that the ethical policy of the journal, as mentioned on the journal’s authors guideline page, was ensured and no ethical approval was required as the study did not collect any sample or questionnaires from animals and humans.
Additional information
Edited by Simon D. Scott.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saha, O., Islam, I., Shatadru, R.N. et al. Temporal landscape of mutational frequencies in SARS-CoV-2 genomes of Bangladesh: possible implications from the ongoing outbreak in Bangladesh. Virus Genes 57, 413–425 (2021). https://doi.org/10.1007/s11262-021-01860-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-021-01860-x