
Abstract
Biomolecular condensates have emerged as an important subcellular organizing principle1. Replication of many viruses, including human respiratory syncytial virus (RSV), occurs in virus-induced compartments called inclusion bodies (IBs) or viroplasm2,3. IBs of negative-strand RNA viruses were recently shown to be biomolecular condensates that form through phase separation4,5. Here we report that the steroidal alkaloid cyclopamine and its chemical analogue A3E inhibit RSV replication by disorganizing and hardening IB condensates. The actions of cyclopamine and A3E were blocked by a point mutation in the RSV transcription factor M2-1. IB disorganization occurred within minutes, which suggests that these molecules directly act on the liquid properties of the IBs. A3E and cyclopamine inhibit RSV in the lungs of infected mice and are condensate-targeting drug-like small molecules that have in vivo activity. Our data show that condensate-hardening drugs may enable the pharmacological modulation of not only many previously undruggable targets in viral replication but also transcription factors at cancer-driving super-enhancers6.
Similar content being viewed by others

Main
RSV is a major cause of respiratory illness in young children, the older people and individuals who are immunocompromised worldwide7,8. Currently, multiple targets are pursued for the development of a safe and effective therapy to treat RSV infections9.
In infected cells, RSV induces the formation of cytoplasmic IBs, in which nucleoprotein (N), phosphoprotein (P), polymerase L, the transcription factor M2-1 and viral genomic RNA are concentrated. We recently demonstrated that IBs are ‘viral factories’ in which viral RNA synthesis occurs3. The morphology of IBs suggests that they are condensates formed by liquid–liquid phase separation (LLPS). A recent study showed that N and P were sufficient to drive the formation of pseudo-IB condensates through LLPS in vitro, both in cells and in biochemical assays10. However, these N–P pseudo-IB condensates are not functional, as they do not shelter RNA synthesis and do not reflect the complexity of IBs in virus-infected cells, which have multiple compartments. Strikingly similar in size and phase organization to the nucleolus condensate11, RSV IBs are multiphasic and contain a sub-compartment called the IB-associated granule (IBAG), which is composed of newly synthesized viral mRNA and M2-13,12. Condensates have emerged as an important subcellular organizing principle1. An important question in anti-viral drug development—and medicinal chemistry more generally—is whether these condensates are druggable. In principle, a drug that dissolved or hardened would prevent viral replication. Neither mechanism has yet been reported.
Chemical analogues without hedgehog antagonism
We previously identified the hedgehog (HH) pathway antagonist cyclopamine (CPM) as a potent inhibitor of RSV replication13. Inhibition of Sonic hedgehog (SHH) signalling is an unwanted feature of CPM as an RSV inhibitor. On the basis of the binding model of the Smoothened (SMO) receptor–CPM ligand binary complex14 (Protein Data Bank (PDB) 4O9R) and data on chemical analogues with enhanced SMO binding and HH signalling antagonism activity15, we designed CPM analogues with a modified A-ring (Extended Data Fig. 1) in order to engineer-out inhibitory HH signalling activity. The A-ring 3′-hydroxyl group was replaced with methoxy (A3M), ethoxy (A3E) and propoxy (A3P) groups, all of which show greatly reduced SHH-mediated signalling (Fig. 1a and Extended Data Fig. 2). The 3′ ring modification in A3E and A3P resulted in near-complete loss of the HH activity (half-maximal inhibitory concentration (IC50) > 20 μM in SHH reporter assays). A3E is more potent against RSV in vitro than A3P (A3E, IC50 = 1.0 ± 0.34 μM; A3P, IC50 = 3 ± 1 μM) (Extended Data Fig. 2b, d, f) and was selected for all further studies. In addition to RSV reference strains, A3E and CPM also inhibit minimally laboratory-experienced strains of RSV (Extended Data Fig. 3). Although the selectivity index of A3E was lower compared with CPM, it consistently ranged above 10. Future studies will need to identify whether chemical modifications can be made to the CPM scaffold that eliminate the SMO binding and HH signalling while maintaining or even improving RSV activity. Of note, the mechanism of RSV inhibition probably remains unchanged as the same of R151K substitution in M2-1 confers resistance to both A3E and CPM (Fig. 1a).

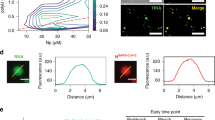
a, The potency of A3E and CPM was determined using RSV–Luc- and RSV–M2-1(R151K)–Luc-infected HEp-2 cells and SHH-induced Gli-dependent luciferase-reporter-expressing NIH3T3 cells. The results are expressed as mean ± s.d. for a representative of three independent experiments performed in triplicate. IC50 concentrations in μM are indicated. b, RSV–M2-1–mGFP-infected HEp-2 cells treated with A3E or CPM for 1 h (see Extended Data Fig. 4). Representative images from four independent experiments are shown. Poly(A) RNA is shown in red and nuclei are shown in blue. Scale bar, 10 μm. White arrowheads indicate IBs.
Disorganization of IBs by A3E and CPM
Formation of IBAGs inside RSV viral factories depends on the de novo synthesis of viral mRNA, which recruits M2-1 and which was proposed to result from phase separation3. We used a recombinant RSV expressing a functional M2-1–mGFP fusion protein to analyse the effect of A3E and CPM on the subcellular localization of M2-1. In untreated cells, M2-1 homogenously distributed in the cytoplasm and concentrated in IBAGs as revealed by FISH (fluorescence in situ hybridization) poly(A)-RNA staining (Fig. 1b and Extended Data Fig. 4). By contrast, IBAGs were no longer visible in compound-treated cells and M2-1–mGFP together with poly(A) RNA was detected throughout the entire IB. Complete loss of the organization of IB and IBAGs was observed after a 1-h treatment with compounds at a concentration equivalent to 0.5× the IC90 (Extended Data Fig. 4). Time-lapse fluorescence imaging revealed that the loss of IB and IBAG partitioning occurred within minutes after the addition of A3E and CPM (Supplementary Videos 1–3). This suggests that both compounds abolished the phase separation between IBs and IBAGs that could be mediated by M2-1–mRNA complexes.
Condensate-hardening small molecules
We hypothesized that RSV viral factories are condensates formed by LLPS10. The dynamic nature of RSV IBs was shown in cells infected with recombinant RSV expressing a fluorescent P protein that had typical liquid-like behaviour showing frequent fusion events between IBs followed by rapid coalescence to form a larger spherical IB (Fig. 2a and Supplementary Video 4). As expected for condensates, IBs disassembled upon osmotic shock and 1,6-hexanediol treatment, which disrupts hydrophobic protein–protein interactions (Fig. 2b, c and Extended Data Fig. 5a, b). To further confirm the liquid nature of IBs, we analysed the fluorescence recovery after photobleaching (FRAP) of IBs in RSV–P–BFP-infected cells. We observed a full and rapid recovery of the fluorescence, which is consistent with the rapid diffusion of P within IBs. By contrast, in compound-treated cells we did not observe any re-distribution of P and fluorescence recovery (Fig. 2d, e). Moreover, IBs were no longer sensitive to osmotic shock, they lost their ability to fuse with each other and exhibited a slight decrease in mobility (Extended Data Fig. 5c, d and Supplementary Videos 5–7). The shape of the IBs, but not the surface area, was also altered in compound-treated cells, which resulted in the partial loss of the characteristic spherical shape of liquid condensates (Extended Data Fig. 5e).

a–g, HEp-2 cells were infected for 24 h with RSV–P–BFP (a–e), RSV–M2-1–mGFP (f) or RSV–M2-1(R151K)–P–BFP (g) and treated with 5 μM CPM, 25 μM A3E or DMSO for 1 h. a, Dynamic behaviour of RSV IBs. Arrowheads indicates a fusion event. Images are representative of ten videos from two experiments. Scale bar, 5 μm. b, c, Hypotonic shock was applied and cells were imaged at the indicated times. Measurements labelled R (recovery) were taken after 5 min of shock followed by 5 min in culture medium. b, The mean ± s.d. number of IBs per image is expressed as the percentage of the pre-shock control. Data are from ten acquisitions in two independent experiments. NS, not significant; ***P < 0.001; Kruskal–Wallis test with two-tailed Dunn’s test for multiple comparisons. c, Representative images. Scale bar, 10 μm. d–g, Hardening of RSV IBs by A3E and CPM is shown using FRAP. The spontaneous re-distribution of fluorescence after photobleaching was recorded, the background and bleaching were corrected for during post-bleach imaging and normalized to the pre-bleach signal. e, g, Data are from 24 FRAP events in two independent experiments. d, f, Representative images of time-lapse microscopy from FRAP experiments are shown for P–BFP (d) and M2-1–mGFP (f). Scale bars, 2 μm. White arrowheads indicate the bleached area.
We further analysed the mobility of M2-1 by performing FRAP on IBAGs in RSV–M2-1–mGFP-infected cells. In untreated cells, photobleached areas of the IBAG structures were readily replenished with M2-1–mGFP within 10 s, revealing rapid diffusion of M2-1 into IBAGs and active trafficking of M2-1 between the IB and IBAGs (Fig. 2f). However, in RSV–M2-1–mGFP-infected and A3E- or CPM-treated cells, M2-1 proteins were no longer able to redistribute into the photobleached area and remained in a hardened state (Fig. 2f). Taken together, our data show that RSV IBs are condensates formed by LLPS that can be hardened by small molecules A3E and CPM. Hardening of IB condensates does not result in the formation of solid fibrillar material as they do not stain positive with thioflavin S coloration16 (Extended Data Fig. 6a). We investigated the permeability of hardened IBs by analysing the diffusion of micro-injected fluorescent dextran beads in infected and treated cells17. The 10-kDa dextran beads, which have a hydrodynamic radius (Rh) of 2–3 nm, fully penetrate the IBs whereas the larger 70-kDa beads (Rh > 6 nm) are excluded from IBs in any experimental condition tested (Extended Data Fig. 6b, c). Notably, the mid-size 40-kDa beads (Rh = 4.5 nm) can enter the hardened IB in treated cells, which suggests that there is an increase in the IB mesh size after treatment with the compounds. This surprising finding warrants further investigation into the biophysical mechanism of IB condensate hardening.
We engineered an RSV–P–BFP recombinant virus that expresses the previously described M2-1(R151K) substitution that confers resistance to CPM13 and analysed IB dynamics and properties in compound-treated cells. In RSV–M2-1(R151K)–P–BFP-infected cells, treatment with CPM or A3E no longer resulted in a loss of roundness, resistance to hypotonic shock or change in the organization of IBs and IBAGs (Extended Data Figs. 7–9). FRAP data show that P mobility in IBs is no longer affected by CPM and A3E treatment (Fig. 2g). Taken together, these data demonstrate that the antiviral effect and the condensate-hardening properties of these small molecules are dependent on the M2-1 protein.
Viruses replicate or assemble in foci in which viral macromolecules locally concentrate, although many—such as positive-strand RNA viruses, including coronaviruses—are not known to form large IB condensates. Notably, small condensates formed by the nucleocapsid of SARS-CoV-2 were recently reported18. Whether smaller viral replication foci can be hardened and inhibited in a manner similar to RSV IB condensates is an interesting avenue of future research.
Efficacy of A3E in the RSV mouse model
To our knowledge, no condensate-targeted compounds have demonstrated in vivo efficacy to date. We therefore analysed whether the RSV condensate-hardening compounds A3E and CPM were able to block RSV replication in a mouse model19. We infected mice with RSV–Luc and treated animals twice daily with various doses of CPM and A3E, starting on the day of infection. Treatment of mice with A3E resulted in significant and dose-dependent inhibition of RSV replication in the lungs (Fig. 3a, b). At the peak of viral infection at 4 days after infection (d.p.i.), the luminescent signal was barely visible in the lungs of treated mice (Fig. 3a, b and Extended Data Fig. 10a). Although typically only minor pathological changes can be seen in the lungs of RSV-A2-infected mice, A3E and CPM reduce the inflammation and exfoliation of lungs of treated animals, which are shown as a combined histopathology score in Fig. 3c. Furthermore, we show that RSV–Luc expressing the resistance substitution (R151K) in M2-1 could not be inhibited by either compound in vivo (P > 0.05) (Fig. 3a, b), which validates M2-1 as the main viral protein targeted by A3E and CPM. Taken together, these data confirm that the hardening of IB condensates in infected cells in vitro translates to antiviral efficacy in RSV-infected mice. The efficacy of A3E in suppressing virus replication is similar but appears to be slightly lower than CPM, which could be due to the lower potency of A3E observed in vitro (Fig. 3a, b). We believe that further investigations using a large-animal disease model of RSV infection can clarify this issue20.

a, b, Mice were infected with RSV–Luc and treated twice daily with CPM (n = 12 mice), A3E (5 mg kg−1, n = 8; 15 mg kg−1, n = 12; 30 mg kg−1, n = 7) or vehicle (n = 27) from 0 to 3 d.p.i. or infected with RSV–M2-1(R151K)–Luc and treated with CPM (15mg kg−1, n = 8), A3E (15 mg kg−1, n = 8) or vehicle (n = 13). a, Bioluminescence reading was done at 4 d.p.i. and representative images of ventral views of infected animals are shown. The average radiance is expressed as the sum of the photons per second from each pixel inside the region of interest per the number of pixels (p s−1 cm−2 sr−1). Normalized bioluminescence results are expressed as mean ± s.e.m. from at least two experiments. *P < 0.05, **P < 0.01, ****P < 0.0001; Kruskall–Wallis test with two-tailed Dunn’s tests for multiple comparisons. c, Histopathological changes in the lungs of mice infected with RSV A2 strain and treated with A3E (15 mg kg−1, n = 6), CPM (15 mg kg−1, n = 3) or vehicle (n = 3). Virus in lung was titred and tissue sections were prepared as described in the Methods. *P < 0.05; one-way Brown–Forsythe and Welch’s ANOVA, followed by two-sided Dunn’s tests for multiple comparisons. Two lung sections per mouse were analysed by three independent pathologists and the mean score was calculated. Data are mean ± s.e.m.
Our in vitro data (Supplementary Videos 2, 3) show that A3E and CPM act on RSV IBs within minutes. We therefore assessed the antiviral efficacy of condensate-hardening compounds in a more realistic setting by comparing RSV replication in mice treated with A3E and CPM at 1, 2 or 3 d.p.i., followed by luminescence measurement at 4 d.p.i. (Extended Data Fig. 10b, c). The reduction in virus replication can be seen when treated up to 2 d.p.i. for both compounds, although significance was not reached for A3E at deferred treatment points.
A fast action is an expected feature of a condensate-hardening drug and was observed in cell culture experiments (Supplementary Videos 1–3). This might be an important advantage of this new class of drugs for acute viral infections that have a limited treatment window. Indeed, RSV causes an acute respiratory disease that is characterized by a peak in viral replication in the lungs21. Patients typically present themselves to physicians several days after the onset of symptoms. Comparative studies with development stage inhibitors of RSV F, L and N proteins should be done to determine the inhibition kinetics of current development-stage RSV inhibitors in vitro and in vivo.
Conclusion
Here we show that condensates formed by a liquid–liquid phase transition can be hardened by small drug-like molecules, resulting in the inhibition of virus replication in vivo. Targeting viral condensates could prove to be valuable for the development of drugs with broad-spectrum activity against future emerging viral pathogens by targeting proteins that are critical for condensate formation and conserved across the virus family. Condensate-targeting drugs may enable the pharmacological modulation of many previously undruggable targets. In recent years, biomolecular condensates have indeed been demonstrated in various physiological processes such as embryonic development, cellular response to stress and pathological protein aggregation in neurodegenerative diseases and cancer.
Methods
Design and synthesis of CPM analogues
CPM (Selleck) was used as the starting material based on the crystal structure of the CPM–SMO complex (PDB 4JKV and PDB 4O9R)14, which shows polar interactions of the A-ring 3′-hydroxyl group with the amino acids in the extracellular domain of the SMO receptor. We designed analogues as shown in Fig. 1a and Extended Data Fig. 1. The synthesis work was performed at WuxiAppTec. To a solution containing CPM (600 mg, 1.46 mM, 1.0 eq) in methanol (5 ml), Boc2O (382.37 mg, 1.75 mM, 402.49 μl, 1.20 eq) was added. The reaction mixture was stirred at 20 °C for 16 h. Thin-layer chromatography (TLC) (petroleum ether/ethyl acetate (PE/EtOAc) = 3/1, Retention factor (Rf) = 0.45, KMnO4) showed no starting material. The solvent was removed to obtain the residue. The residue was purified by column chromatography on silica gel (PE/EtOAc = 10/1) to give an intermediate compound (550 mg, 1.07 mM, 73.62% yield) as a white solid, which was named CPM-IM-1 (intermediate product 1). Next, to a solution of CPM-IM-1 (250 mg, 488.54 μM, 1.0 eq) in THF (4 ml), NaH (39.08 mg, 977.08 μM, 60% purity, 2.0 eq) was added at 20 °C. The reaction mixture was stirred at 60 °C for 20 min. MeI (346.72 mg, 2.44 mM, 152.07 μl, 5.0 eq) was added to the reaction mixture and stirred at 60 °C for 2 h. TLC (PE/EtOAc = 3/1, Rf = 0.70, KMnO4) showed no starting material. The reaction mixture was diluted with water (40 ml), extracted with EtOAc (25 ml ×2), washed with brine (30 ml), dried over Na2SO4 and concentrated to give the crude product. The crude product was purified by column chromatography on silica gel (PE/EtOAc = 10/1) to give intermediate product 2 (CPM-IM-2; 250 mg, 475.50 μM, 48.67% yield) as a white solid. Next, to a solution of CPM-IM-2 (150 mg, 285.3 μM, 1.0 eq) and 2,6-lutidine (91.71 mg, 855.90 μM, 99.68 μl, 3.0 eq) in DCM (3.00 ml), TMSOTf (95.12 mg, 427.95 μM, 77.33 μl, 1.50 eq) was added at 0 °C. The reaction mixture was stirred at 0 °C for 30 min. TLC (PE/EtOAc = 3/1, Rf = 0.00, KMnO4) showed no starting material. The reaction mixture was diluted with saturated NaHCO3 (30 ml), extracted with EtOAc (30 ml ×2), washed with brine (30 ml), dried over Na2SO4 and concentrated to give the crude product. The crude product was purified by prep-HPLC (NH4HCO3) to give A3M (13.0 mg, 30.54 μM, 10.70% yield) as a white solid. A3E and A3P were synthesized with the same procedure, shown in Extended Data Fig. 1.
Cells
HEp-2 cells (ATCC, CCL-23) were maintained in Dulbecco’s modified Eagle’s medium (DMEM). ATCC number CCL-23 is derived from a HeLa cell contaminant and is recommended to grow RSV. BSRT7/5 cells—BHK-21 cells that constitutively express the T7 RNA polymerase22—were maintained in Glasgow’s MEM19. HH-signalling-pathway Gli-dependent luciferase-reporter-expressing NIH3T3 cells (BPS Bioscience, 60409) were maintained in DMEM. Cells were grown in medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) supplemented with penicillin–streptomycin solution. In addition, 0.5 mg ml−1 of geneticin was added to BSRT7/5 cells and Gli-dependent luciferase-reporter-expressing NIH3T3 cells. Cells were grown in an incubator at 37 °C in 5% CO2. Cell lines were not authenticated. BSRT7/5 and HEp-2 cells tested negative for mycoplasma using a MycoAlert PLUS Mycoplasma Detection Kit (Lonza).
Virus strains and recombinant virus rescue
RSV Long strain (ATCC, VR-26) was used for infection assays. The recombinant viruses, RSV–Luc, which expresses firefly luciferase and RSV–M2-1–mGFP, which expresses M2-1 fused to monomeric GFP, were prepared as described previously19. Recombinant RSV expressing the Luciferase reporter gene and encoding a Arg-to-Lys substitution at position 151 in the M2-1 protein (RSV–M2-1(R151K)–Luc) was engineered using the pACNR-rHRSV-Luc vector (GenBank accession, KF713491.1) as a template to amplify seven fragments covering the whole RSV genome. The fragments were assembled along with the p15A-Chl vector fragment using Gibson Assembly Master Mix (NEB, E2611). Recombinant RSV–M2-1(R151K)–Luc (GenBank accession, MW039343), RSV–P–BFP (GenBank accession, MT994243) and RSV–M2-1(R151K)–P–BFP (GenBank accession, MT994242) were rescued by reverse genetics and amplified in HEp-2 cells as described previously19.
RSV inhibition assays
HEp-2 cells were seeded at 5 × 104 cells per well in 96-well plates the day before and infected with 104 plaque forming units (PFU) of RSV–Luc in the presence of various concentrations of compounds. CPM (Selleck, S1146), CPM analogues and GDC-0449 (also known as Vismodegib; Selleck, S1082) were solubilized in DMSO at 8 mM, 8 mM and 10 mM, respectively. Compounds were further diluted serially in DMEM containing 0.25% DMSO to final concentrations ranging from 20 μM to 0.0005 μM. In cell and virus control wells, 0.25% DMSO was added. Compound dilutions were pre-incubated with viral suspensions for 5 min at 37 °C before addition to the cell monolayers in 96-well plates. Plates were incubated at 37 °C for 24 h for RSV–Luc before analysis. Luminescent readings were performed with a Bright Glo Luciferase System (Promega, E2610) and a Bioteck Synergy H1 plate reader. Relative light units indicate the luciferase activity relative to the mean control value (expressed as a percentage). The IC50 was defined as the compound concentration required to achieve a 50% reduction in the maximal virus replication. In addition, cytotoxicity assays were done with the CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7570) after incubation with the compounds to test the cell viability. The 50% cytotoxic concentration (CC50) was defined as a 50% reduction in luminescence compared to control wells. IC50 and CC50 values were calculated by fitting the data to a sigmoidal curve equation in GraphPad software (GraphPad Prism 5).
Minimally laboratory-experienced RSV isolates
Primary RSV isolates were obtained at the Hannover Medical School, Germany, in 2013 and 2016 from children with respiratory tract infections and with a multiplex RT–PCR-confirmed diagnosis of RSV infection23. HEp-2 cells were inoculated with nasal swab, bronchial lavage fluid or throat swab material for 4 h at 37 °C. After a change to fresh medium, cells were incubated and, if necessary, passaged for several days until strong syncytia formation was visible. the supernatant and cells were collected, and cell-associated virus was released by three freeze–thaw cycles in liquid nitrogen. Cell debris was removed from the media by centrifugation at 1,000g and aliquots of the supernatant were snap-frozen in liquid nitrogen and stored at −80 °C. Viral RNA was isolated, reverse transcribed and RSV subtypes (RSV A GA2, RSV A ON1 and RSV B) were determined by Sanger sequencing of the G protein gene (or next-generation sequencing of the complete viral genome) and aligned to known sequences found using Nucleotide BLAST.
Assays to test HH antagonism of CPM analogues
The screening of compounds for HH antagonism was conducted at BPS Bioscience using the Gli-luciferase reporter system24. In brief, Gli-dependent luciferase-reporter-expressing NIH3T3 cells, stably transfected with a Gli-dependent firefly luciferase expression plasmid, were seeded into white 96-well microplates and cultured overnight. After 24 h, the medium was changed to Opti-MEM containing diluted compounds and the cells were incubated for an additional 2 h, followed by addition of 1 μg ml−1 recombinant mouse SHH protein. Untreated cells were used as control. After treatment for 24 h, cells were lysed and luciferase assay was performed using ONE-Step luciferase assay system (BPS bioscience, 60690). Luminescence was measured using a luminometer (BioTek SynergyTM 2 microplate reader). Reporter assays were performed in triplicate for each concentration and the luminescence intensity (L) was analysed using Graphpad Prism. The luminescence intensity in the absence of a compound was used as a positive control (Lp) and scored as 100%. The signal recorded in the absence of cells (Lb) was scored as 0%. The percentage luminescence in the presence of each compound was calculated according to the following equation: Luminescence (%) = (L − Lb)/(Lp − Lb). The values of the percentage of luminescence for various concentrations of the same compound were then plotted using nonlinear regression analysis followed by calculation of the IC50 value.
Quantification of IB and IBAG disorganization following compound treatment
HEp-2 cells were grown on glass coverslips and infected with RSV–M2-1–mGFP or RSV-M2-1(R151K)–P–BFP at a multiplicity of infection (MOI) of 1. At 24 h after infection, cells were treated with CPM or A3E at the indicated concentrations for 1 h. FISH was performed as previously described3. In brief, cells were fixed with 4% formaldehyde in PBS (v/v) for 10 min at 4 °C and endogenous biotin was blocked in PBS–1% BSA (w/v) supplemented with free streptavidin (4 μg ml−1) for 1 h. Coverslips were incubated in hybridization mix (2× SSC, 300 mM NaCl and 30 mM sodium citrate), 10% dextran (w/v), 20% formamide (v/v), 1 mg ml−1 herring sperm DNA and 1 μM of poly(dT) probe in a humidified chamber at 37 °C for 3 h. After serial washes, probes were detected by incubating the cells with streptavidin–Alexa Fluor 647 conjugate (8 μg ml−1) in PBS–1% BSA (w/v) and then stained with rabbit polyclonal anti-N antibody3. z-stack image acquisitions of approximately 50 infected cells per condition were performed using a WLL Leica SP8 microscope with a 63× oil-immersion objective and a numerical zoom of ×2. The specific N antibody labelling was used for automatic detection of IBs in ImageJ software using the Analysis Particles function and a size threshold at >3 μm2. The presence of IBAG(s) was then assessed manually based on M2-1–mGFP (if relevant) and poly(A) RNA signals.
Time-lapse microscopy and photobleaching experiments
Live-cell imaging and FRAP experiments were done using HEp-2 cells seeded in Ibidi μ-dishes with a polymer coverslip bottom that were infected with recombinant viruses (RSV–P–BFP, RSV–M2-1(R151K)–P–BFP or RSV–M2-1–mGFP) at high MOI for 20 h to 24 h. Image acquisition was performed using an Olympus FV3000 inverted confocal microscope with a 60× oil-immersion objective and a ×2.5 numerical zoom. Cells were maintained in a climate-controlled chamber (37 °C, 5% CO2) during imaging. To analyse IB dynamics, images were acquired every 30 s for 15–30 min using cells treated for 1 h with 5 μM CPM or 25 μM A3E, or mock-treated (DMSO). Images and videos are representative of ten videos from two experiments. Ten 15-min-long videos from two independent experiments were analysed to quantify fusion events and IB mobility. Image editing was performed using both ImageJ and Icy software. IBs were detected using the Spot Detector plugin of the Icy software and fusion events were counted manually. Maximum velocities were obtained using the Spot Detector plugin with a size-filtering option and the Track Manager plugin of Icy software.
To visualize IBAG dynamics after CPM and A3E treatment, images were acquired every 6 s. After 5 min, the chemical compound (5 μM CPM or 25 μM A3E) was added and image acquisition was carried out for 20 min. Videos are representative of nine videos from three independent experiments.
FRAP acquisition was performed 1 h after addition of 5 μM CPM, 25 μM A3E or DMSO (mock, control). All FRAP experiments were realized using the same settings: 6 s pre-bleach, 5 ms bleach and 60 s post-bleach at a frame rate of 1 image every 126 ms. Bleaching was performed in a circular region at 100% and 80% laser intensity for GFP and BFP, respectively. Target IBs were close to other IB controls, which were used to correct for the loss of fluorescence due to photobleaching. The average fluorescence intensity as a function of the time of every bleached region was obtained using the Icy software. Background intensity was estimated by measuring a region outside the cell as far as possible from the target IB. Normalization of the recovery curves was performed using the easyFRAP, a MATLAB stand‐alone application25. For each experimental condition, two individual experiments were performed in which 12 IBs were analysed. To obtain the FRAP images displayed in Fig. 2, one image was acquired 5 s before bleaching and regions of interest were bleached with full power laser beam for 10 ms. Images were then acquired every 30 s (after a first image at 10 s). Image editing was performed using both ImageJ and Icy software.
Characterization of RSV IB area and shape
HEp-2 cells grown on glass coverslips were infected with RSV–P–BFP at MOI = 1 for 24 h and exposed to the indicated treatments (DMSO, CPM or A3E) for 1 h. Then, cells were fixed with PBS–4% formaldehyde (v/v) for 10 min at room temperature and permeabilized with PBS containing 1% BSA (w/v) and 0.1% Triton X-100 (v/v) for 10 min. Cells were incubated for 1 h with Hoechst 33342 (1 μg ml−1) and after washing in PBS, coverslips were mounted in ProLong Diamond antifade reagent (Thermofisher). z-stack image acquisitions of 100 cells per condition from 2 independent experiments were performed using a WLL Leica SP8 microscope with a 63× oil-immersion objective and a numerical zoom of ×2. The shape and area measurements of RSV IBs were performed with the ImageJ software using the Analysis Particles function. After adjusting the threshold and converting the images to black and white, this command enables the automatic detection of IBs, calculation of the area and the roundness using the formula \(\,4\,\frac{a}{{\rm{\pi }}{b}^{2}\,}\) (where a corresponds to the area and b to the major axis) of each IB. For multiple comparisons between different groups of treatments a Welch’s ANOVA was performed followed by a Games–Howell post hoc test using the R statistical language (https://www.r-project.org/).
Sensitivity of IBs to osmotic shock and to 1,6-hexanediol treatment
HEp-2 cells were seeded on Ibidi μ-Dish polymer coverslips and infected with RSV–P–BFP or RSV–M2-1(R151K)–P–BFP at high MOI. At 24 h after infection, cells were exposed to 5 μM CPM or 25 μM A3E for 1 h at 37 °C. Image acquisition was performed using an Olympus FV3000 inverted confocal microscope with a 60× oil-immersion objective and a numerical zoom of ×2.5. Cells were maintained in a climate-controlled chamber (37 °C, 5% CO2) during multi-position imaging. Five individual positions were studied per experiment and one image was acquired before treatment in two independent experiments. Hypotonic shock was performed by incubating the cells in 10% MEM diluted in water (v/v) for 5 min. To study a possible recovery of IBs after the shock, fresh 100% MEM was added, and the cells were imaged every 1 min for 5 min. To assess 1,6-hexanediol sensitivity, cells were incubated with 10% 1,6-hexanediol (w/v) and imaged every 2 min for 20 min after treatment. Image thresholding and IB detection was performed automatically in Icy software using the Otsu threshold clustering algorithm from the Best Threshold plugin and the Spot Detector plugin by defining a minimum (0,8 μm2) and maximum (50 μm2) ROI size.
Microinjection experiments
HEp-2 cells seeded on Ibidi μ-Dish polymer coverslips were infected with RSV–P–BFP for 24 h and exposed to CPM, A3E or DMSO at the indicated concentrations for 1 h at 37 °C. Cell microinjections were carried out with a FemtoJet microinjector (Eppendorf) mounted on an Olympus IX73 inverted microscope using Femtotips II needles (Eppendorf). Tetramethylrhodamine–dextran (40 kDa and 70 kDa, Sigma-Aldrich) and Texas-Red–dextran (10 kDa, Sigma-Aldrich) were prepared in injection buffer (48 mM K2HPO4 4.5 mM KH2PO4, 14 mM NaH2PO4, pH 7.2) at final concentrations of 6 mg ml−1. The 10-kDa, 40-kDa and 70-kDa dextrans have a hydrodynamic radius of approximately 2–3 nm, 4–5 nm and >6 nm, respectively17. Cells were imaged 2–5 min after microinjection using a 63× oil-immersion objective and average fluorescence intensity values of every region (cytoplasm, condensates and background) were obtained using the Icy software. Average fluorescence intensity of every region was corrected by subtracting the background (a region outside the cell).
Thioflavin S staining
HEp-2 cells were grown on glass coverslips, infected with RSV–P–BFP for 24 h and then treated with CPM, A3E or DMSO at the indicated concentrations at 37 °C for 1 h. Cells were then fixed with 4% formaldehyde in PBS (v/v) and permeabilized with PBS containing 1% BSA (w/v) and 0.1% Triton X-100 (v/v) for 10 min at room temperature. Thioflavin S (Sigma-Aldrich) was dissolved in water at 1% (w/v) and filtered before use. Cells were incubated for 1 h at room temperature with 0.01% thioflavin S in 10% FBS and 0.5% Tween-20 in PBS (v/v). After serial washes with 0.5% Tween-20 in PBS (v/v), cell nuclei were stained with Hoechst 33342 (1 μg ml−1). Mouse PrP amyloid fibrils were adsorbed on fixed cells and treated in parallel as a positive control of amyloid fibrils at a concentration of 1 μM. Image acquisition was performed using a WLL395 Leica SP8 microscope with a 63× oil-immersion objective and a numerical zoom of ×4 and image editing was performed using the Icy software.
Flow-cytometry-based assay to quantify minimally laboratory-experienced RSV
HEp-2 cells were seeded at 2 × 104 cells per well in 96-well plates the day before virus inoculation. Cells were infected at MOI = 1 in the presence of the respective compound. After 2 h, the supernatant was removed and fresh medium containing the same concentration of the compound was added. At 24 h after inoculation, cells were washed once with PBS, and detached by trypsin treatment. Cells were washed, centrifuged at 400g, resuspended in fixation buffer (0.5% PFA and 1% FBS in PBS) and incubated at 4 °C for at least 30 min or stored overnight. Subsequently, cells were permeabilized with 0.1% saponin and 1% FBS in PBS on ice for 20 min. Purified mouse anti-RSV phosphoprotein hybridoma supernatant was diluted 1:500 in permeabilization buffer and incubated with the cells on ice for 45 min26. Cells were then washed with 1% FBS in PBS once and afterwards incubated with goat anti-mouse-Alexa 488 secondary antibody (ThermoFisher) in permeabilization buffer in the dark on ice for 1 h. After two additional washes with 1% FBS in PBS, cells were resuspended in fixation buffer and stored at 4 °C overnight. Samples were analysed using a 99% attenuation filter in a BD Accuri C6 Flow Cytometer. Results were analysed using FlowJo V10. FSC-A versus SSC-A was used to gate on living cells. Living cells that had a higher FL1-A signal compared to stained but uninfected cells were considered RSV-positive. For each virus infection in the presence of a compound, infection was normalized to infected, DMSO-treated control cells.
Mouse infection and compound administration
Female BALB/c mice (approximately 8 weeks of age) were purchased from the Centre d’Elevage R. Janvier. Mice were housed under standard conditions with air filtration in a specific-pathogen-free animal facility using unbleached tissue nesting material, and water and food were provided ad libidum. Mice were acclimatized for 1 week before experiments started. For infection experiments, mice were housed in cages inside stainless-steel separate isolation cabinets that were ventilated under negative pressure with high-efficiency particulate-filtered air. Before infection, mice were anaesthetized using a mixture of ketamine and xylazine (1 mg and 0.2 mg per mouse, respectively) and infected with 50 μl of PBS containing 6 × 104 PFU of RSV–Luc or RSV–M2-1(R151K)–Luc through the intranasal inoculation route. For compound administration to animals, compounds were dissolved in a sodium phosphate/citrate buffer (pH 3) containing 10% 2-hydropropyl-β-cyclodextrin (w/v). Compounds were administered by intraperitoneal injection at the indicated doses in a volume of 200 μl twice a day. The control group received vehicle only.
In vivo luminescence measurements
For in vivo imaging, mice were anaesthetized with ketamine and xylazine, but for daily imaging experiments, anaesthesia was induced using isoflurane in a well-ventilated room and a scavenging system. Calculations of sample size and randomization were not carried out in these experiments. In brief, mice were placed in the chamber (XGI-8, Caliper Life Sciences), the anaesthesia unit was turned on with a flow of 100% oxygen at a rate of 1.5–2 l min−1 mixed with around 4–5% (v/v) isoflurane delivered to the anaesthesia chamber. Then, the deeply anaesthetized mice were injected by intranasal injection with 50 μl of PBS containing d-luciferin at 0.75 mg kg−1. After 2 min, mice were placed with their back on a plate in the IVIS 200 imaging system (Xenogen, Perkin Elmer) with a nose cone providing 100% oxygen mixed with around 1.5–2% (v/v) isoflurane. Living Image software (v.4.0, Caliper Life Sciences) was used to measure luciferase activity. Bioluminescence images were acquired for 1 min with f/stop = 1 and binning = 8. A digital false-colour photon-emission image of the mouse was generated, and photons were counted within a constant region of interest corresponding to the surface of the chest encompassing the whole-airway area. Photon emission was measured as radiance in p s−1 cm−2 sr−1.
Histopathology of RSV-infected mice
Female BALB/c mice were housed in individually ventilated cages in the biosafety level-2 laboratories of WuXi AppTec after quarantine. Animal care and use followed the WuXi IACUC-approved animal use protocol (IACUC no. ID01-QD029-2020v1.0).
Compounds (CPM or A3E) were administered at a dose of 15 mg kg−1 intraperitoneally twice daily for 4 days, from 0 d.p.i. Mice were anaesthetized using a mixture of zoletil 50 and xylazine (30 mg kg−1 and 6 mg kg−1, respectively) and inoculated through intranasal injection with around 105 PFU in 50 μl of RSV A2 strain. The control group received vehicle only. At 4 d.p.i., mice were euthanized, and the lung tissue was collected and snap-frozen in Hanks’ balanced salt solution in a volume equal to tenfold the tissue weight (w/v) for further assays. Virus titration was performed as previously described13. The supernatant of tissue homogenates or virus stock was used to infect the HEp-2 cells in 12-well plates. After a 4-h incubation, cells were washed and subsequently overlaid with 0.625% low-melting agarose in DMEM supplemented with antibiotics and 2% FBS, and then incubated for 96 h. Subsequently, cells were fixed with 4% PFA for 30 min at room temperature, washed, blocked with BSA at room temperature for 1 h, and incubated for around 2–3 h at room temperature with mouse anti-RSV fusion protein monoclonal antibody (Abcam, ab94968) in 1× TBS, followed by a washing step with PBS–0.02% Tween-20. Antigen–antibody complexes were revealed by incubating the cells for 2–4 h at room temperature with a secondary goat anti-mouse HRP-conjugated antibody (Abcam, ab6728) in 1× TBS. Cells were then washed with PBS–0.02% Tween-20 and overlaid with 4CN (4-chloro-1-naphthol) and H2O2 for 0.5 h. The plates were then rinsed with water and dried for counting foci. Final RSV titres are expressed as log10-transformed PFU per g lung.
Histopathological analysis of mice was carried out at WuxiAppTec. Lobules of lung tissue were taken, fixed in 4% PFA for 24 h before being transferred to a 70% ethanol solution. Subsequently, the samples were paraffin-embedded and cut into 5-μm thick sections. Sections were stained with haematoxylin and eosin, and examined under a wide-field microscope by three independent pathologists, who were blinded to the treatment group of the animals. Lung inflammation and exfoliation was recorded and scored semiquantitatively. The degree of pathological changes was graded on a minimal, mild, moderate and marked scale corresponding to the numbers 0 to 3.
Ethics statement
The in vivo work of this study was carried out in accordance with INRAE guidelines in compliance with European animal welfare regulations. The protocols were approved by the Animal Care and Use Committee at the ‘Centre de Recherche de Jouy-en-Josas’ (COMETHEA) under relevant institutional authorization (‘Ministère de l’éducation nationale, de l’enseignement supérieur et de la recherche’, authorization number 2015100910396112v1 (APAFIS#1487)). All experimental procedures were performed in a biosafety level-2 facility.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All experimental data shown in Figs. 1–3 and Extended Data Figs. 1–10 are available from the corresponding authors upon reasonable request. Sequences of newly described viruses are accessible from the GenBank under accession numbers MW039434, MT994243 and MT994242. Source data are provided with this paper.
Code availability
No new custom computer code or algorithm was used to generate the results reported in the paper.
References
Wheeler, R. J. & Hyman, A. A. Controlling compartmentalization by non-membrane-bound organelles. Phil. Trans. R. Soc. Lond. B 373, 20170193 (2018).
Wileman, T. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu. Rev. Microbiol. 61, 149–167 (2007).
Rincheval, V. et al. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 8, 563 (2017).
Nikolic, J. et al. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 8, 58 (2017).
Guseva, S. et al. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 6, eaaz7095 (2020).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Nam, H. H. & Ison, M. G. Respiratory syncytial virus infection in adults. Br. Med. J. 366, l5021 (2019).
Pneumonia Etiology Research for Child Health (PERCH) Study Group. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: the PERCH multi-country case–control study. Lancet 394, 757–779 (2019).
Heylen, E., Neyts, J. & Jochmans, D. Drug candidates and model systems in respiratory syncytial virus antiviral drug discovery. Biochem. Pharmacol. 127, 1–12 (2017).
Galloux, M. et al. minimal elements required for the formation of respiratory syncytial virus cytoplasmic inclusion bodies in vivo and in vitro. mBio 11, e01202-20 (2020).
Feric, M. et al. Coexisting liquid phases underlie nucleolar subcompartments. Cell 165, 1686–1697 (2016).
Richard, C. A. et al. RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog. 14, e1006920 (2018).
Bailly, B. et al. Targeting human respiratory syncytial virus transcription anti-termination factor M2-1 to inhibit in vivo viral replication. Sci. Rep. 6, 25806 (2016).
Weierstall, U. et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 (2014).
Peluso, M. O. et al. Impact of the Smoothened inhibitor, IPI-926, on smoothened ciliary localization and Hedgehog pathway activity. PLoS ONE 9, e90534 (2014).
Barron, R. M. et al. PrP aggregation can be seeded by pre-formed recombinant PrP amyloid fibrils without the replication of infectious prions. Acta Neuropathol. 132, 611–624 (2016).
Wei, M. T. et al. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 9, 1118–1125 (2017).
Iserman, C. et al. Genomic RNA elements drive phase separation of the SARS-CoV-2 nucleocapsid. Mol. Cell 80, 1078–1091.e6 (2020).
Rameix-Welti, M. A. et al. Visualizing the replication of respiratory syncytial virus in cells and in living mice. Nat. Commun. 5, 5104 (2014).
Blodörn, K. et al. A bovine respiratory syncytial virus model with high clinical expression in calves with specific passive immunity. BMC Vet. Res. 11, 76 (2015).
Kirsebom, F., Michalaki, C., Agueda-Oyarzabal, M. & Johansson, C. Neutrophils do not impact viral load or the peak of disease severity during RSV infection. Sci. Rep. 10, 1110 (2020).
Buchholz, U. J., Finke, S. & Conzelmann, K. K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73, 251–259 (1999).
Blockus, S. et al. Labyrinthopeptins as virolytic inhibitors of respiratory syncytial virus cell entry. Antiviral Res. 177, 104774 (2020).
Mullor, J. L., Sánchez, P. & Ruiz i Altaba, A. Pathways and consequences: Hedgehog signaling in human disease. Trends Cell Biol. 12, 562–569 (2002).
McNally, J. G. Quantitative FRAP in analysis of molecular binding dynamics in vivo. Methods Cell Biol. 85, 329–351 (2008).
Haid, S., Grethe, C., Bankwitz, D., Grunwald, T. & Pietschmann, T. Identification of a human respiratory syncytial virus cell entry inhibitor by using a novel lentiviral pseudotype system. J. Virol. 90, 3065–3073 (2016).
Acknowledgements
We thank T. Mitchison for discussions, interpretation of the data and reading the manuscript, H. Rezaei for mouse PrP amyloid fibrils and discussions, D. Descamps and M. Bauducel for help with animal work, C. Sizun for NMR assays, J. Fix for virus amplification, G. Hansen and M. Wetzke for collection of primary RSV isolates, H. Wang for technical assistance in DNA recombination and D. Xu for logistic support and helpful advice; W.-L. (Billy) NG for reviewing the SI chemistry; staff of the MIMA2 platform for access to the IVIS200 station, which was acquired with funds from the Ile de France région (SESAME); and staff of the Cymages platform for access to the Leica SP8 microscope and Olympus FV3000 inverted confocal microscope, which was supported by grants from the region Ile-de-France. This work was supported by grants from the Natural Science Foundation of China Youth Project (31900147) to J.C., Fondation Air Liquide grant to J.-F.E. and R.A., the 111 Project (B16030), ATIP-AVENIR INSERM program and the Fondation Del Duca-Institut de France grant to J.R.-B. and M.-A.R.-W., a Sino-German Helmholtz International Lab grant (10000089395401) to Y.Z. and T.P., and People’s Livelihood Technology Project of Qingdao City (17-3-3-2-nsh) to Z.W.
Author information
Authors and Affiliations
Contributions
J.R.-B., J.C., V.R., M.G., J.-F.E., M.-A.R.-W. and R.A. designed experiments, analysed data and wrote the manuscript. R.A. initiated and designed the overall RSV drug discovery programme and designed the chemical analogues. M.-A.R.-W., J.-F.E., T.P. and R.A. reviewed the manuscript. R.A. and J.C. designed, performed and analysed the experiments, and analysed the data relative to the effects of the drugs on RSV replication and HH antagonism. H.Z., M.D. and X.Z. participated in the antiviral and HH compound testing. T.P., S.H. and S.M.S. designed, performed and analysed the inhibition assay with minimally laboratory-experienced RSV. J.R.-B., V.R., J.-F.E. and M.-A.R.-W. designed the experiments and analysed the data relative to the effects of the drugs on condensates in infected cells. J.R.-B. performed the experiments. V.R. performed experiments and provided expertise to analyse condensate disorganization and A.M.-J. provided support and expertise for the dextran micro-injection experiments. R.A., M.G. and R.L.G. designed and analysed the animal experiments. M.G., R.L.G., A.D. and F.H. infected and handled mice for animal efficacy studies. Y.Z. and Z.W. provided critical logistic and infrastructure support. Z.W., Y.Z., T.P., J.-F.E., M.-A.R.-W. and R.A. obtained funding for the study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.A. and J.J.C. are inventors of patents 2016100874979.4 (China), 10,035,816 B2 (USA), 6588156 (Japan) and patent application 17749894.6 (EU) held by Shandong University. The other authors declare no competing interests.
Additional information
Peer review information Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Synthetic route of the CPM analogues.
The detailed synthesis protocol is described in the Supplementary Methods. The IDs of the end products are A3M (A-ring, 3′-methoxy-cyclopamine; compound 4), A3E (A-ring, 3′-ethoxy-cyclopamine, compound 6) and A3P (A-ring, 3′-propoxy-cyclopamine, compound 8). The name of A3E (3′-ethoxy-cyclopamine) according to IUPAC nomenclature is 3S,3′R,3a′S,6aS,6bS,6′S,7a′R,9R,11aS,11bR-3-ethoxy-3′,6′,10,11b-tetramethyl-1,2,3,3a′,4,4′,5′,6,6a,6b,6′,7,7′,7a′,8,11,11a,11b-octadecahydro-3′H-spiro[benzo[a]fluorene-9,2′-furo[3,2-b]pyridine].
Extended Data Fig. 2 Biological activity of CPM chemical analogues in vitro.
a, c, e, g, Potency of compounds to inhibit SHH signalling was tested in Gli-dependent luciferase-reporter-expressing NIH3T3 cells. Data are expressed as the percentage ± s.d. of the DMSO control. Data are representative of two experiments performed in triplicate. b, d, f, h, Potency of compounds to inhibit RSV replication was tested in RSV–Luc-infected HEp-2 cells. Luciferase activity was measured at 24 h after infection and expressed as the percentage ± s.d. of the DMSO control. The cytotoxicity of compounds was measured in parallel in either HEp-2 or NIH3T3 reporter cells after treatment for 24 h (data not shown) or 72 h. IC50 and CC50 values were calculated with GraphPad software. Data shown are representative of two (b, f) or three (d, h) experiments performed in triplicate.
Extended Data Fig. 3 Ability of A3E and CPM to inhibit minimally laboratory-experienced RSV.
a, Inhibition of minimally laboratory-experienced RSV was tested in HEp-2 cells infected for 24 h with two RSV A or one RSV B isolates and analysed by flow cytometry as described in the Methods. The results are expressed as mean ± s.d. from three independent experiments performed in duplicate. b, c, IC50 values are calculated from three independent experiments performed in duplicate. CC50 values are from a representative experiment out of three experiments performed in triplicate. The selectivity index (SI) is calculated as SI = CC50/IC50.
Extended Data Fig. 4 Disorganization of IB and IBAG architecture in RSV–M2-1–mGFP-infected and compound-treated cells.
HEp-2 cells were infected with RSV–M2-1–mGFP for 24 h and then treated for 1 h with A3E and CPM at the indicated concentrations and stained to detect poly(A) RNA and N protein as described in the Methods. a, Representative images from two independent experiments are shown. Poly(A) RNA (red), N (white), M2-1–mGFP (green) and nuclei (blue) were visualized. A merged image of red, green and blue channels is shown (merge). Scale bars, 10 μm. b, The presence of IBAGs within IBs was quantified and indicated as IBAG (+) when visible and IBAG (−) when absent (described in the Methods) for approximately 50 infected cells per condition from two independent experiments.
Extended Data Fig. 5 RSV IBs lose velocity and the ability to fuse but remain sensitive to 1,6-hexanediol.
a, b, HEp-2 cells infected with RSV–P–BFP for 24 h were treated for 1 h with DMSO, CPM (5 μM) or A3E (25 μM) and then exposed to 10% 1,6-hexanediol (Hex), imaged at 10 min and quantified (see Methods). Data are from 10 acquisitions captured in two independent experiments. Representative images are shown in a. Scale bar, 10 μm. The mean ± s.d. number of IBs per image is expressed as the percentage of the pre-treatment control. **P < 0.01; two-tailed Wilcoxon signed-rank test. c, d, HEp-2 cells were infected with RSV–P–BFP for 24 h and treated for 1 h with DMSO, CPM (5 μM) or A3E (25 μM) and then imaged as described in the Methods. Fusion events were quantified as described in the Methods. Results are expressed as mean ± s.d. from 10 videos (15 min) from two independent experiments (c). Maximum velocities were obtained by automatic tracking of IBs from 10 videos (15 min) from two independent experiments (see Methods). Box plots show the median (centre line) and the first and third quartiles (upper and lower hinges). Statistical analysis of maximum velocities was done using Kruskal–Wallis tests followed by Dunn’s test for multiple comparisons (d). *P < 0.05; ***P < 0.001. e, HEp-2 cells were infected for 24 h with RSV–P–BFP and treated with CPM (5 μM), A3E (25 μM) or DMSO as control for 1 h. IBs shape was analysed as described in the Methods. Box plots indicate the median (centre line) and the first and third quartiles (upper and lower hinges). **P < 0.01; Welch’s ANOVA followed by a Games–Howell post hoc test.
Extended Data Fig. 6 A3E and CPM do not induce the formation of fibrillar solids but increase the mesh size of IBs.
a, HEp-2 cells infected with RSV–P–BFP for 24 h and treated with CPM (5 μM) or A3E (25 μM) for 1 h, then fixed and stained with ThS. Purified mouse PrP amyloid fibrils adsorbed on fixed cells and treated in parallel were used as positive control. IBs are shown by P–BFP fluorescence (blue) and ThS is shown in green. Representative images from two independent experiments are shown. Scale bar, 10 μm. b, c, Fluorescent dextran beads were selected based on their approximate hydrodynamic radius (Rh) and micro-injected into HEp-2 cells that were infected with RSV–P–BFP for 20–24 h and treated for 1 h with A3E (25 μM) or CPM (5 μM). Cells were imaged 2–5 min after micro-injection under a wide-field microscope. Representative images from two independent experiments are shown in c. Scale bar, 2 μm. The fluorescent dextran signal inside IBs relative to the cytoplasm was quantified as described in the Methods. Data are mean ± s.e.m. of the signal ratio from at least 25 IBs from two independent experiments. ****P < 0.0001; ns, not significant; Kruskal–Wallis test followed by Dunn’s test for multiple comparisons.
Extended Data Fig. 7 Variation in the size and shape of IBs in compound-treated RSV-infected cells.
a, b, HEp-2 cells were infected with RSV–P–BFP or RSV–M2-1(R151K)–P–BFP for 24 h and treated for 1 h with A3E (25 μM), CPM (5 μM) or DMSO at 24 h after infection. Cells were fixed, stained and analysed by confocal microscopy as described in the Methods. The size (a) and roundness (b) of IBs were quantified. Results of 50 cells from two independent experiments are shown. Box plots indicate the median (centre line) and the first and third quartiles (upper and lower hinges). Statistical significance analysis of size and roundness data was performed using Welch’s ANOVA followed by a Games–Howell test for multiple comparisons. **P < 0.01.
Extended Data Fig. 8 IBs in RSV–M2-1(R151K)–P–BFP-infected and compound-treated cells are still susceptible to hypotonic shock.
a, b, HEp-2 cells were infected for 24 h and treated with A3E (25 μM), CPM (5 μM) or DMSO for 1 h. Hypotonic shock was applied for 10 min and cells were imaged at the indicated times and the signals were quantified. Measurements labelled ‘Rec’ (recovery period) were taken after 5 min of shock followed by 5 min in culture medium. a, Representative images from two independent experiments and ten acquisitions are shown. Scale bar, 10 μm. b, Data are expressed as the mean ± s.d. number of IBs per image and compared to pre-shock values. Statistical significance analysis was performed using Kruskal–Wallis tests followed by two-sided Dunn’s test for multiple comparisons; **P < 0.01, ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 9 IB and IBAG architecture is unaffected by compound treatment in cells infected with the RSV–M2-1(R151K) mutant virus.
a, b, HEp-2 cells were infected with RSV–M2-1(R151K)–P–BFP for 24 h and then treated for 1 h with A3E (25 μM) or CPM (5 μM) followed by staining for poly(A) RNA and N protein as described in the Methods. a, Representative images from two independent experiments are shown. Poly(A) RNA, N and P–BFP are shown in red, white and blue, respectively. Merge of poly(A) RNA, N and P–BFP or poly(A) RNA and P–BFP are shown in the fifth and fourth column, respectively. Scale bar, 10 μm. b, The presence of IBAGs within IBs was quantified and scored as IBAG (+) when visible and IBAG (−) when absent from approximately 50 infected cells per condition from two independent experiments (see Methods).
Extended Data Fig. 10 Time course and treatment window for A3E and CPM in RSV–Luc-infected mice.
a, Time course of RSV–Luc infection in compound-treated mice. Mice were infected intranasally with 5 × 104 PFU ml−1 RSV–Luc and treated with the indicated compounds twice daily from 0 to 3 d.p.i. and bioluminescence readings were obtained at 3, 4, 5 and 6 d.p.i. One representative of two time-course experiments performed is shown. The mean ± s.e.m. radiance is expressed as the sum of the photons per second from each pixel inside the region of interest per the number of pixels (p s−1 cm−2 sr−1). Vehicle group, n = 10 mice; all treatment groups, n = 5 mice. b, Experimental design to determine the A3E and CPM treatment window for RSV–Luc-infected mice. Viral replication in lungs was determined by bioluminescence imaging as described in a and the Methods. Mice were infected with 5 × 104 PFU ml−1 RSV–Luc, treated with compounds at 15 mg kg−1 twice daily for the indicated time periods and imaged at 4 d.p.i. c, Results are expressed as the percentage of RSV replication compared to vehicle control and are shown as mean ± s.e.m. for a specified number of RSV–Luc-infected animals. Vehicle group, n = 27 mice, n = 4 experiments; CPM 0–3 d.p.i., n = 12 mice, n = 3 experiments; CPM 1–3 d.p.i., n = 4 mice, n = 1 experiment; CPM 2–3 d.p.i., n = 4 mice, n = 1 experiment; CPM 3 d.p.i., n = 4 mice, n = 1 experiment; A3E 0–3 d.p.i., n = 12 mice, n = 2 experiments; A3E 1–3 d.p.i., n = 4 mice, n = 1 experiment; A3E 2–3 d.p.i., n = 4 mice, n = 1 experiment; A3E 3 d.p.i., n = 4 mice, n = 1 experiment. Statistical significance analysis was performed using Kruskall–Wallis tests followed by two-sided Dunn’s test for multiple comparisons. *P < 0.05, **P < 0.01, ****P < 0.0001.
Supplementary information
Supplementary Information
This file contains details of the chemical synthesis of A3M, A3E and A3P, including a List of compounds, synthesis scheme and images of analytical data.
Supplementary Video 1
IBAG dynamics in compound-treated cells. Time-lapse microscopy of IBAGs in HEp-2 cells infected with RSV-M2-1-mGFP. At 24 hpi, images were taken every 6 s in a CO2-controlled chamber heated at 37 °C, with an Olympus FV3000 inverted confocal microscope. After five minutes, cells were treated with DMSO (mock, Supplementary Video 1), 5 μM CPM (Supplementary Video 2) or 25 μM A3E (Supplementary Video 3) and subsequently images were taken for another 20 min. The resulting videos were visualized using Image J software (10 fps). A representative video from three independent experiments in which 3 videos were acquired is shown. Scale bar 10 μm.
Supplementary Video 2
IBAG dynamics in compound-treated cells. Cells were treated with 5 μM CPM; see legend for Supplementary Video 1.
Supplementary Video 3
IBAG dynamics in compound-treated cells. Cells were treated with 25 μM A3E; see legend for Supplementary Video 1.
Supplementary Video 4
IB dynamics. Time-lapse microscopy of IBs in HEp-2 cells infected with RSV-P-BFP. At 24 hpi, cells were imaged every 30 s in a CO2-controlled chamber heated at 37 °C, with an Olympus FV3000 inverted confocal microscope. The resulting videos were visualized under the Image J software (10 pfs). A representative video from 10 videos from 2 independent experiments is shown. Scale bar 10 μm.
Supplementary Video 5
IB dynamics and mobility. Time-lapse microscopy of IBs in HEp-2 cells infected with RSV-P-BFP. At 24 hpi, cells were treated for 1 h with DMSO (mock, Supplementary Video 5), 5 μM CPM (Supplementary Video 6) or 25 μM A3E (Supplementary Video 7) and subsequently imaged every 30s for 15 min in a CO2-controlled chamber heated at 37 °C, with an Olympus FV3000 inverted confocal microscope. The resulting videos were visualized under Image J and ICY software (10 pfs). A representative video from 10 videos from 2 independent experiments is shown. Scale bar 10 μm.
Supplementary Video 6
IB dynamics and mobility. Cells were treated with 5 μM CPM; see legend for Supplementary Video 5.
Supplementary Video 7
IB dynamics and mobility. Cells were treated with 25 μM A3E; see legend for Supplementary Video 5.
Source data
Rights and permissions
About this article

Cite this article
Risso-Ballester, J., Galloux, M., Cao, J. et al. A condensate-hardening drug blocks RSV replication in vivo. Nature 595, 596–599 (2021). https://doi.org/10.1038/s41586-021-03703-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03703-z
This article is cited by
-
Phosphorylation regulates viral biomolecular condensates to promote infectious progeny production
The EMBO Journal (2024)
-
Drug repurposing screen identifies lonafarnib as respiratory syncytial virus fusion protein inhibitor
Nature Communications (2024)
-
Cuproptosis-Related Ferroptosis genes for Predicting Prognosis in kidney renal clear cell carcinoma
European Journal of Medical Research (2023)
-
Screening antivirals with a mCherry-expressing recombinant bovine respiratory syncytial virus: a proof of concept using cyclopamine
Veterinary Research (2023)
-
Biomolecular condensates: insights into early and late steps of the HIV-1 replication cycle
Retrovirology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
