
Abstract
Fusiform rust disease, caused by the endemic fungus Cronartium quercuum f. sp. fusiforme, is the most damaging disease affecting economically important pine species in the southeast United States. Unlike the major epidemics of agricultural crops, the co-evolved pine-rust pathosystem is characterized by steady-state dynamics and high levels of genetic diversity within environments. This poses a unique challenge and opportunity for the deployment of large-effect resistance genes. We used trait dissection to study the genetic architecture of disease resistance in two P. taeda parents that showed high resistance across multiple environments. Two mapping populations (full-sib families), each with ~1000 progeny, were challenged with a complex inoculum consisting of 150 pathogen isolates. High-density linkage mapping revealed three major-effect QTL distributed on two linkage groups. All three QTL were validated using a population of 2057 cloned pine genotypes in a 6-year-old multi-environmental field trial. As a complement to the QTL mapping approach, bulked segregant RNAseq analysis revealed a small number of candidate nucleotide binding leucine-rich repeat genes harboring SNP associated with disease resistance. The results of this study show that in P. taeda, a small number of major QTL can provide effective resistance against genetically diverse mixtures of an endemic pathogen. These QTL vary in their impact on disease liability and exhibit additivity in combination.
Similar content being viewed by others

Introduction
Endemic pathosystems of plant disease are characterized by steady-state dynamics between host and pathogen populations, in which negative frequency-dependent selection maintains genetic polymorphism and ensures that neither player gains the upper hand (Ennos 2015; Tellier and Brown 2007). This contrasts with epidemic pathogens that cause catastrophic local extinction events and large fluctuations of population size in both the host and the pathogen. In agricultural plant breeding, much attention has been focused on epidemic pathogens and the identification of specific host resistance genes that confer resistance against the predominant race of the pathogen in the local production environment (Saintenac et al. 2013; Vossen et al. 2003). The traditional experimental approach for identifying these gene-for-gene interactions is geared toward epidemic crop pathogens since it involves isolating a pure strain of the predominant pathotype from the production environment and testing it for virulence against a panel of known host resistance genes (Keen 1990).
In endemic pathogens of forest trees, millions of years of coevolution result in a large degree of genetic variability, both in the host and the pathogen (Thompson and Burdon 1992). Studies of endemic fungal pathogens of forest trees have indicated little or no discernible population structure, making it difficult to adequately sample wild pathogen populations (Hamelin et al. 1994; Mercière et al. 2017). In these pathosystems, the host population may adapt to the pathogen through increased tolerance to the disease, reducing the fitness benefit of qualitative resistance genes (Roy and Kirchner 2000). Nonetheless, effective qualitative resistance is highly sought after in production forestry, since endemic diseases result in significant losses of merchantable timber and other forest products through mortality or decreased wood quality (Mercière et al. 2017; Anderson et al. 1986; Powers et al. 1974). Gene-for-gene interactions discovered in laboratory settings using single-strain inoculation experiments should be interpreted within the context of endemic forest pathogens rather than from the agricultural paradigm, since there is little chance that the single-race isolate will be present in any given environment. New population-level approaches for identifying gene-for-gene interactions in these pathosystems should be focused on effectors rather than pathotypes, since in the absence of reproducible pathotypes, it is the allele frequency of the cognate effector that defines the efficacy of the host resistance gene.
The pine-rust pathosystem is an ideal platform for studying genetic disease resistance against co-evolved endemic pathogens. Pinus taeda L. (loblolly pine) is the most widely planted forest tree species in the world, established on over 13 million hectares in the southern United States for fiber, biomass, and wood production (Prestemon and Abt 2002). The species is used extensively in Africa, Asia, and South America (Schultz 1999). Fusiform rust disease, caused by the endemic fungus Cronartium quercuum (Berk.) Miyabe ex. Shirai f. sp. fusiforme (abbreviated Cqf hereafter), is the most important disease affecting pine species such as P. taeda in the southern United States (Kubisiak et al. 2005; Wilcox et al. 1996). Annual losses from fusiform rust disease range between $40 and $100M (Anderson et al. 1986; Cubbage et al. 2000; Powers et al. 1974). The host-parasite interaction between Cqf and various species of pine dates back as early as the Jurassic period, preceding the split of the Laurasia supercontinent (Millar and Kinloch 1991; Wilcox et al. 1996). Monokaryotic (1N) basidiospores are released from telia on the underside of oak leaves in the early spring and are wind-dispersed to nearby pines. Infection of young pine trees by Cqf occurs through cotyledons, needles, and soft stem tissue. It results in the formation of spindle-shaped woody galls (Supplementary Fig. 1), which reduces the structural integrity of the wood. Galls formed on the main stem in the first 5 years of the tree’s life cycle often result in mortality from stem girdling or wind breakage, but galls formed on branches are usually tolerated. Dikaryotic (N + N) aeciospores are released from the galls in the early spring and are wind-dispersed to nearby oaks where they can germinate on the underside of oak leaves to complete the life cycle (Czabator 1971). Due to the large geographic deployment area and long-lived nature of P. taeda, durable genetic resistance to fusiform rust is one of the most important breeding objectives for the species (Isik and McKeand 2019).
Studies reporting genomic mapping of resistance loci in P. taeda date back to 1996, and in all cases, the objective was identifying resistance genes that confer resistance to single-race isolates of Cqf (Amerson et al. 2015; Isik et al. 2012; Wilcox et al. 1996). In the first mapping study in which the Fr1 resistance QTL was identified, two single-race isolates were used in a controlled inoculation experiment. Using a genomic map consisting of several hundred RAPD markers, a QTL was identified for resistance against one isolate but not the other, leading the authors to conclude there was contrasting virulence between the isolates against the Fr1 gene (Wilcox et al. 1996). The mapping of the other eight named resistance loci, Fr2 through Fr9 was described in Amerson et al. (2015). In these experiments, seven different pine families were tested with a set of five single-gall isolates in controlled inoculations, resulting in the naming of an additional eight resistance loci, most of which were clustered on a single linkage group (LG) (Amerson et al. 2015). In the case of Fr1, the resistance QTL was only observed using specific rust isolates, and the localization of the gene was approximate since the genetic map only consisted of several hundred RAPD markers to cover a genome greater than 22 Gb in size (Neale et al. 2014). These factors, coupled with the expensive and time-consuming RAPD genotyping procedure, made it difficult to incorporate Fr1 and the other Fr QTL as targets for marker-assisted selection.
The fitness benefit of pathotype-specific disease resistance genes is a function of the genetic diversity of the pathogen. Wild populations of Cqf are characterized by low LD, high allelic diversity, and extensive variation in genome size. Several studies found that the genetic diversity of Cqf within local regions was equivalent to the diversity among regions (Anderson et al. 1986; Burdine et al. 2007; Kubisiak et al. 2005). The lack of evidence for clear population structure, together with the evidence for differential avirulence profiles among the handful of Cqf isolates tested (Amerson et al. 2015; Wilcox et al. 1996), suggests a multitude of avirulence alleles may be present in wild populations. This contrasts with the major fungal pathogens of agricultural crops such as Puccinia graminis or Magnaporthe oryzae, which are characterized by a relatively small number of predominant races that affect large production areas (Fang et al. 2017; Saintenac et al. 2013).
A significant gap exists in our knowledge of how genetic resistance functions in co-evolved forest pathosystems. On the one hand, genetic resistance in forest trees at the population level has long been explained by a polygenic model of host resistance in which the inheritance of disease resistance is controlled by many alleles throughout the genome, each with a small effect (Isik et al. 2012). Alternatively, some examples of the gene-for-gene interaction paradigm have been demonstrated in tree species, resulting in a proposed model in which the host may have multiple pathotype-specific resistance genes corresponding to different pathogen avirulence alleles (Devey et al. 1995; Wilcox et al. 1996). In this study, we use a bulked inoculum strategy to identify race-nonspecific broad-spectrum resistance (RNS-BSR) QTL (Li et al. 2020). A small number of RNS-BSR resistance genes have been identified in agricultural crops (Deng et al. 2017; Wang et al. 2015), but have not yet been identified in any forest tree species. We propose that major host resistance QTL harboring one to many single genes may confer effective immunity against a diverse group of Cqf isolates, provided the allele frequency of its cognate effector is high in the fungal population. Other resistance QTL may be more specific since their cognate effectors are at lower frequency in the pathogen population. By averaging over the genetic variability of the pathogen, QTL that confer RNS-BSR relevant to production forestry should immediately become apparent. This perspective on the allele frequency of the effector is lost in single-strain inoculation experiments. Such experiments may be successful in identifying gene-for-gene interactions but yield little information about the frequency of these gene-for-gene interactions in real environments.
The primary question we addressed in this study relates to the genetic architecture of RNS-BSR in a conifer species, P. taeda. Can environmentally stable, high-level disease resistance measured in the progeny of a P. taeda parent be explained by the segregation of single large-effect RNS-BSR QTL? Secondly, are RNS-BSR QTL that are discovered in controlled inoculation experiments effective and durable in real forest environments? Finally, if there are expressed genes associated with RNS-BSR, are they similar to other known disease resistance genes? In order to discover associations with RNS-BSR in the transcriptome, we used a modified version of bulked segregant RNASeq (Liu et al. 2012) to interrogate allelic variation in the gene space for associations with the disease phenotype.
Materials and methods
QTL discovery populations
Since the goal of this work was mapping RNS-BSR QTL, parents 4 and 9 were selected as resistance donors due to their superior rust resistance breeding values and high rank stability across multiple environments. These parents were compared to the parents used in the mapping of the named Fr genes using analysis of multi-environmental progeny trials in the southern United States (Supplementary Table 1). From these results, it was clear that the disease resistance of the parents used in the original mapping experiments, including the parent 10-5 which is known to segregate for the Fr1 resistance gene (Wilcox et al. 1996), was mediocre or below average. The one exception to this is parent A, which was identified as a heterozygote for the Fr2 resistance gene (Amerson et al. 2015); parent A was ranked in the top 50, but significantly below parents 4 and 9. Parent A has no known pedigree relationships with parents 4 or 9.
Two full-sib families were produced by crossing parents 4 and 9 to rust-susceptible parents 202 and 313, respectively (Supplementary Fig. 3). In both crosses, the rust-resistant parents 4 and 9 were used as a male. About 1500 seeds from each cross were harvested 18 months after the mating. Seeds were sown at the USDA Forest Service Resistance Screening Center in Asheville, NC, in May of 2018 for artificial inoculation (see details in “Artificial inoculation of QTL discovery population” section). In August 2018, about 1000 full-sib seedlings from each family were individually labeled and sampled for DNA and RNA extraction. These full-sib families will hereafter be referred to as E4 and E9.
QTL validation population
A clonally propagated population was used to validate the field-level efficacy of the QTL. This population was established in the early 2000s, and is described elsewhere (Shalizi and Isik 2019). Briefly, the Atlantic Coastal Elite population consisted of three disconnected eight-parent diallels among 24 parents, which resulted in 76 full-sib families. Each full-sib family was challenged with a bulked Cqf inoculum at the USDA Forest Service Resistance Screening Center in Asheville, NC. The spore density used in the assay was 50,000 spores/ml. From the original 76 full-sib families, 23 highly susceptible families were removed from the population. About 46 rust-gall-free progeny from the remaining 53 full-sib families were clonally propagated via rooted cutting techniques and planted across eight test environments in the southeastern United States (Shalizi and Isik 2019). The QTL validation population was scored 6 years after test establishment for tree height, stem diameter, stem form, and the presence/absence of fusiform rust disease. The disease incidence recorded at 6 years in the field (eight locations) represents random wild environmental inoculum.
The QTL discovery populations (full-sib families E4 and E9) shared extensive pedigree connectivity with the QTL validation population described above (Supplementary Fig. 3). Parent 4 shared two grandparents with a total of 406 clones, and shared one grandparent with a total of 556 clones. Parent 9 had 221 offspring clones in the validation population. These offspring were distributed among six nested full-sib families.
Artificial inoculation of QTL discovery populations
Collection and preparation of the fungal inoculum were performed by USDA Forest Service Resistance Screening Center personnel using standard in-house protocols (K. McKeever, personal communication). The Cqf inoculum used in the study was composed of a mixture of 150 different single-gall isolates. These isolates were collected from five regions in the southeast United States known to have the highest fusiform rust incidence. Within each region, three collections were made, with each collection site being located at least 16 km away from any other collection site. Within each collection site, aeciospores from ten individual galls on separate trees were collected. Each sample was tested for spore viability using germination tests, and samples with 15% or less germination were discarded. Viable aeciospore samples from each collection site were then mixed in equal volumetric proportions to produce a single collection, which was then put through a stepwise process of desiccations and packaged under vacuum in glass vials. In order to produce the basidiospores used in the inoculation, desiccated aeciospores from three collections per region were resuspended in sterile water at a concentration of 1.2 × 10−3 g/ml, for a total of 15 aeciospore resuspensions representing the five regions. These resuspensions were mixed in equal proportions and used to inoculate the underside of oak leaves. Once telia appeared on the undersides of the leaves (2–3 weeks after inoculation), the leaves were clipped and suspended over acidified water (pH = 2) in order to release the basidiospores. The acidified water was filtered through a Millipore system, and the basidiospores were captured on filter paper. These spores were then resuspended in sterile water at a concentration of 100,000 basidiospores/ml. Empirical dose-response studies found the concentration of 100,000 basidiospores/ml minimized the rate of escapes, or known susceptible seedlings not developing disease due to a lack of pathogen challenge, to 5% or below (Kuhlman et al. 1997).
Artificial fungal inoculations were performed at the USDA Forest Service Resistance Screening Center in Asheville, NC using standard in-house protocols. Briefly, spore suspensions were sprayed over 3-month-old seedlings using the concentrated basidiospore inoculation system of Mathews and Rowan (1972) at a concentration of 100,000 basidiospores/ml. Seven months after inoculation, the seedlings were observed for the presence or absence of galls. Disease incidence was recorded as a binary response variable for individually labeled full-sib seedlings. The seedlings were then moved to greenhouse facilities at the Horticultural Field Lab at North Carolina State University, Raleigh, USA, for continued observation. A second observation was made on the population 10 months after inoculation to confirm the original disease incidence data and to ensure that disease symptoms that may not have been apparent at 7 months were accurately recorded.
Marker genotyping and SNP filtering
In the QTL discovery population, genomic DNA was isolated from needle tissue of seedling progeny from the two full-sib families, as well as two replicates of each of the four parents used in the crosses. The samples were genotyped with the Pita50K Axiom array developed for P. taeda in October 2019 (Caballero et al., In Review). In total, 920 full-sib progeny from family E9 and 1071 full-sib progeny from family E4 were genotyped. In-house scripts were used to ascertain the parental genotypes and to compare observed with expected progeny genotype ratios. The criteria for SNP inclusion in the genetic map were threefold: (1) the SNP genotype must be identical for both replicates of the same parent, (2) the SNP had to be heterozygous in one or both parents, and (3) the progeny genotype ratios must not differ significantly from 1:1 (segregation distortion). Following SNP filtering, 6019 SNP were retained for linkage mapping in family E9, and 8552 SNP were retained in family E4 (Supplementary Table 2). All SNP filtering was performed in the R programming environment using customized in-house scripts.
In the clonal validation population (clonal field trials), 2057 clonal varieties were successfully assayed with the Pita50K Axiom array. The SNP for these clones were filtered to include only those SNP that had a minor allele frequency >0.01 and were placed on the linkage map generated from the QTL discovery populations. This resulted in a total of 10,040 SNPs used in GWAS of the validation population.
RNA sample collection
Prior to the inoculation, two random samples of 100 seedlings of each family were taken by removing a single needle from each seedling and immediately placing the tissue on dry ice, followed by storage in a −80 °C freezer. This sample was taken to provide a random sample of the population prior to the disease challenge, which was used to construct the PacBio reference transcriptome and to estimate allele frequencies from read count data. Seven months after inoculation, three samples of diseased individuals and three samples of non-diseased individuals were taken from each family (50 seedlings per sample or 150 seedlings in each category). A single needle from each seedling was collected and immediately placed on dry ice, followed by storage in a −80 °C freezer. At the 10-month disease assessment, one additional diseased bulk of 50 individuals and one additional non-diseased bulk of 50 individuals were taken from both families, using the same procedure outlined above. RNA from all samples was extracted simultaneously, using conventional methods via the Qiagen RNEasy MiniPrep kit.
Linkage maps, QTL analysis, and model fitting
Linkage maps were produced using the two-way pseudo-testcross design for both families. Briefly, prior to linkage mapping, the genotype of each parent at each marker was ascertained. For each cross, a linkage map was produced using markers in either backcross configuration (AB:BB or BB:AB), resulting in separate maps for the maternal and paternal genomes. Markers in the intercross configuration (AB:AB) were used to generate the sex averaged map for each LG, but these were dropped from the dataset prior to QTL analysis since the linkage phase of heterozygous genotypes for which both parents are heterozygous is unknown in an outbred F1 cross. The consensus map combining the linkage maps from all four parents was generated through linear programming methods. Details of these procedures are presented in the Supplementary Materials.
All QTL analysis was conducted using the R package R/QTL (Broman and Sen 2009). Interval mapping was conducted via the Expectation Maximization algorithm, using a logistic regression model implemented in the scanone function of R/QTL. LOD thresholds for declaring QTL were determined using 1000 permutations of the phenotypic data. Peaks that surpassed the LOD threshold were declared as putative QTL, and their genomic positions under a conditional model (with more than one QTL) were determined using the refineqtl function. For identified QTL, genotype probabilities for each individual were estimated by calc.genoprob. Individuals were assigned to two genotype classes (Rr, rr) in family E9, and assigned to four genotype classes (rr/rr, Rr/rr, rr/Rr, Rr/Rr) in family E4. These genotype classes were used as categorical variables in generalized linear models. The amount of variance explained by each QTL was estimated using the fitqtl function in R/QTL, which utilizes a simple transformation of the conditional LOD score. Additive effects for QTL haplotypes and specific contrasts were estimated using the GLIMMIX procedure of SAS/STAT software v9.1 (SAS Institute, Cary NC 2013). In these models, the susceptible haplotype (either one- or two-QTL susceptible haplotype) was declared as the reference level.
Modeling the effect of QTL haplotypes
Disease occurrence was recorded as a binary variable; yi = 1 if the seedling progeny had a gall on the stem, and yi = 0 if the progeny was gall-free. The disease outcome is a realization of the random variable Yi with the probability of π for 1 outcome and the probability 1 − π for 0 outcome. The response variable Yi is distributed according to a Bernoulli distribution with the expectations E(Yi) = μi = πi and Var(Yi) = σ2 = nπi(1 − πi) (Collett 2002). We modeled the effect of each QTL haplotype (the odds of disease outcome) using the following logistic regression model:
Where Yij is the log-odds of the ith seedling in the jth family being 1, β0j represents the fixed intercept or the mean disease incidence for family j, βn is the logit-scaled regression parameter (fixed effect) for the average effect of the nth QTL haplotype, xn is an indicator variable taking on values of 0 or 1 for 0 or 1 alleles of the nth QTL haplotype. The odds ratios and the least square means (probability scale) were calculated using the lsmeans option of SAS GLIMMIX procedure (SAS Institute Inc. 2019). The number of haplotype effects in the model was 2q where q was the number of QTL discovered in the family. The design matrix for the haplotype effects was estimated from the QTL probabilities in the output of calc.genoprob in R/QTL. For each individual, the probability of both QTL genotypes at each QTL was estimated from local marker data in the QTL regions using calc.genoprob, and the multi-locus haplotype with the most support from the data (the highest probability) was declared as the individual’s putative haplotype. Interaction terms were explored in the models, but they did not improve the model fit in either family and were dropped from the final analysis.
Bulked segregant RNASeq analysis
Contingency tables were used to identify polymorphisms associated with disease. Variants called against the PacBio assembly for each family were analyzed separately using a customized R pipeline. For each polymorphism, a 2 × 2 matrix consisting of the summation of the reference allele counts X and alternate allele counts Y across diseased bulks i = 1,…, n and resistant bulks j = 1,…, m was computed, and the log of the count ratio was obtained as follows:
The odds ratio under the null hypothesis was derived by multiplying the allele frequency observed in the random bulks by the total number of reads in each disease category. Because the random bulks were sampled prior to disease challenge, the read counts observed in these samples could not have been influenced by pathogenesis, disease-induced programmed cell death, or any other interaction with or contamination by Cqf. Fisher’s exact test, via the native R function fisher.test, was used for testing the null hypothesis of independence between rows and columns of the matrix. p values were adjusted using Bonferroni correction.
Since this mapping exercise sought to identify variants in tight linkage to dominant resistance genes, any SNP that were fixed in the diseased bulks but polymorphic in the resistant and random bulks were automatically considered potential candidates for fine mapping. In this context, all diseased individuals are expected to be homozygous recessive (rr) at the resistance locus, while non-diseased individuals would either be heterozygous (Rr) for the resistance allele or homozygous recessive (rr), depending on whether the non-diseased individual inherited the resistance allele or was an escape, respectively. In a bulk sample composed of non-diseased or randomly selected individuals, both alleles should be expected at the resistance locus. In a sample of diseased individuals, either a single allele (the susceptible allele) or a high frequency of the susceptible allele would be expected.
Genome-wide association analysis
Experimental-design adjusted predictions of clones were obtained from a generalized linear mixed model and were used in genome-wide association analysis (see details in the Supplementary Materials). The GWAS function within the R package rrBLUP was used as a mixed-model platform for association analysis (Endelman 2019). The following model was used for the association analysis:
With y representing a (2057 × 1) vector of probability-scaled clonal predictions for fusiform rust incidence. X represents a (2057 × 3) design matrix of ones relating the principal component loadings of each clone on the first three principal components of the kinship matrix to the response vector y, and β represents a (6171 × 1) vector of principal component loadings for each of the 2057 clones on the first three principal components of the kinship matrix, Z is the design matrix for the random effects, in this case relating the additive genetic values for the 2057 clones to the response vector y, g is a (2057 × 1) vector of additive genetic values for each clone, S was a (2057 × 10,040) marker design matrix taking on values of −1, 0, 1 for the minor allele homozygote, heterozygote, and major-allele homozygote, respectively, τ was a (10,040 × 1) vector of fixed marker effects, and ε is a (2057 × 1) vector of residual errors. The random genetic background effect had the expectations g~N(0, Kσ2) with K representing the realized genomic relationship matrix calculated from SNP markers using the observed allele frequencies in the validation population (VanRaden 2008). The residual errors were distributed \(\varepsilon \sim N\left( {0,\,\bf{I}\sigma _e^2} \right)\). The loadings in β were determined from an eigenvalue decomposition of K.
RNASeq library preparation, sequencing, variant calling, sequence annotation and phylogenetic analysis of the NLR gene family
Detailed procedures used for RNASeq are described in the Supplementary Materials. Briefly, PacBio ISOSeq libraries were produced from needle tissue of 100 randomly selected seedlings from each family prior to the inoculation in order to produce family-specific reference transcriptomes. After the inoculation, Illumina libraries were prepared from multiple bulked samples of the two disease categories in each family, and the short-read data were aligned against the corresponding family-specific PacBio reference transcriptome. Variants were called using FreeBayes (Garrison and Marth 2012), and the digital read count data from the Illumina data were used in bulked segregant analysis. Details pertaining to sequence annotation and genomic alignment of the PacBio reference transcriptomes, as well as phylogenetic analysis of the NLR transcripts discovered in both families, are presented in Supplementary Materials.
Results
Disease incidence
The first observation of the disease outcome in the QTL discovery populations was taken 7 months postinoculation. There was a large contrast in overall disease incidence between the two families. In family E4, 26% of the seedlings were diseased. In family E9, 71% of the seedlings were diseased. The populations were observed again at 10 months postinoculation. The mean incidence increased slightly to 28% in family E4 but it remained the same in family E9.
Genetic maps
Both families had the expected number of LGs (12) for the maternal and paternal genomes. For family E4, the map consisted of 6835 markers with an average spacing of 0.6 cM and an individual map length of 2024 cM per genome, with a maximum gap of 8.8 cM. The map for family E9 had 4623 markers with an average spacing of 1.5 cM and an individual map distance of 3353 cM, with a maximum gap of 23 cM. The consensus map combined the maps from both families, and consisted of 10,204 markers distributed across 12 LGs, with a total map distance of 3137 cM and an average spacing of 0.31 cM (Supplementary Table 4).
LG assignments from our consensus map for the Pita2.01 reference genome contigs were compared to previously published LG assignments (De La Torre et al. 2019). A total of 7187 contigs were placed on our consensus map, out of which only 2.8% mapped to two or more LGs. A total of 15 contigs were placed on three LGs; notably, all these contigs had the prefix “super” (e.g, “super2789”, “super2905”). Out of these 7187 contigs, 1761 were also placed in the De La Torre et al. (2019) consensus map. Agreement between the two consensus maps was generally high, with an average correlation between the map positions of 0.92 (Supplementary Fig. 2). The increased genetic resolution of our map, due to larger population size, was readily apparent. Large regions lacking any apparent recombination in the De La Torre et al. (2019) map were resolved into unique positions in our map. To make our analyses directly comparable with previously published work, we renamed our LGs to be consistent with the nomenclature in De La Torre et al. (2019), and re-oriented the marker order for those LGs in which the marker order was inverted in our map relative to the order in the De La Torre map.
QTL analysis
In family E4, a double QTL peak was observed on the paternal complement of LG2 (Fig. 1) with a LOD score >20 from the output of one-dimensional QTL scanning via the scanone function of the R/QTL package (Broman et al. 2003). The QTL locations, as well as the overall model likelihood, were improved using the function refineQTL, which utilizes a multi-QTL model to adjust the location and effect estimates for each QTL (Broman et al. 2003).The two-QTL model was a better fit to the data than the one QTL model, resolving the doublet into two single, separate QTL which together explained 31% of the variance (Fig. 1). The first and second QTL each explained 23% of the phenotypic variance. The 95% Bayesian credible intervals for QTL1 and QTL2 were 0–0 and 29.3–29.4 cM, respectively (Table 1 and Fig. 2). The large population size in each family and the large number of markers contributed to narrow confidence intervals for the QTL locations.

Three loci, one on paternal LG7 of family E9 and two on paternal LG2 of family E4 showed large effects on disease outcome. Paternal linkage map is displayed with LG identifiers ending in “.P”. Map positions are given along the x-axis of each subpanel in centimorgans (cM). The inset on the top right shows the improvement of the model likelihood under the two QTL model implemented in refineQTL function of R/QTL compared to the one QTL scanone model for the two peaks on LG2, with LOD values approaching 70 under the two-QTL model. The LOD profile over the maternal linkage map is not shown, since no QTL were discovered on maternal LGs.

Genetic distance (cM) is shown on the ruler to the left of the figure. 1-LOD confidence intervals for QTL are shown in red. Closely linked markers validated for MAS are shown in blue text. Cyan Map of LG2 harboring the QTL discovered in family E4 (GRID1, GRID2). The paternal LG2 map from family E4 is shown to the right of the consensus map of LG2. Blue Map of LG7 harboring the QTL discovered in family E9 (GRID3). The paternal LG7 map from family E9 is shown to the right of the consensus map of LG7.
We modeled the effects of the linked QTL on LG2 on the basis of two-locus haplotypes. QTL haplotype effects (Rr/rr, rr/Rr, Rr/Rr) for disease resistance in family E4 were significantly different from the double-susceptible haplotype (rr/rr) (Table 2). The relative frequencies of the single- and double-resistant haplotypes in the population suggest the resistance alleles at the two QTL are linked in repulsion on LG2 of parent 4. Individuals inheriting the resistance allele at either QTL (Rr/rr or rr/Rr) had a disease probability of 0.19. Individuals with both resistance alleles (Rr/Rr), representing recombinant haplotypes, had a disease probability of 0.007 (Table 2). Only one out of 156 individuals carrying both resistance alleles was diseased. Individuals inheriting neither resistance allele (rr/rr) in family E4 had a disease probability of 0.92. The average effects for the two single-resistant haplotypes were not significantly different from each other. These loci are hereafter termed GRID1 and GRID2 (General Rust Immunity Determinant). One small QTL peak on maternal LG7 was also observed in the results of scanone which marginally surpassed the significance threshold (not shown). However, this peak only explained 0.5% of the variation, so was dropped from the model.
In family E9, a single QTL discovered on LG7 of the paternal genome explained 14% of the variance, with a LOD of 29.8 (Fig. 1). The 95% Bayesian credible interval for the QTL on LG7 extended from 47.1 to 47.4 cM (Table 1 and Fig. 2). This QTL is hereafter referred to as GRID3. The average effect for this QTL was highly significant (Table 2), although not the same magnitude as GRID1 or GRID2. Individuals in family E9 that inherited the resistance allele at this QTL had a disease probability of 0.54, while individuals that did not inherit the resistance allele at this QTL had a disease probability of 0.89 (Table 2).
GWAS validation
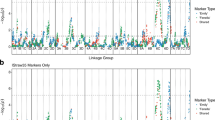
Significant associations were observed on LG2 and LG7 in the same genomic regions as the QTL from the discovery population (Fig. 3A). A single SNP marker (PitaSNP268432) at 184.6 cM on LG7, surpassed the 5% false discovery rate threshold. This marker was not mapped to the paternal complement of LG7 for family E9 in the QTL analysis. A marginally significant SNP marker (PitaSNP092601) located at 189.9 cM had a LOD value of 26.02 in scanone, and was mapped to the paternal complement of LG7 for family E9 (Table 3).

A Genome-wide association analysis results for 2057 clonal genotypes from the multi-environment trial. The −log(p-value) for a false discovery rate of 0.05 is shown as a horizontal red dashed line. Markers are ordered according to genetic position on the consensus map along each LG. B Close-up of the region of LG2 harboring significant associations with rust resistance. The region between 15 and 20 cM contains markers that were mapped to the distal end of LG2 on the paternal linkage map of parent 4 of family E4, in proximity to GRID1. The region around 30 cM contains markers that were mapped in proximity to GRID2 on the paternal linkage map of LG2. C QQ plot showing close agreement with the null hypothesis for the vast majority of markers included in the GWAS, while a small number of SNP in LD with the resistance QTL deviate from the expectation.
On LG2, two regions contained markers significantly associated with rust resistance, the first region around 20 cM, and the second from 28.6 to 33 cM (Fig. 3B). A total of nine SNP loci were significantly associated with resistance on LG2, but only two of these were mapped to the paternal complement of LG2 for family E4 in the QTL analysis. These two SNP loci had LOD values of 15.1 and 21.5 in scanone (Table 3). A close agreement between the expected and observed p values was evident for the vast majority of SNP, with minimal inflation caused by long-range LD or population structure (Fig. 3C).
Principal component decomposition of the kinship matrix revealed four large subclusters in the validation population (Fig. 4). Within each subcluster, a relatively large number of highly resistant and a smaller number of highly susceptible clones was evident, suggesting that disease resistance was not stratified by population structure. This indicates that the parents contributing the rust resistance alleles were used in many crosses that spanned all four large-scale strata within the validation population.

Loadings of each clone on the first two principal components of the kinship matrix revealed four large subpopulation clusters. Within each cluster, both highly resistant (incidence ~0) and highly susceptible (incidence >0.25) clones were identified. This was likely caused by the diallel mating design, in which the parents contributing the resistance genes were used in multiple crosses that spanned all large-scale population strata. The average proportion of “susceptible” clones in each full-sib family was 0.24. Probability-scaled rust incidences are presented in the third row and the third column to show how a similar ratio of resistant to susceptible clones was observed in each cluster.
Bulked segregant RNASeq
The average sequence coverage of the PacBio reference transcriptomes was around 163x for both families (Supplementary Materials). This amount of sequence coverage was associated with >98% agreement between SNP identified in RNAseq and whole-genome-sequence datasets in humans (Piskol et al. 2013). In the transcriptome SNP dataset for family E4, ten out of 71,848 SNP exhibited complete separation on the basis of disease status, and had odds ratios of infinity (Fig. 5A). These SNP were fixed in the diseased bulks but polymorphic in the resistant and random bulks, and had an intermediate minor allele frequency in the random sample (Fig. 5/B). Three out of ten of these SNP were discovered within NLR genes (Table 4).

A Distribution of LOD values for variants discovered in RNAseq dataset from family E4. Cutoff line for SNP candidacy is shown as a dashed red line. Only 11 SNP surpassed this threshold in family E4. B Example of read count data for a SNP with a LOD value of ~2.9 (left panel) and a LOD value of infinity (right panel). C Amino acid sequence alignment in the NB-ARC domain of the three candidate genes from family E4 (tx_69, tx_8565, tx_12122) and the one candidate gene from family E9 (tx_5752).
In family E9, 65,849 variants were available for analysis out of which 11 SNP had odds ratios of infinity, showing a complete separation based on disease status. One of these was found within an NLR gene, while another was found within a partial NLR gene that only had a leucine-rich repeat domain (Table 4).
Phylogenetic analysis of NLR genes
The transcriptome datasets for family E4 and E9 contained a total of 231 and 288 NLR gene sequences, respectively. To determine the relationship between the candidate RNS-BSR genes and the rest of the NLR gene complement, the highly conserved NB-ARC domain from each protein was used in a multiple sequence alignment. Two of the candidate genes discovered in family E4, tx_69 and tx_8565, were highly homologous in this domain and clustered together within a small clade containing one other protein, transcript tx_5551 (Fig. 5C and Supplementary Fig. 4). All of the proteins within this clade were discovered in family E4. The third candidate gene in E4, transcript tx_12122, did not share significant sequence homology with the other two candidate genes in family E4.
Sequence alignment and characterization
The alignments of the candidate genes against the Pita2.01 reference genome revealed differences in the NLR gene complement between the two families. Some isoforms (including tx_69 and tx_8565) were detected in one family but not the other, and a large degree of variation in gene structure was observed. A description of the candidate NLR genes discovered in the PacBio reference transcriptomes is provided in the Supplementary Materials. The alignment between the PacBio contigs discovered in family E4 harboring SNP significantly associated with rust resistance against the Pita2.01 reference genome is shown in Supplementary Fig. 5.
Discussions
We present the first evidence for putative RNS-BSR QTL in a conifer genome. Some evidence for efficacy of the previously identified Fr loci against bulked inocula was obtained (Isik et al. 2012), but the genetic map used to localize the Fr loci was based on 311 markers and fewer than 100 individuals in each cross (Amerson et al. 2015; Echt et al. 2011). This genetic resolution is not high enough to rule out the existence of multiple linked resistance QTL, and makes the determination of the number of genes segregating in each family very difficult. Our demonstration of one-to-many relationships between host resistance QTL and fungal isolates is supported by high mapping resolution in the QTL discovery populations, high-depth coverage of the expressed gene content, and independent validation in a clonally replicated field trial. Moreover, the field-level efficacy of the QTL mapped here are much higher than the previously named Fr resistance genes (Supplementary Table 1); presumably, this is because the resistance QTL segregating in E4 and E9 may have less specific resistance spectra, or recognize effectors that are common in the pathogen population. Importantly, both of the LGs harboring rust resistance QTL have been associated with resistance in previous studies. LG2 was previously associated with rust resistance QTL; it is the same LG believed to harbor six Fr loci (Amerson et al. 2015). However, since parent 4 has no known pedigree relationships with any of the other parents used in Amerson et al. (2015) study, and since parent 4 shows significantly superior field-level rust resistance to the parents used in the Amerson study (Supplementary Table 1), we infer that the QTL segregating in family E4 are distinct from the those identified in previous studies. LG7, harboring GRID3 in this study, was previously associated with a rust resistance marker in a GWAS (Cumbie et al. 2020); however, the predicted map position of GRID3 is ~100 cM away from the predicted map position of SNP2374 reported in Cumbie et al. (2020) study.
This discovery may help to answer long-standing questions about the efficacy of qualitative resistance in complex forest pathosystems. Resistance QTL may be variable in terms of their resistance spectra and exhibit additivity with respect to their impacts on disease liability. The long-term maintenance of such effective qualitative resistance alleles in the host population is likely facilitated by multiple factors. These factors include the broad host range of the pathogen, the abundance of susceptible trees in natural environments, the tolerance of the pine hosts to the disease, and the fact that aeciospores released from galls can reinfect oak but not pine (Czabator 1971). These factors would act in concert to reduce the likelihood of large-scale epidemics caused by the sudden emergence of a virulent pathogen genotype within a local environment and maintain polymorphism at large-effect resistance loci in the host population. Conversely, these are the same factors that would maintain the efficacy of these resistance genes over the long term, since the broad host range and high levels of disease tolerance reduce the selection pressure for virulence in the pathogen population (Roy and Kirchner 2000).
For a resistance gene to be classified as a broad spectrum, its protein must confer resistance against two or more pathogens or pathotypes (Li et al. 2020; Ning and Wang 2018; Xiao et al. 2001). From the results presented here, we infer that parent 4 carries two QTL with similar broad-spectrum properties, and parent 9 carries one QTL. In family E4, these QTL behave additively with respect to the odds of disease outcome in the population, suggesting that their resistance spectra and/or modes of action are not identical. The evidence suggests that the QTL segregating in family E9 is quantitatively different from the QTL segregating in family E4 (Table 1), and we infer that this difference is a function of different resistance spectra and/or mode of action. Finally, the candidate genes detected in the transcriptome are similar to other known plant resistance genes (Dangl and Jones 2001), suggesting that RNS-BSR against fusiform rust of pines may be mediated through NLR resistance genes.
One likely explanation for the difference in disease incidence between the two families is that the resistance allele at GRID3 in family E9 has a narrower resistance spectrum, since it recognizes an effector protein that is present at a lower frequency in the pathogen population than the effectors recognized by GRID1 and GRID2. In family E4, the additivity between the resistance alleles at GRID1 and GRID2 suggests that they have recognition spectra that are not identical. The disease incidence of the double-resistant GRID1/GRID2 individuals was striking, with only one out of 156 individuals in this class being diseased. The additivity between the resistance alleles at these QTL suggests that the probability of infection may be a multiplicative function of the allele frequencies of the virulence alleles in the pathogen population.
Despite a population history of high selection pressure for rust resistance in the clonally replicated validation population (Shalizi and Isik 2019), polymorphism at the resistance loci was maintained. This is likely due to the fact that a lower spore density (~50,000 spores/ml) was used in the initial screening for the validation population, which was only half the spore density used in the QTL discovery experiment. This probably resulted in a higher rate of escapes, which would have preserved the genetic variation within families. It is important to note that the inoculum used to screen the validation population was different from the inoculum used in the QTL discovery experiment. Furthermore, the disease recorded at the eight test sites was derived from wild environmental inoculum. Nonetheless, the same genomic regions were associated with rust resistance in both populations. The principal component decomposition of the kinship matrix revealed the existence of four large sub-groups, all of which contained both highly susceptible and highly resistant clones (Fig. 4). The SNP with significant associations all had a moderate to high minor allele frequency (Table 2), which reflects the low effective population size and the nested family structure of the population. The QQ plot showed a small number of significant p values and a close agreement with the null hypothesis for the vast majority of SNP, suggesting that the population’s history of selection did not severely limit the genetic resolution of the analysis (Fig. 3C). The known pedigree connectivity, combined with the overall agreement between the genomic locations of the QTL found in the discovery population and the marker-trait associations detected in the validation population, suggests that all three resistance alleles detected in GWAS are identical-by-descent with the alleles detected in the QTL analysis.
The bulked segregant analysis, although not providing conclusive proof of function for the candidate genes, indicated that these resistance QTL might harbor genes that are members of the NLR superfamily. In family E4, the proportion of PacBio contigs annotated as NLR-type resistance genes was only 0.006; but in the set of candidate SNP showing a complete separation with disease status, this proportion was 0.30. A similar enrichment was observed for family E9. The large magnitude of this enrichment, combined with the high depth of coverage of the expressed gene content (~163x), suggests that the identification of these transcripts as the causal factors of RNS-BSR observed in this study is not due to chance alone. In family E4, which had threefold lower rust incidence than family E9, two of the three candidate proteins clustered into a clade that was private to family E4 (Supplementary Fig. 3). This suggests that the candidate genes in E4 may have a similar function, and this function may be lacking from family E9. More experimentation will be required to develop SNP markers within these candidate genes to determine their linkage relationships with the QTL on LG2 and LG7. Since the family-specific reference transcriptomes were derived from a single time-point, there may have been other expressed sequences in the transcriptome that played a role in conferring resistance that was not represented in the PacBio ISOSeq libraries. The fact that multiple SNP were discovered on the candidate transcripts but only one was associated with resistance in each case suggests there were other highly homologous expressed sequences in the genome, which is expected of duplicated gene families such as NLR (Van Ghelder et al. 2019; Wegrzyn et al. 2014). This fact makes the SNP discovered in bulked segregant RNAseq particularly valuable, since they could help identify the causal gene in a group of multiple duplicated genes and/or pseudogenes. However, since Illumina data were used for SNP discovery, and there were other SNP on each PacBio contig that were not associated with disease, we can only conclude that the fragment of the transcript encompassed by the paired-end reads aligning to the SNP site were associated with disease resistance.
Because of the large number of gametes sampled within each full-sib family, the genetic map produced in this study is one of the highest resolution linkage maps ever produced for a conifer species (De La Torre et al. 2019; Sewell et al. 1999; Westbrook et al. 2015). A total of 10,204 markers of the Pita50K Affymetrix array were genotyped on two populations of ~1000 individuals each, which undoubtedly contributed to the narrow confidence intervals for the QTL detected in this study. The high resolution of our map is evident in the comparison between map positions of Pita2.01 contigs from our consensus map and the map published in de la Torre et al. (2019) (Supplementary Table 2); large blocks of contigs that were placed at the same position in the De La Torre et al. (2019) map could be uniquely positioned in our map due to a large number of recombination events. The same genomic regions underlying the QTL were associated with field-level rust resistance, confirming both the accuracy of the consensus map as well as the efficacy and durability of the QTL across a broad sample of environments in the southeastern United States (Shalizi and Isik 2019).
The demonstration of one-to-many relationships between host resistance QTL and fungal genotypes shows how simply inherited qualitative genetic resistance can deliver effective levels of immunity, despite the genetic variability of the pathogen. In crop systems, resistance alleles with such strong effects would not be expected to retain efficacy due to the inevitable adaptation in the pathogen population, resulting in resistance breakdown (Janzac et al. 2009; Li et al. 2003). However, in endemic systems such as the pine-rust pathosystem, the selection pressure for virulence is reduced due to physiological tolerance and the abundance of susceptible genotypes of multiple species in the environment, leading to the persistence of large-effect qualitative resistance genes in the host population. Further experimentation will be conducted to understand the pathogen side of the interaction and validate the efficacy spectrum hypothesis presented here.
Data availability
Raw .fastq files from the NovaSeq6000 instrument, as well as subreads.bam files from the PacBio Sequel instrument used to produce the family-specific reference transcriptomes were deposited at NCBI sequence read archive under BioProject accession number PRJNA719490. QTL datasets formatted for import into R/QTL were deposited at the Dryad depository and can be located using this https://doi.org/10.5061/dryad.mcvdnck0x. Family-specific PacBio reference transcriptomes were deposited at the Dryad depository and can be located using this https://doi.org/10.5061/dryad.jsxksn08x. Variants discovered in the bulked segregant RNAseq analysis were deposited at the Dryad depository as .vcf files, and can be located using this url: https://doi.org/10.5061/dryad.hhmgqnkgg.
Change history
23 July 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41437-021-00458-1
References
Amerson HV, Nelson CD, Kubisiak TL, Kuhlman EG, Garcia SA (2015) Identification of nine pathotype-specific genes conferring resistance to fusiform rust in loblolly pine (Pinus taeda L.). Forests 6(8):2739–2761. https://doi.org/10.3390/f6082739
Anderson RL, McClure JP, Cost N, Uhler RJ (1986) Estimating fusiform rust losses in five southeast states. South J Appl Forestry 10(4):237–240. https://doi.org/10.1093/sjaf/10.4.237
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl, vol. 46. Springer.
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19(7):889–890. https://doi.org/10.1093/bioinformatics/btg112
Burdine CS, Kubisiak TL, Johnson GN, Nelson CD (2007). Fifty‐two polymorphic microsatellite loci in the rust fungus, Cronartium quercuum f. sp. fusiforme.Molecular Ecology Notes. 7(6):1005-8.
Caballero M, Lauer E, Bennett J, Zaman S, McEvoy S, Acosta J, Jackson C, Townsend L, Eckert A, Whetten R, Loopstra C, Holliday J, Mandal M, Wegrzyn J, Isik F (In Review) Towards genomic selection in Pinus taeda (Pinaceae): integrating resources to support array design in a complex conifer genome. Appl Plant Sci (in print)
Collett D (2002) Modelling binary data, 2nd edn. CRC Press.
Cubbage FW, Pye JM, Holmes TP, Wagner JE (2000) An economic evaluation of fusiform rust protection research. South J Appl Forestry 24(2):77–85. https://doi.org/10.1093/sjaf/24.2.77
Cumbie WP, Huber DA, Steel VC, Rottmann W, Cannistra C, Pearson L, Cunningham M (2020) Marker associations for fusiform rust resistance in a clonal population of loblolly pine (Pinus taeda, L.). Tree Genet Genomes 16(6):86. https://doi.org/10.1007/s11295-020-01478-4
Czabator FJ (1971) Fusiform rust of southern pines: a critical review. Southern Forest Experiment Station, Forest Service, U.S. Department of Agriculture.
Dangl JL, Jones JDG (2001) Plant pathogens and integrated defence responses to infection. Nature 411(6839):826–833. https://doi.org/10.1038/35081161
De La Torre AR, Wilhite B, Neale DB (2019) Environmental genome-wide association reveals climate adaptation is shaped by subtle to moderate allele frequency shifts in loblolly pine. Genome Biol Evolution 11(10):2976–2989. https://doi.org/10.1093/gbe/evz220
Deng Y, Zhai K, Xie Z, Yang D, Zhu X, Liu J, Wang X, Qin P, Yang Y, Zhang G, Li Q, Zhang J, Wu S, Milazzo J, Mao B, Wang E, Xie H, Tharreau D, He Z (2017) Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 355(6328):962–965. https://doi.org/10.1126/science.aai8898
Devey ME, Delfino-Mix A, Kinloch BB, Neale DB (1995) Random amplified polymorphic DNA markers tightly linked to a gene for resistance to white pine blister rust in sugar pine. Proc Natl Acad Sci USA, 92(6):2066–2070. https://doi.org/10.1073/pnas.92.6.2066
Echt CS, Saha S, Krutovsky KV, Wimalanathan K, Erpelding JE, Liang C, Nelson CD (2011) An annotated genetic map of loblolly pine based on microsatellite and cDNA markers. BMC Genet 12(1):17. https://doi.org/10.1186/1471-2156-12-17
Endelman J (2019) rrBLUP: Ridge Regression and Other Kernels for Genomic Selection (4.6.1) [Computer software]. https://CRAN.R-project.org/package=rrBLUP
Ennos RA (2015) Resilience of forests to pathogens: an evolutionary ecology perspective. Forestry: Int J For Res 88(1):41–52. https://doi.org/10.1093/forestry/cpu048
Fang X, Snell P, Barbetti MJ, Lanoiselet V (2017) Races of Magnaporthe oryzae in Australia and genes with resistance to these races revealed through host resistance screening in monogenic lines of Oryza sativa. Eur J Plant Pathol 148(3):647–656. https://doi.org/10.1007/s10658-016-1122-4
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. http://arxiv.org/abs/1207.3907
Hamelin RC, Doudrick RL, Nance WL (1994) Genetic diversity in Cronartium quercuum f.sp. Fusiforme on loblolly pines in southern U.S. Curr Genet 26(4):359–363. https://doi.org/10.1007/BF00310501
Isik F, Amerson HV, Whetten RW, Garcia SA, McKeand SE (2012) Interactions of Fr genes and mixed-pathogen inocula in the loblolly pine-fusiform rust pathosystem. Tree Genet Genomes 8(1):15–25. https://doi.org/10.1007/s11295-011-0416-0
Isik F, McKeand SE (2019) Fourth cycle breeding and testing strategy for Pinus taeda in the NC State University Cooperative Tree Improvement Program. Tree Genet Genomes 15(5):70. https://doi.org/10.1007/s11295-019-1377-y
Janzac B, Fabre F, Palloix A, Moury B (2009) Constraints on evolution of virus avirulence factors predict the durability of corresponding plant resistances. Mol Plant Pathol 10(5):599–610. https://doi.org/10.1111/j.1364-3703.2009.00554.x
Keen NT (1990) Gene-for-gene complementarity in plant-pathogen interactions. Annu Rev Genet 24(1):447–463. https://doi.org/10.1146/annurev.ge.24.120190.002311
Kubisiak TL, Amerson HV, Nelson CD (2005) Genetic interaction of the fusiform rust fungus with resistance gene Fr1 in loblolly pine. Phytopathology 95(4):376–380. https://doi.org/10.1094/PHYTO-95-0376
Kuhlman EG, Amerson HV, Jordan AP, Pepper WD (1997) Inoculum density and expression of major gene resistance to fusiform rust disease in loblolly pine. Plant Dis 81(6):597–600. https://doi.org/10.1094/PDIS.1997.81.6.597
Li H, Sivasithamparam K, Barbetti MJ (2003) Breakdown of a Brassica rapa subsp. Sylvestris single dominant blackleg resistance gene in B. napus rapeseed by leptosphaeria maculans field isolates in Australia. Plant Dis 87(6):752–752. https://doi.org/10.1094/PDIS.2003.87.6.752A
Li W, Deng Y, Ning Y, He Z, Wang G-L (2020) Exploiting broad-spectrum disease resistance in crops: from molecular dissection to breeding. Annu Rev Plant Biol 71(1):575–603. https://doi.org/10.1146/annurev-arplant-010720-022215
Liu S, Yeh C-T, Tang HM, Nettleton D, Schnable PS (2012) Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 7(5):e36406. https://doi.org/10.1371/journal.pone.0036406
Matthews FR, Rowan SJ (1972) Improved method for large-scale inoculations of pine and oak with Cronartium fusiforme. Plant Dis Rep. http://agris.fao.org/agris-search/search.do?recordID=US201302232103
Mercière M, Boulord R, Carasco-Lacombe C, Klopp C, Lee Y-P, Tan J-S, Syed Alwee SSR, Zaremski A, De Franqueville H, Breton F, Camus-Kulandaivelu L (2017) About Ganoderma boninense in oil palm plantations of Sumatra and peninsular Malaysia: ancient population expansion, extensive gene flow and large scale dispersion ability. Fungal Biol 121(6):529–540. https://doi.org/10.1016/j.funbio.2017.01.001
Millar CI, Kinloch BB (1991) Taxonomy, phylogeny, and coevolution of pines and their stem rusts. In: Hiratsuka Y, Samoil JK, Blenis PV, Crane PE, Laishley BL (eds). Rusts of pine: proceedings of the 3rd IUFRO Rusts of Pine Working Party Conference, September 18–22, 1989. Forestry Canada, Banff, Alberta, Northwest Region, North Forestry Centre, Edmonton, Alberta. Information Report NOR-X-317, p. 1–38. https://www.fs.usda.gov/treesearch/pubs/31858
Neale DB, Wegrzyn JL, Stevens KA, Zimin AV, Puiu D, Crepeau MW, Cardeno C, Koriabine M, Holtz-Morris AE, Liechty JD, Martínez-García PJ, Vasquez-Gross HA, Lin BY, Zieve JJ, Dougherty WM, Fuentes-Soriano S, Wu L-S, Gilbert D, Marçais G, Langley CH (2014) Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol 15(3):R59. https://doi.org/10.1186/gb-2014-15-3-r59
Ning Y, Wang G-L (2018) Breeding plant broad-spectrum resistance without yield penalties. Proc Natl Acad Sci USA 115(12): 2859–2861. https://doi.org/10.1073/pnas.1801235115
Piskol R, Ramaswami G, Li JB (2013) Reliable identification of genomic variants from RNA-Seq Data. Am J Hum Genet 93(4):641–651. https://doi.org/10.1016/j.ajhg.2013.08.008
Powers HR, McClure JP, Knight HA, Dutrow GF (1974) Incidence and financial impact of fusiform rust in the south. J Forestry 72(7):398–401. https://doi.org/10.1093/jof/72.7.398
Prestemon JP, Abt RC (2002) Southern forest resource assessment highlights: the Southern Timber Market to 2040. J Forestry 100(7):16–22. https://doi.org/10.1093/jof/100.7.16
Roy BA, Kirchner JW (2000) Evolutionary dynamics of pathogen resistance and tolerance. Evolution 54(1):51–63. https://doi.org/10.1111/j.0014-3820.2000.tb00007.x
Saintenac C, Zhang W, Salcedo A, Rouse MN, Trick HN, Akhunov E, Dubcovsky J (2013) Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341(6147):783–786. https://doi.org/10.1126/science.1239022
Schultz RP (1999). Loblolly--the pine for the twenty-first century. New forests. 17(1):71-88.
Sewell MM, Sherman BK, Neale DB (1999) A consensus map for loblolly pine (Pinus taeda L.). I. Construction and integration of individual linkage maps from two outbred three-generation pedigrees. Genetics 151(1):321–330
Shalizi MN, Isik F (2019) Genetic parameter estimates and GxE interaction in a large cloned population of Pinus taeda L. Tree Genet Genomes 15(3):46. https://doi.org/10.1007/s11295-019-1352-7
Tellier A, Brown JKM (2007) Stability of genetic polymorphism in host–parasite interactions. Proc R Soc B: Biol Sci 274(1611):809–817. https://doi.org/10.1098/rspb.2006.0281
Thompson JN, Burdon JJ (1992) Gene-for-gene coevolution between plants and parasites. Nature 360(6400):121–125. https://doi.org/10.1038/360121a0
Van Ghelder C, Parent GJ, Rigault P, Prunier J, Giguère I, Caron S, Stival Sena J, Deslauriers A, Bousquet J, Esmenjaud D, MacKay J (2019) The large repertoire of conifer NLR resistance genes includes drought responsive and highly diversified RNLs. Sci Rep. 9(1):1–13. https://doi.org/10.1038/s41598-019-47950-7
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91(11):4414–4423. https://doi.org/10.3168/jds.2007-0980
Vossen EVD, Sikkema A, Hekkert B, te L, Gros J, Stevens P, Muskens M, Wouters D, Pereira A, Stiekema W, Allefs S (2003) An ancient R gene from the wild potato species Solanum bulbocastanum confers broad-spectrum resistance to Phytophthora infestans in cultivated potato and tomato. Plant J 36(6):867–882. https://doi.org/10.1046/j.1365-313X.2003.01934.x
Wang J, Qu B, Dou S, Li L, Yin D, Pang Z, Zhou Z, Tian M, Liu G, Xie Q, Tang D, Chen X, Zhu L (2015) The E3 ligase OsPUB15 interacts with the receptor-like kinase PID2 and regulates plant cell death and innate immunity. BMC Plant Biol 15(1):49. https://doi.org/10.1186/s12870-015-0442-4
Wegrzyn JL, Liechty JD, Stevens KA, Wu L-S, Loopstra CA, Vasquez-Gross HA, Dougherty WM, Lin BY, Zieve JJ, Martínez-García PJ, Holt C, Yandell M, Zimin AV, Yorke JA, Crepeau MW, Puiu D, Salzberg SL, Dejong PJ, Mockaitis K, Neale DB (2014) Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 196(3):891–909. https://doi.org/10.1534/genetics.113.159996
Westbrook JW, Chhatre VE, Wu L-S, Chamala S, Neves LG, Muñoz P, Martínez-García PJ, Neale DB, Kirst M, Mockaitis K, Nelson CD, Peter GF, Davis JM, Echt CS (2015) A consensus genetic map for pinus taeda and pinus elliottii and extent of linkage disequilibrium in two genotype-phenotype discovery populations of pinus taeda. G3: Genes, Genomes, Genet 5(8):1685–1694. https://doi.org/10.1534/g3.115.019588
Wilcox PL, Amerson HV, Kuhlman EG, Liu BH, O’Malley DM, Sederoff RR (1996) Detection of a major gene for resistance to fusiform rust disease in loblolly pine by genomic mapping. Proc Natl Acad Sci USA, 93(9):3859–3864. https://doi.org/10.1073/pnas.93.9.3859
Xiao S, Ellwood S, Calis O, Patrick E, Li T, Coleman M, Turner JG (2001) Broad-spectrum mildew resistance in arabidopsis thaliana mediated by RPW8. Science 291(5501):118–120. https://doi.org/10.1126/science.291.5501.118
Acknowledgements
Funding for this project was provided by USDA-NIFA award 2019-67013-29169 “Genomic Selection in Forest Trees: Beyond Proof of Concept”. We would like to thank the Genomic Science Laboratory of North Carolina State University for library preparation and sequencing, the Bioinformatic Research Center for cluster computing resources. Kathleen (Katie) McKeever and Brenda Minton at the USDA Forest Service Resistance Screening Center in Asheville, NC, helped for plant rearing and fungal inoculations. We thank all the staff, students, and members of The North Carolina State University Cooperative Tree Improvement Program. We thank April Meeks, Doug Dobson, Syamsudin Slamet, Nasir Shalizi, Colin Jackson, and Jaesoon Hwang. The first author thanks his Ph.D. committee, James Holland, Ross Whetten, and Chris Maltecca, for intellectual discussions and advice, as well as his friends and family for their infinite support. We thank John MacKay (Oxford University Plant Sciences) for providing suggestions to improve the manuscript. We are also grateful to the associate editor of Heredity journal and two anonymous reviewers for providing valuable suggestions to improve the manuscript during the review process.
Author information
Authors and Affiliations
Contributions
EL conceived and designed the experiments, organized and performed the research activities, analyzed the QTL and RNASeq data, drafted this manuscript, and served as senior personnel on USDA-NIFA award 2019-67013-29169. FI conceived the experiments, and led the Pita50K SNP array marker discovery as part of USDA-NIFA award 2016-67013-24469, served as PI on USDA-NIFA award 2019-67013-29169, assisted in analysis of data, the recording of phenotypic data, and provided extensive revisions in drafting this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Dario Grattapaglia
Supplementary information
Rights and permissions
About this article

Cite this article
Lauer, E., Isik, F. Major QTL confer race-nonspecific resistance in the co-evolved Cronartium quercuum f. sp. fusiforme–Pinus taeda pathosystem. Heredity 127, 288–299 (2021). https://doi.org/10.1038/s41437-021-00451-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-021-00451-8
This article is cited by
-
Low-density AgriSeq targeted genotyping-by-sequencing markers are efficient for pedigree quality control in Pinus taeda L. breeding
Tree Genetics & Genomes (2023)
-
Multiple-trait analyses improved the accuracy of genomic prediction and the power of genome-wide association of productivity and climate change-adaptive traits in lodgepole pine
BMC Genomics (2022)
-
Genetic linkage between the training and selection sets impacts the predictive ability of SNP markers in a cloned population of Pinus taeda L.
Tree Genetics & Genomes (2022)
