The Evidential Statistics of Genetic Assembly: Bootstrapping a Reference Sequence
 Yukihiko Toquenaga
Yukihiko Toquenaga Takuya Gagné
Takuya Gagné- 1Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Japan
- 2Graduate School of Biological Sciences, University Tsukuba, Tsukuba, Japan
The reference sequences play an essential role in genome assembly, like type specimens in taxonomy. Those references are also samples obtained at some time and location with a specific method. How can we evaluate or discriminate uncertainties of the reference itself and assembly methods? Here we bootstrapped 50 random read data sets from a small circular genome of a Escherichia coli bacteriophage, phiX174, and tried to reconstruct the reference with 14 free assembly programs. Nine out of 14 assembly programs were capable of circular genome reconstruction. Unicycler correctly reconstructed the reference for 44 out of 50 data sets, but each reconstructed contig of the failed six data sets had minor defects. The other assembly software could reconstruct the reference with minor defects. The defect regions differed among the assembly programs, and the defect locations were far from randomly distributed in the reference genome. All contigs of Trinity included one, but Minia had two perfect copies other than an imperfect reference copy. The centroid of contigs for assembly programs except Unicycler differed from the reference with 75bases at most. Nonmetric multidimensional scaling (NMDS) plots of the centroids indicated that even the reference sequence was located slightly off from the estimated location of the true reference. We propose that the combination of bootstrapping a reference, making consensus contigs as centroids in an edit distance, and NMDS plotting will provide an evidential statistic way of genetic assembly for non-fragmented base sequences.
1. Introduction
Assume that you got multiple non-fragmented base sequences assembled from data generated with next-generation sequencing (NGS) or more advanced methods. We further assume that we do not have available reference sequences for the material. QUAST (http://bioinf.spbau.ru/quast) and similar tools would recommend choosing longer sequences as plausible ones. But the length of the sequence itself does not guarantee how the sequences resemble or correspond to the correct sequence. Here we propose an evidential statistical method for inferring true sequence by bootstrapping and Nonmetric Multidimensional Scaling (NMDS) plotting with the assembled non-fragmented sequences.
1.1. Background
In evidential statistics, we never seek the true model for a specific data set. Instead, we choose models supported by the given data set (Edwards, 1992; Royall, 1997). The information-theoretic approach also neglects to chase the true model for increasing the prediction ability of selected models (Burnham and Anderson, 1998; Konishi and Kitagawa, 2004; Akaike et al., 2007). Even in Bayesian statistics, the true model is believed to be included in their models with parameter distributions. But the true model for a specific data set still plays an essential role in biology using base sequence data.
DNA or RNA sequencing is rather conservative. It relies on reference sequences often obtained with decades-old sequence techniques (e.g., Sung, 2017). Assume that you get short segments of sequences (called “reads”) with NGS methods. Then you have to align them correctly for constructing the whole sequence. Reads are inherently erroneous, and you will have to use several approaches to reconstruct the entire sequence. The reconstructed sequences could be divergent. But those that resemble the reference are promising candidates. Here the reference sequences play the role of type specimens in taxonomical identification (Ballouz et al., 2019).
But it is well known that different assembly programs return different assembly results for a given read data (e.g., Salzberg et al., 2012). Researchers use reference sequences or check annotation of known genes for correcting resultant sequences (Sung, 2017; Sohn and Nam, 2018). High variation in sequence results among assembly programs is mainly caused by applying only heuristic approaches, such as the base-by-base approach, de Bruijn graph, and String graph (Sung, 2017; Sohn and Nam, 2018). Random searching is the core for those approaches, but somehow some assembly programs return the same contig data (both contents and order of sequences) for a given read data set. Others return different contigs for a given data in multiple trials.
For example, the insect mitochondrial genome is a compact circular molecule typically 15–18 kb in size (Cameron, 2014). Recently two genome sequences of mitochondria of Acanthoselides obtectus were proposed: one with 16,130 bp (Yao et al., 2017) but the other with 26,613 bp (Sayadi et al., 2017). The latter sequence includes repetitive spacer sequences (Figure 1). Usually, repetitive or duplicated sequences are targets to collapse by assemblers. But the researchers published the longer mitochondria claim that they used a new and reliable long-read technique, and hence, the repetitive sequences are real (Sayadi et al., 2017). From now on, we will have to deal with the two references for mtDNA of A. obtectus.
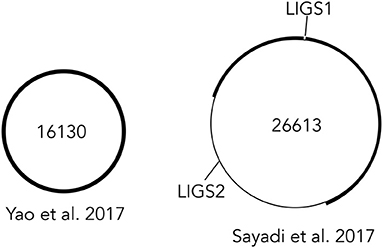
Figure 1. Two schematic mtDNA of Acanthoscelides obtectus with different lengths. There are short (1) and long (2) long intergenic spacers (LIGS1 and LIGS2) in the longer one. Other than the spacers, two mtDNAs are identical.
What will happen if the reference sequence is not reliable or not exist at all? How can we choose promising ones among divergent reconstructed sequences? Conventionally, researchers believe that the reference sequences are correct. But the references are also samples obtained at some time and location with a specific method. How can we measure the reliability of a reference sequence? Conversely, can we measure the reliance of an assembly method by assembling random reads generated with the reference sequence itself? The random read generation is what we call the bootstrapping of the reference sequence. We propose that bootstrapping can evaluate and characterize assembly programs by using distance measurements that play essential roles in the evidential statistics (Lindsay, 2004).
1.2. Preliminary Analysis With the phiX174 Reference
We tried to reconstruct the genome sequence of a very simple life, phiX174, an Escherichia coli bacteriophage. (Figure 2). Several read data sets for phiX174 are available on the internet (e.g., https://github.com/gigascience/galaxy-bgisoap/tree/master/test-data/phiX174). In our preliminary examination with one of the sample read data, 13 free and frequently used assembly programs (all programs in Table 1 except Unicycler) fairly well reconstructed the reference sequence; defect proportion was <5%. But none of them returned the perfect sequence of the reference. Most defects were concentrated at the origin (locus = 0) of the circular phiX174 genome.
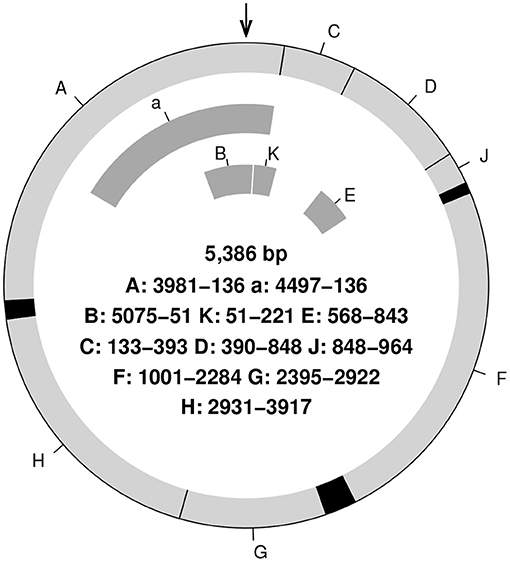
Figure 2. A schematic genome structure and gene sizes of phiX174. There are 11 genes, and black regions are intergenic spacers. The arrow indicates the origin. Note that there is a 4-base overlap between genes A and C.
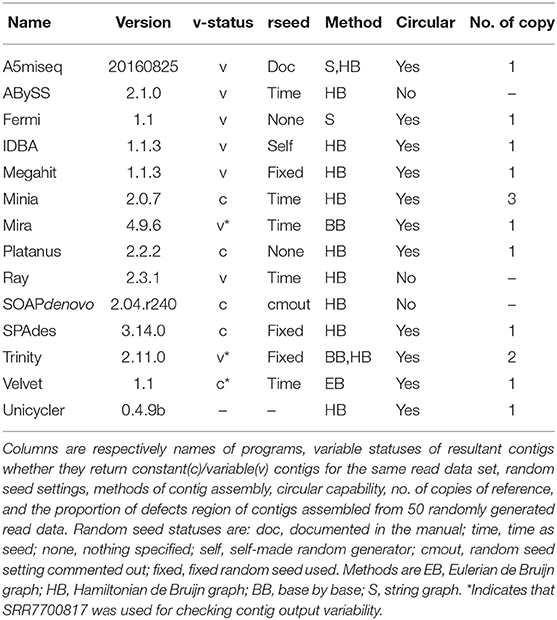
Table 1. List of assembly programs and their characteristics.
We obtained similar results when we reconstructed random reads generated from the reference with a random read simulator, ART (art-illumina Q version) ver. 2.5.8 (Huang et al., 2011). None of the assembly software perfectly reconstructed the reference. Increasing the coverage did not change the results. Again defects were concentrated at the locus zero of the reference. But those results might be caused by an artifact; the random reads were generated, assuming that the reference genome was linear. Both edges inevitably had minimal coverage that caused concentrated defects at the head or tail of the sequence. We should prepare reads generated from the reference assumed to be circular. We also want to exclude all errors specific to NGS methods while generating random read data.
1.3. Flow of This Article
We first show how to obtain hypothetical read data sets for bootstrapping the phiX174 reference sequence in the following sections. Next, we introduce and characterize 14 free assembly programs. Then we explain how to analyze resultant contig sequences. We also introduce the way of constructing consensus sequences from the resulting contigs. The consensus sequences were then plotted in an edit distance space with an NMDS method. For the best-performing assembly program, we tried to reconstruct mtDNAs of A. obtectus in Yao et al. (2017) and Sayadi et al. (2017). Results are reported according to the same order. We discuss the possibility of estimating the true reference sequence from the NMDS plots of the consensus and the reference sequences based on the evidential statistics.
2. Materials and Methods
2.1. Read Simulators
We surveyed 24 sequence simulators (Alosaimi et al., 2020) and found that only two of them capable of generating reads for circular references. One of them adopts GUI, so we have to use the other one, GemSIM ver. 1.6 (McElroy et al., 2012). But GemSIM cannot specify random seeds. Moreover, GemSIM cannot stop errors specific to sequencers. We had to generate hypothetical random reads without any INDEL and sequence-specific errors. So we made an Illumina read simulator free from any kinds of errors by ourselves with Ruby's programming language (ringreads.rb, ref.rb, and doRingreads.rb).
The simulator accepts a FASTA file of a circular reference DNA sequence. A random read generation started from a location randomly selected within the reference. Then the read was extended to the prespecified read length. Next, a new starting point was located apart from the endpoint of the read with the extent of an insertion length. Then the “paired” read with the same length was generated, but this new read was transformed to its reverse complement. In this way, paired-end reads separated with the given insertion length were generated. This procedure was repeated for the circular genome until the minimal coverage for each locus exceeded the pre-specified coverage value. The Ruby scripts are available at: https://tivoli.ska.life.tsukuba.ac.jp/~toque/to9ue/ringreads.
2.2. Assembly Programs
We collected 14 free assembly programs (see Table 1). All programs were installed from source codes or binary distributions. We used Mac OS X 10. 14.6 on a Mac mini (2018) and iMac (Retina 5K, 27-inch, 2020) as our computational environment. We obtained source codes of all software irrespective of the ways of installation. We focused on program performance only at the contig construction level because our target genome of phiX174 was short enough, and each reconstructed sequence was almost always a single contig. We did not have to apply any polishing processes, either. Other than specifying lengths of reads and insertion, we used default parameter settings for each assembly program. For Unicycler, we applied normal model, https://github.com/rrwick/Unicycler.
For all programs except Unicycler, we checked whether each program returned the same contig for the same read data set for multiple trials. For this purpose, we used reads of Homo sapience chromosome 3 (30CJCAAXX_4_[12].fq.gz) available at: http://sjackman.ca/abyss-activity. Please consult the link for parameter settings for k-mers and insertion lengths. Mira, Velvet, and Trinity could not handle the chromosome 3 reads, so we used a smaller data set of Human Mitochondrial DNA from Postmortem Brain and Blood (SRR7700817 in SRX4559088) for those three programs.
An assembly program was judged as constant only if both the order and the content of contigs were the same in multiple assembly trials. The program was judged as variable otherwise, or it was judged as variable even if only the order of the same contig sets was different. We also checked random seed settings specified in the source codes of the assembly programs. Methods for contig reconstruction were examined for each assembly program based on the description in Sohn and Nam (2018), Sung (2017), software manuals, and publications on the software (Chevreux et al., 1999; Boisvert et al., 2010; Grabherr et al., 2011; Kajitani et al., 2014; Coli et al., 2015; Li et al., 2015). Only 11 out of 14 assembly programs could handle reads generated from the circular reference (Table 1). So we applied randomly generated read data only for those 11 assembly programs. Please consult doASSEMBLER_NAME.rb at: https://tivoli.ska.life.tsukuba.ac.jp/~toque/to9ue/ringreads that provide parameters for assembly programs. For Unicycler, we performed assembly with and without polishing with the pilon algorithm using “–no_pilon” option.
2.3. Bootstrapping Reads From the Reference
We generate 50 random sets of reads from the phiX174 reference (accession no. NC_001422), in each of which the data structure was the same as paired-end data of an available read data set (https://github.com/gigascience/galaxy-bgisoap/tree/master/test-data/phiX174); read length = 90, insert length = [200, 500], and the minimum coverage = 20. For each random read data, we tried to assemble contigs with the 11 assembly programs. We aligned the resultant contigs to the reference sequence and examined unique sequences among the 50 contigs for each assembly program. The number of reads for insertion length of 200 ranged from 3,216 to 3,812. Those for insertion length of 500 ranged from 3,132 to 3,964 (Figure 3).
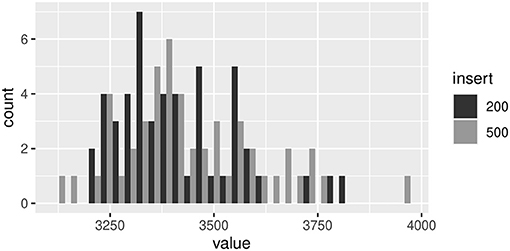
Figure 3. Frequency distributions of the number of random reads for 200 and 500 insertion lengths.
2.4. Analyzing Contig Sequences
If a resultant contig is different from the reference and a little longer, we would have two possibilities (Figure 4). One is that defective parts are scattered within the contig. The other is that defects are concentrated at a specific region, as in the preliminary experimental results in which we treated the reference as a linear genome. If we apply BLAST search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) for the known 11 genes of the phiX174, we would not be able to reconstruct several genes in the former case. On the contrary, we could not reconstruct only a couple of genes in the latter case. We performed BLAST searches for the 11 genes for the resultant contigs generated with the 11 assembly software to distinguish the two scenarios. We used the rBLAST library (https://github.com/mhahsler/rBLAST) for it.
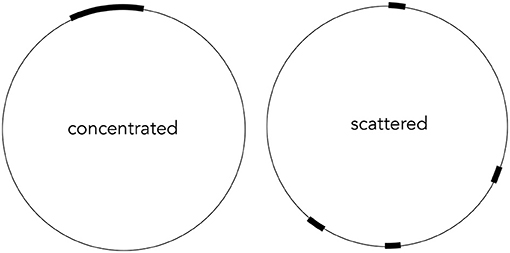
Figure 4. Two hypothetical patterns of defect locations. One is concentrated at a location, but defect locations are scattered in the other.
The BLAST search of genes, which adopts the Smith-Waterman algorithm, can not correctly determine whether a given contig succeeded in reconstructing the reference because it arbitrarily inserts gaps or deletion for sequence comparison. What we want to do is precisely compare a contig and the reference without any insertion and deletions. To do so, we used the diffobj library of R (https://cran.r-project.org/web/packages/diffobj/index.html). Functions of the diffobj work just like the diff command of UNIX. We can pinpoint the defective parts among the contig with this functionality. But we should apply the diff operation to two sequences that start at the same origin.
Resultant contigs started from 5” to 3” arbitrarily. So we first have to align all contigs to the direction of the phiX174 reference. Then we have to find the true origin corresponding to the locus zero of the reference because assembly programs returned linear contigs starting from arbitrary origins. To do so, we have to use some distance measurements for comparing two sequences. We applied the Levenshtein edit distance [LEDx, y(i, j)] defined by three operations: deletion, insertion, and substitution (Equation 1).
where 1(xi≠yj) is the indicator function equals to zero when xi = yj and unity otherwise. LEDx, y(i, j) is the distance between the first i characters of x and the first j characters of y. Normalized Levenshtein edit distance can be obtained by dividing the raw edit distance with max[length(x), length(y)]. We used the stringsim function provided by the stringdist library (https://cran.r-project.org/web/packages/stringdist/index.html) for R ver. 3.6.3. (R Core Team, 2018).
To find the true locus zero in a contig, we chose a tentative origin randomly within a linear contig. We decided on another random starting point if the similarity between the contig and the phiX174 reference was higher than a pre-specified threshold. Otherwise, we chose the next origin by bit-wise walking to the right or left direction. We chose the direction so that the similarity between the contig and the reference increased. We stopped the process and defined the location as the locus zero if we attained the maximum similarity to the reference. We applied diffobj functions against the reference and the contig starting from the locus zero.
2.4.1. Consensus Sequence
Consensus sequences that we want to reconstruct differ from those obtained with conventional bioinformatics software, such as DECIPHER for R (Wright, 2016). Conventional applications insert gaps for reconstructing consensus sequences after the alignment of sequences of the same length. But our consensus sequences should have no gaps. As a result, the size of our consensus sequences was indefinite before the reconstruction.
To construct such a consensus sequence of assembly software from 50 contigs of randomly generated read data, we made a program equipped with GPU genetic algorithm (GPU-GA) to search the nearest neighborhood to all 50 contigs within a Levenshtein edit distance space. For GPU-GA calculation, we made an R library named gpuga in which We applied OpenCL (https://www.khronos.org/opencl/) for applying to GPU hardware including non-NVIDIA products. Note that you should use R ver. 3.x for running gpuga. The gpuga package is available at https://tivoli.ska.life.tsukuba.ac.jp/~toque/to9ue/ringreads.
We first listed up the longest common substrings among the 50 contigs. We used those common substrings for masking from INDEL operations during GA calculations. Next, we copied each of the 50 contigs 20 times for constructing the initial population of 1,000 bit-strings, each of which represents its specific contig sequence. We let evolve the bit string population for 100 generations with setting 0.0001 and 0.002 respectively for mutation and crossing over rates per bit. The fitness value is the sum of edit distance from the initial 50 contigs. After 100 generations, we applied bit-shift to the evolved bit-string population and then tried another ten generations of evolution to avoid being trapped in local optima.
After obtaining the consensus sequences of assembly programs, we reconstructed NMDS plots of sequences for examining relative locations against the reference. According to Ponciano and Taper (2019), we can obtain reliable estimates of the generating (true) model by plotting candidate models in a distance space with NMDS methods. A critical difference from Ponciano and Taper (2019) is that we do not have parametric generating functions for reconstructing contigs from the reference, and we cannot apply estimating methods for neg-cross and neg-selfentropies. But if we can assume that h2 = 0 in Equation 9 (Ponciano and Taper, 2019), we can estimate the true reference location as the origin (0,0) in the reconstructed NMDS spaces.
Multiple NMDS plots may be derived from the same data. In our preliminary examination, a general NMDS method applicable for sequence data (nmds, Taguchi and Oono, 2005) could not converge to a common spatial configuration. Dr. Mark L. Taper kindly recommended using mds function in smacof library (https://cran.r-project.org/web/packages/smacof/vignettes/smacof.pdf) for NMDS plottings, based on his experience in Ponciano and Taper (2019), compared to other NMDS functions available in R. Different NMDS functions adopt different stress functions being minimized. We checked that 2D NMDS plots created with metaMDS function in vegan library (https://www.rdocumentation.org/packages/vegan/versions/2.4-2/topics/metaMDS), isoMDS (https://www.rdocumentation.org/packages/MASS/versions/7.3-51.6/topics/isoMDS), and sammon (https://www.rdocumentation.org/packages/MASS/versions/7.3-51.6/topics/sammon) in MASS library were rotationally symmetric with that created with the mds function of smacof library.
2.5. Reconstructing mtDNA of A. obtectus
We created random reads from the sequence data of mtDNA of A. obtectus for both references respectively proposed in Yao et al. (2017) (accession no. KX825864) and Sayadi et al. (2017) (accession no. MF925724). We adopted the same lengths of reads (90) and insertion (200 or 500) and the coverage (20) as for the phiX174. Then we applied for the best assembly program in the phiX174 trial for reconstructing the mtDNA of A. obtectus. We analyzed the resultant contigs in a similar way as those for the phiX174 data sets. But the mtDNA sequences are more than three times longer than that of phiX174, which hindered analyzing methods, such as using R diffobj libraries.
3. Results
3.1. Assembly Performance
Most contigs generated with assembly software were longer than the reference sequence. After the BLAST searches, most contigs had a pattern with defects concentrated on a small region. In other words, each contig contained an almost perfect copy of the reference. Analyses with diffobj searches confirmed this result, and the proportion of discrepancy equals to the following equation.
where Lr and Lc are lengths of the reference and a contig, respectively.
Contigs generated by assembly methods consist of two groups based on the size: monomer and polymer ones (Figure 5). Monomer contigs are those with <6,200 bases. Polymer contigs include those with two- or threefold lengths of the reference. In the following, we explain the state of contigs for each assembly method.

Figure 5. Schematic linear representation of resultant contigs assuming that defect concentrated at the end of gene A. A monomer contig consisted of nearly a whole part of the reference (black) and a region mixing the rest of the reference and defects (gray). Trinity contigs included another complete reference. Minia contigs contained two perfect copies of the reference.
3.1.1. Polymer Contigs
Trinity returned all different contigs against 50 random read data sets, but the size of all of them was 10,795 bases. Each contig contained a perfect and an imperfect reference sequence concatenated in a line. Each imperfect reference had an extra paste margin, and its location was scattered all over among the 11 genes. The distribution was weakly biased to gene H (Chisq = 11.724, df = 6, P = 0.06841, Figure 6).
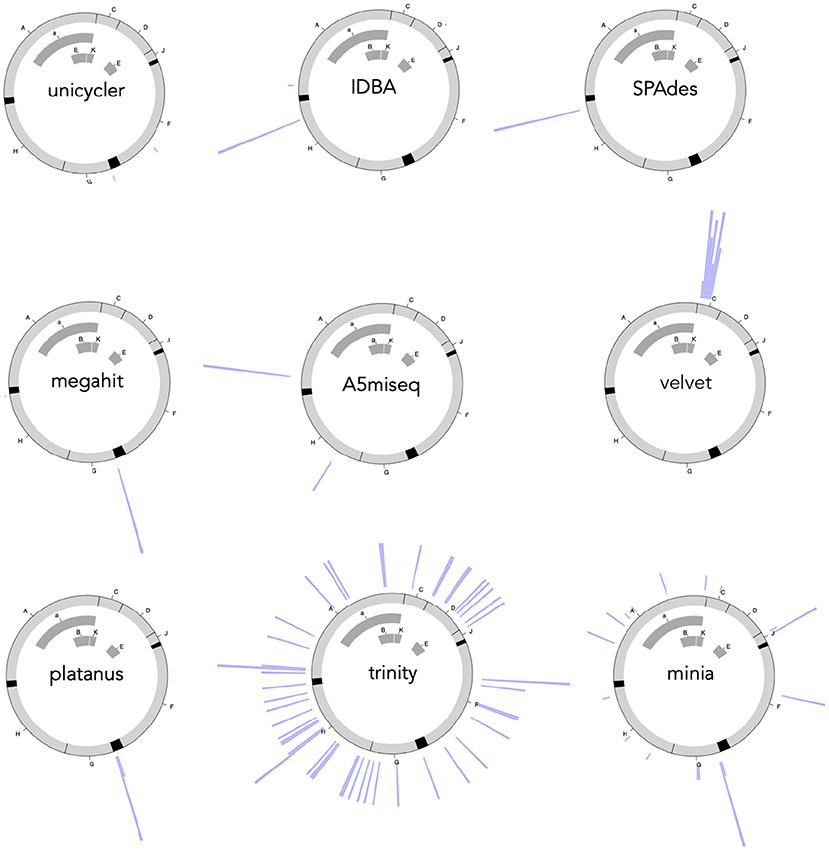
Figure 6. Circular histograms of defect locations on the reference for the nine assembly software.
Minia returned 24 unique contigs with all of which had 16,189 bases. Each contig consisted of two complete and one incomplete copy of the reference sequence concatenated linearly. The defect parts were scattered among the incomplete reference but heavily biased to gene G compared to the reference (Chisq = 33.595, df = 6, P = 8.055E-06, Table 2, Figure 6).
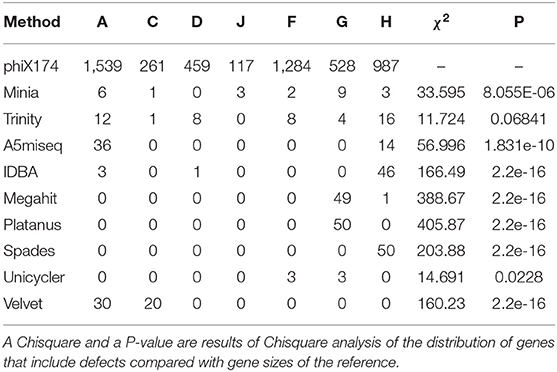
Table 2. Distribution of defects among the seven gene regions. For reference, gene sizes are indicated.
3.1.2. Monomer Contigs
A5miseq returned 11 unique contigs ranging from 5,461 to 5,465 bases. Fourteen and 36 contigs, respectively, had defects at genes A and H (Chisq = 56.996, df = 6, P = 1.831e-10, Figure 6). On the contrary, fermi returned four contigs for each random read data set. There were 191 unique contigs among them, ranging from 5,454 to 5,473 bases. Only 33 contigs among them passed BLAST searches for the reconstruction of 11 genes. But those 33 contigs had defects at small portions within gene D. Other contigs failed to reconstruct one of the genes of a (A), K, C, and D (not E) in the BLAST searches. One contig failed gene a (A). Fifty-two contigs failed gene K. Ninety-one contigs failed gene C. 15 contigs failed gene D (not E). Because of the high failure rate, we did not conduct diffobj analyses for Fermi contigs.
IDBA returned only three unique contigs ranging from 5,417 to 5,427 bases. Defects of each contig concentrated at genes H, A (not a), and E (and D), respectively (Chisq = 166.49, df = 6, P = 2.2e-16, Figure 6). Megahit also returned only three unique contigs with the common base length of 5,417. Defects were heavily concentrated at genes G and H (Chisq = 388.67, df = 6, P = 2.2e-16, Figure 6). Platanus returned seven unique contigs ranging from 5,430 to 5,436 bases. Defects were heavily concentrated at gene G (Chisq = 405.87, df = 6, P = 2.2e-16, Figure 6). SPAdes returned a single unique contig of 5,441 bases with a defect at gene H (Chisq = 203.88, df = 6, P = 2.2e-16, Figure 6).
Unicycler returned eight unique contigs ranging from 5,380 to 5,386. Those with 5,386 bases, which is the size of the reference genome, completely reconstructed the reference. Others failed to reconstruct genes F and G (Chisq = 14.691, df = 6, P = 0.0228). In summary, 44 out of 50 (88%) contigs were the perfect copy of the reference sequence. Velvet returned 39 unique contigs. All of them were with 5,416 bases and failed to reconstruct narrow regions of gene K overlapped with the end regions of genes A, a, and the beginning of gene C (Chisq = 160.23, df = 6, P = 2.2e-16). The performance of Unicycler did not change a lot when we even stopped the polishing process with the pilon algorithm; 39 out of 50 (78%) contigs were still the perfect copy of the reference.
Mira returned 49 unique contigs with 5,803–6,164 bases. Thirty-four of them completely reconstructed all 11 genes. Discrepancy regions for the 34 contigs were scattered all around the reference genome. Among the rest of 15 contigs, two failed to reconstruct gene A other than the region of gene a. Five could not reconstruct gene a. Six and two could not reconstruct genes F and H, respectively. Because of the high rate of failure, we did not conduct diffobj analyses for Mira contigs.
3.2. Consensus Sequence
We reconstructed consensus sequences for those assembly methods that returned almost perfect copy or copies of the reference; we excluded those of Mira and Fermi because those software returned variable contigs with variable defects. Table 3 shows defect starting locations and similarities against the reference for those consensus contigs. As expected, the consensus sequences resemble each methods' majorities, and their similarity to the reference is more than 0.986. That means the number of defects or extra bases was about 75 at most. There was no specific region for defects; incomplete reconstructions occurred at genes A, C, D, G, and H.
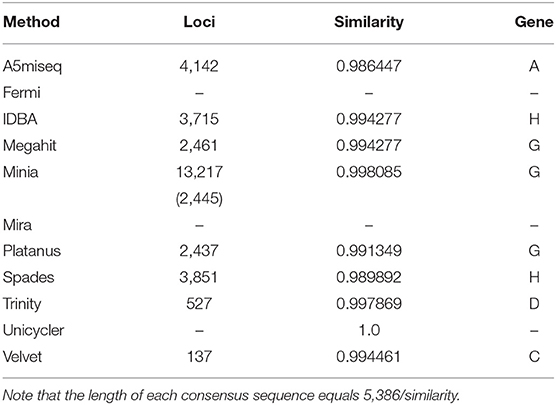
Table 3. The starting location of defects and similarity to the reference for each consensus sequence.
Figure 9 shows the NMDS plot of monomer consensus contigs with the reference. The mds function of smacof library returned the same plot for multiple trials. As expected, the reference (and Unicycler) location was close to the origin (0,0) in the NMDS plot. Megahit, velvet, and IDBA were moderately apart from the origin. On the contrary, A5miseq, SPAdes, and platanus were fairly apart from the origin. Especially, A5miseq was isolated from the cluster of the other six programs.
3.3. mtDNA of A. obtectus
Unicycler, the best performer in the assembly programs, returned a unique contig for each random data set. It reconstructed the same sequence with accession no. KX825864 in 38 out of 50 (76%) data sets. Unicycler returned the sequence with the same length (16,130) with the reference. For the six data sets of the rest, the sequence length was 16,129. For the two of the rest, the sequence length was 16,127. For the last data set, the length was 16,126.
But Unicycler returned a longer and multiple much shorter (a 10th of the longer one at most) contigs for random data sets created from the reference of MF925724. The length of the long contig ranged from 16,576 to 17,284 (Figure 7), which was enough longer than that of the reference of KX825864. The number of short contigs ranged from one to eight (mode = 5, Figure 8). Results were similar even we applied mode = bold to obtain the minimum number of contigs.
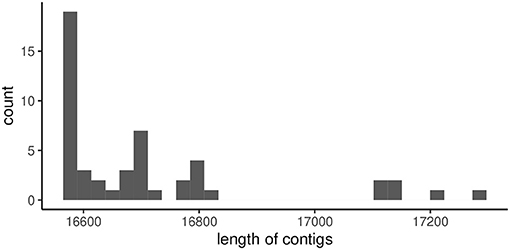
Figure 7. The length of longer contigs generated by Unicycler for random read data generated from MF925724 reference.
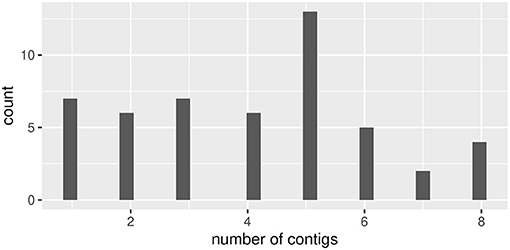
Figure 8. The number of short contigs generated by Unicycler for random read data generated from MF925724 reference.
4. Discussion
Reconstructed contigs contained only a slightly incomplete reference genome of phiX174. Assembly programs were good at assembling the length of 5.4 ks bases but merely failed to glue the final contigs' edges. For making a ring from the resultant linear contig, software inserted extra bases (the left panel of Figure 4). Interestingly, there is not much freedom for the gluing positions for the software that reconstructed monomer contigs. Contrarily, defect locations were scattered among the genome for those generating polymer contigs (Figure 4). But the occurrences of the defective regions were not proportional to the size of genes (Table 2).
Unicycler almost correctly reconstructed the reference. It is not so surprising because this assembly software is specially developed for a circular genome. Interestingly, its backend software, SPAdes, could not achieve similar performance. It is also interesting that SPAdes returned a unique answer for all data set, but Unicycler returned variable solutions. I am not sure the monomorphic behavior of SPAdes is caused by the fact that its random seed is fixed to 42, an enigmatic number (e.g., Adams, 1980), for random seeds at several places in its source codes. SPAdes, IDBA, megahit A5miseq, velvet, and Platanus returned quite a similar sequence to the reference. Defective locations were highly concentrated: genes H, A, and G for the programs. Velvet specifically had defects at gene C. These defect specificity does not seem to be related to assembly methods nor random seed specification (Table 1).
Minia and Trinity returned polymer contigs. Interestingly, the two programs' contigs included two and one complete copy of the reference with an incomplete one. Those copies were concatenated linearly. The enlargement of the contigs occurred by a quite different mechanism from what happened in the mitochondrial genome of A. obtectus (Sayadi et al., 2017; Yao et al., 2017); no regions like the repetitive spacer sequences were detected in contigs for Minia and Trinity (Figure 5). The mtDNA structure rather resembles that of monomer contigs, but the defects of the latter were much shorter than LIGSs in the mtDNAs.
Unicycler fairly succeeded in reconstructing the mtDNA of A. obtectus for Yao et al. (2017). The success rates dropped only 12% for the reference with the threefold reference. But the longer mtDNA of A. obtectus proposed by Sayadi et al. (2017) could not be reconstructed correctly with Unicycler. Resultant contigs could not converge to a single contig. Moreover, the longest contigs were much longer than the reference of Yao et al. (2017). Including LIGS added different features to the reference genome even though the LIGS are entirely separated from the known gene regions. The skewness of Figure 7 to the shorter contigs (the left direction) may indicate a bias for estimating shorter contigs, also criticized in Sayadi et al. (2017).
Evidential statistics uses the evidence functions (e.g., likelihood ratios and consistent information criteria) to quantify the strength of evidence in the data (Royall, 1997; Lele, 2004). If each reconstructed contigs plays the role of a model in a statistical sense, distances among contigs or those from the reference can be a statistical loss function (Lindsay, 2004). If we take the linear or circular DNA/RNA sequences as models, the Levenshtein edit distance can be an appropriate distance measurement among the models and references. The consensus sequence or the centroid in the edit distance for contigs generated by assembly programs is a statistical representation of the specific assembly methods. The consensus sequence of Unicycler coincided with the reference for the short mtDNA of A. obtectus (KX825864) as well as phiX174.
Other than Unicycler, reconstructed contigs created by the assembly programs showed a variation in distance from the reference. How can this variation be evaluated? The behavior of consensus sequences may propose two contrasting understandings. One is simply the bias of the assembly programs or incorrectness of the reference itself. This possibility is convincing from the biased defective locations (Figure 6). The other interpretation might arise if we take the consensus sequence as a tentative true model. Figure 9 represents plots for the reference and consensus contigs in the reconstructed NMDS space. As we expected, the reference and the consensus contig of Unicycler have their position close to the origin (0,0).
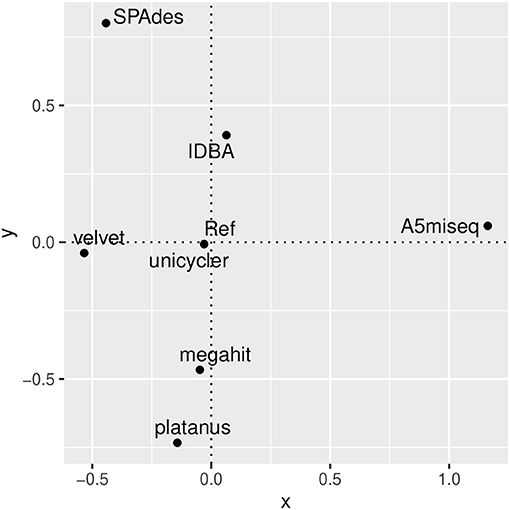
Figure 9. Non-metric multidimensional scaling (NMDS) plot in the edit distance space of the reference and monomer consensus contigs reconstructed with the seven assembly programs using the mds function of smacof library. Note that Unicycler was located at the same place with the reference (Ref).
These results may suggest a way of finding the true reference sequence when you assembly a novel but none-fragmented genome. Analyzing with multiple assembly programs, you should reconstruct consensus contigs of each assembly program. Then you plot the consensus contigs in a reliable NMDS plot. If there is no consensus contig near the origin, create candidate sequences by taking centroids of those consensus contigs until you obtain a sequence sufficiently close to the origin. But the reference (and that of Unicycler) was not located precisely at the origin in the NMDS plot. Because the reference is another sample from the true model, the origin indicated by the NMDS methods might show the true reference locations. Once we accept the consensus contigs' variation in the bootstrapping of a reference, we will need an alternative representation of the references.
Conventionally, a reference sequence has been represented as a single base string. We should express the uncertainty of the reference for incorporating the variable parts in the reference, as we see in our bootstrapping results. Although some researchers claim a necessity for being aware of the stochastic aspects at each base locus (e.g., O'Rawe et al., 2015), assembly algorithms incorporating stochastic locus have never been proposed. A regular expression using the IUPAC nucleotide code (e.g., Paris and Després, 2012) might be an alternative way to express the stochasticity of sequence loci. The flexibility of each locus is expressed with one of the 15 patterns in the IUPAC code. On the contrary, the flexibility of length is characterized by rules of regular expression. The major drawback of using such regular expressions is the lack of standard measurements of distances against given sequences or regular expressions. It might be a little bit rude to apply Levenshtein edit distance against those regular expressions. Approximate regular expression matching is proposed at most (Belazzougui and Raffinot, 2013).
We proposed an evidential statistics approach consists of three steps; bootstrapping a non-fragmented base sequence, reconstructing consensus sequence from the assembled ones, and plotting the consensus sequences with NMDS. In this new approach, we still obey the one-base-for-one-locus rule. Those consensus sequences are centroids of bootstrapped references and can be taken as approximations of the regular expression with the IUPAC nucleotide code. They might be more evidential for a given read data set than a legacy reference obtained with hopefully reliable but an old sequence method. The proposed method relies on a strong assumption in which we have already got non-fragmented sequences for inferring the true reference. But once you could obtain non-fragmented sequences, and if we can improve the analyzing method using R diffobj libraries more efficiently, our method can be applicable for much longer genomes than that of phiX174.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/_001422; https://www.ncbi.nlm.nih.gov/genbank/KX825864; https://www.ncbi.nlm.nih.gov/genbank/MF925724.
Author Contributions
YT designed this project and made R and ruby scripts for assembling and analyzing contigs and wrote the manuscript. TG created gpuga library and R and python scripts with which he reconstructed consensus sequences for simulated contigs. All authors contributed to the article and approved the submitted version.
Funding
This study was partly supported by JSPS KAKENHI Grant Numbers 17H04612 and 18K06410.
Conflict of Interest
TG is employed by Alphadrive Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Drs. M.L. Taper and J.M. Ponciano for giving us to contribute a paper for this feature on evidential statistics. We thank Dr. T. Tanaka for the valuable discussion on the current status of the assembly of read data sets generated with NGS. We thank Hikaru Kuwabara, who did preliminary experiments assuming the phiX174 reference was linear. We are grateful to members of the TOQUE lab. at Univ. Tsukuba for their debate and helpful ideas at the early stage of our manuscript. Finally, we thank Dr. H. Yasue for providing us the information on assembly failures of some read data sets.
References
Akaike, H., Amari, S., Kabashima, Y., Kitagawa, G., and Shimodaira, H. (2007). Akaike Information Criterion AIC: Modeling, Prediction and Knowledge Discovery (in Japanese). Tokyo: Kyoritsu.
Alosaimi, S., Bandiang, A., van Biljon, N., Awany, D., Thami, P. K., Tchamga, M. S., et al. (2020). A broad survey of DNA sequence data simulation tools. Brief. Func. Genom. 19, 49–59. doi: 10.1093/bfgp/elz033
Ballouz, S., Dobin, A., and Gillis, J. A. (2019). Is it time to change the reference genome? Genome Biol. 20, 1–9. doi: 10.1186/s13059-019-1774-4
Belazzougui, D., and Raffinot, M. (2013). Approximate regular expression matching with multi-strings. J. Discr. Algorith. 18, 14–21. doi: 10.1016/j.jda.2012.07.008
Boisvert, S., Laviolette, F., and Corbeil, J. (2010). Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comp. Biol. 17, 1519–1533. doi: 10.1089/cmb.2009.0238
Burnham, K. P., and Anderson, D. R. (1998). Model Selection and Multimodel Inference: A Practical Infromation-Theoretic Approach. New York, NY: Springer. doi: 10.1007/978-1-4757-2917-7_3
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Chevreux, B., Wetter, T., and Suhai, S. (1999). Genome sequence assembly using trace signals and additional sequence information, in Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB), Vol. 99 (Leipzig), 45–56.
Coli, D., Jospin, G., and Darling, A. E. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from illumina miseq data. Bioinfomatics 31, 587–589. doi: 10.1093/bioinformatics/btu661
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Huang, W., Li, L., Myers, J., and Marth, G. (2011). Art: a next-generation sequencing read simulator. Bioinformatics 28, 593–594. doi: 10.1093/bioinformatics/btr708
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Lele, S. R. (2004). Evidence functions and the optimality of the law of likelihood, in The Nature of Scientific Evidence, eds M. L. Taper and S. R. Lele (Chicago, IL: Chicago Press), 191–216. doi: 10.7208/chicago/9780226789583.003.0007
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). Megahit: an ultra-fast single-node solution for large and com- plex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Lindsay, B. G. (2004). Statistical distances as loss functions in assessing model adequacy, in The Nature of Scientific Evidence, eds M. L. Taper and S. R. Lele (Chicago, IL: Chicago Press), 439–487. doi: 10.7208/chicago/9780226789583.003.0014
McElroy, K. E., Luciani, F., and Thomas, T. (2012). Gemsim: general, error-model based simulator of next-generation sequencing data. BMC Genomics 13:74. doi: 10.1186/1471-2164-13-74
O'Rawe, J. A., Ferson, S., and Lyon, G. J. (2015). Accounting for uncertainty in DNA sequencing data. Trends in Genet. 31, 61–66. doi: 10.1016/j.tig.2014.12.002
Paris, M., and Després, L. (2012). Data production and analysis in population genomics. Methods in molecular biology, in In Silico Fingerprinting (ISIF): A User-Friendly In Silico AFLP Program, eds F. Pompanon and A. Bonin (New York, NY: Springer), 55–64. doi: 10.1007/978-1-61779-870-2_4
Ponciano, J. M., and Taper, M. L. (2019). Model projections in model space: a geometric interpretation of the AIC allows estimating the distance between truth and approximating models. Front. Ecol. Evol. 7:413. doi: 10.3389/fevo.2019.00413
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Salzberg, S. L., Phillippy, A. M., Zimin, A., Puiu, D., Magoc, T., Koren, S., et al. (2012). Gage: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 22, 557–567. doi: 10.1101/gr.131383.111
Sayadi, A., Immonen, E., Tellgren-Roth, C., and Arqvist, G. (2017). The evolution of dark matter in the mitogenome of seed beetles. Genome Biol. Evol. 9, 2697–2706. doi: 10.1093/gbe/evx205
Sohn, J.-I., and Nam, J.-W. (2018). The present and future of de novo whole-genome assembly. Brief. Bioinformatics 19, 23–40. doi: 10.1093/bib/bbw096
Sung, W.-K. (2017). Algorithms for Next-Generation Sequencing. Boca Raton, FL: CRC Press. doi: 10.1201/9781315374352
Taguchi, Y., and Oono, Y. (2005). Relational patterns of gene expression via non-metric multidimensional scaling analysis. Bioinformatics 21, 730–740. doi: 10.1093/bioinformatics/bti067
Wright, E. (2016). Using decipher v2.0 to analyze big biological sequence data in r. R J. 8, 352–359. doi: 10.32614/RJ-2016-025
Keywords: NGS, PhiX174, consensus sequences, bootstrapping, nonmetric multidimensional scaling
Citation: Toquenaga Y and Gagné T (2021) The Evidential Statistics of Genetic Assembly: Bootstrapping a Reference Sequence. Front. Ecol. Evol. 9:614374. doi: 10.3389/fevo.2021.614374
Received: 06 October 2020; Accepted: 01 July 2021;
Published: 26 July 2021.
Edited by:
Dapeng Wang, University of Oxford, United KingdomReviewed by:
Xiaofan Zhou, South China Agricultural University, ChinaRayan Chikhi, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2021 Toquenaga and Gagné. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukihiko Toquenaga, toque@biol.tsukuba.ac.jp
†Present address: Takuya Gagne, Alphadrive Co., Ltd., Tokyo, Japan