
Abstract
Immunoglobulins (Ig) play an important role in the immune system both when expressed as antigen receptors on the cell surface of B cells and as antibodies secreted into extracellular fluids. The advent of high-throughput sequencing methods has enabled the investigation of human Ig repertoires at unprecedented depth. This has led to the discovery of many previously unreported germline Ig alleles. Moreover, it is becoming clear that convergent and stereotypic antibody responses are common where different individuals recognise defined antigenic epitopes with the use of the same Ig V genes. Thus, germline V gene variation is increasingly being linked to the differential capacity of generating an effective immune response, which might lead to varying disease susceptibility. Here, we review recent evidence of how germline variation in Ig genes impacts the Ig repertoire and its subsequent effects on the adaptive immune response in vaccination, infection, and autoimmunity.
Similar content being viewed by others

Introduction
Adaptive immune responses carried out by B and T cells are central in the body’s fight against pathogens, and they are also implicated in autoimmune diseases. Both types of adaptive immune cells recognise antigenic determinants by specific receptors – the B-cell receptor (BCR) and the T-cell receptor (TCR). Whereas functionality of a BCR is dependent on complex formation with the CD79a/b co-receptor, in this review, we refer to BCR as the antigen-binding, cell-surface anchored immunoglobulin without this co-receptor. Similarly, we refer to the TCR as the antigen-binding receptor without the CD3 co-receptor. To be able to recognise the vast plethora of existing antigens, an immense diversity of both BCRs and TCRs is required. The collection of BCRs (or TCRs) in an individual is called the BCR (or TCR) repertoire. Immune repertoire diversity is mainly obtained by somatic gene rearrangements, so-called VDJ recombination, where one variable (V), one diversity (D) and one joining (J) gene segment combine in a supposedly stochastic fashion [1]. Note that D gene segments are only present in the heavy chain locus and are lacking in the kappa and lambda light chain loci. Although the majority of rearranged Ig sequences contain only one D gene segment, VDDJ recombination has also been reported [2]. BCRs may further diversify by somatic hypermutation, which introduces point mutations in certain hotspots.
While the TCR only exists as a membrane-bound form, the antigen-binding component of BCR can be produced also as water-soluble Ig molecules – antibodies – that exert effector functions in extracellular fluids (serum, mucosa). Each B cell has its unique BCR and its specificity is determined by heavy and light chain pairing, V(D)J recombination, combinatorial and junctional diversity and somatic hypermutation. The BCR of a naive B cell is in its germline configuration without any somatic hypermutation. For a naive B cell to become activated, it needs to bind an antigen and the activated B cell can then give rise to effector B cells and memory B cells. The repertoire of a specific B-cell population can be studied separately by sorting the target population; for example, sorting naive B cells allows the exploration of a naive BCR repertoire.
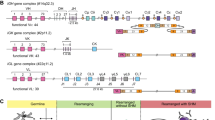
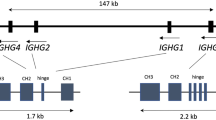
In humans, the genes encoding BCRs and antibodies are located on Ig loci found on three chromosomes (chr). The heavy chain locus is located on chr14 (14q32.33) [3], the kappa chain locus is located on chr2 (2p11.2) and the lambda chain locus is located on chr22 (22q11.2) [4] (Fig. 1). Apart from the heavy, kappa and lambda loci, a number of Ig genes can also be found in clusters or alone on other human chromosomes (chr1, chr2, chr 8, chr 9, chr10, chr 15, chr16, chr18, chr21, chr22, chrY) [5, 6]. Such orphon Ig genes are generally perceived to be non-functional and have been described as pseudogenes or open-reading frames (ORFs) [6]. It is conceivable that genes outside the main Ig loci can also contribute to BCR formation. The findings of LAIR1 and LILRB1 gene sequences in rearranged BCRs in malaria-infected patients and also healthy individuals suggest that such a mechanism indeed is operative [7,8,9,10]. The LAIR1 and LILRB1 genes are located outside the IGH locus, yet templated insertions of LAIR1 and LILRB1 sequences were found to be introduced into rearranged IGH genes by various modalities. Both LAIR1 and LILRB1 encode receptors that bind to P. falciparum repetitive interspersed families of polypeptides (RIFINs)-variant surface antigens. These RIFINs antigens contribute to malaria pathogenesis by enterocyte aggregation. The non-canonically generated antibodies bind to P. falciparum-infected enterocytes, and the findings suggest that such non-conventional antibodies play a role in immune evasion. Importantly, these findings demonstrate that BCR repertoires are influenced directly by non-Ig genes.

A In humans, the main immunoglobulin loci are located on chromosomes 14 (heavy), 2 (kappa) and 22 (lambda). Each of the main Ig loci contains clusters of V, J and C genes, and the heavy locus also contains a cluster of D genes. The organisation of genes within each locus is different. The production of a functional transcript requires V(D)J recombination. This figure is schematic only and does not show all genes present in the loci. B Different V(D)J combinations, as well as various pairings of heavy and light chain, produce a diverse set of antibodies/BCRs with a variety of paratopes with the capacity to recognise different antigens. The binding capacity of antibodies can be further enhanced by somatic hypermutation, which introduces point mutations in mutational hotspots of the V genes.
Available information on the composition and sequence variation in the human Ig loci remains incomplete. This is due to a large number of highly similar and sometimes duplicated genes [11, 12], which render the correct assembly of the Ig loci from short sequences challenging. Advances in high-throughput sequencing methods and the development of specialised software tools have expanded our knowledge of Ig genes [13,14,15,16,17]. There is a high degree of genetic variation in the Ig loci. The numbers of allelic variants in databases for inferred variants from adaptive immune receptor repertoire sequence data [18, 19] and in the IMGT/GENE-DB database [20] have rapidly increased in the past few years. The genetic variation in Ig genes likely results from adaptation to pathogens. Consequently, functional effects of germline variation in Ig genes should be found when studying immune responses to pathogens and, therefore, may also be involved in autoimmune diseases. Germline, as used in the context of this review, refers to the unmutated configuration of a V gene, regardless of whether it is rearranged or not.
Crosstalk between B cells and T cells shapes antibody responses
The shaping of the BCR repertoire after antigen encounter is orchestrated by T cells [21, 22]. An antigen bound by a BCR is endocytosed by a B cell and it gets processed into peptide fragments. These peptide fragments then bind to MHC class II molecules, which are transported to the cell surface for display to T cells. Upon TCR recognition of the peptide-MHC II complex, T cells become activated and provide help to B cells. Interaction of B cells with T cells typically takes place in organised lymphoid structures (e.g., spleen, lymph node). As a consequence of B-T-cell interaction, B cells may localise to germinal centres where BCR sequence diversification via somatic hypermutations as well as isotype switching takes place. The T cell - B cell crosstalk impacts both cell types; both T cells and B cells will undergo clonal expansion and some of the cells will persist as memory cells making up the memory BCR repertoire. A subset of post-germinal centre B cells will differentiate into effector cells (plasmablasts and plasma cells) that secrete soluble antibodies. In germinal centres, the B-cell clones with BCRs that have the highest affinity will be selected over those with lower BCR affinity in a process called antibody affinity maturation. It follows from this scheme that a BCR’s ability to bind antigen in the germline configuration is essential for the mounting of a B-cell response (Fig. 2). Therefore, the outcome of this response is highly influenced by the genetic variation affecting the sequence of the BCR itself and the number of circulating naive B cells with a BCR that can bind a given antigen [23].

The extent of antigen recognition by B cells has an impact on their activation, which in turn can influence the effectiveness of the immune response to a specific antigen.
Lessons from studies in mice
The notion that there is extensive genetic variation in Ig loci has been fuelled by comparative analysis of Ig genes in inbred mouse strains. Analysis of IgM- and IgG-associated VDJ rearrangements in C57BL/6 and BALB/c strains showed presence of 99 and 164 functional variable heavy (VH) genes in the genomes of the two strains, respectively [24]. Remarkably, only five VH sequences were common to both strains. Subsequent analysis of five additional mouse strains representing major subspecies revealed further large variation, and the majority of inferred germline sequences were unique to a single strain [25]. Based on this finding, it was suggested that the mouse Igh loci are complex mosaics of short haplotype blocks of disparate origins. The entire Igh V region sequence of the C57BL/6 strain has been established [26]. However, it is clear from the comparative studies that the Igh locus of C57BL/6 is unable to serve as a map of genes for other strains, and further that the large genetic variation across mouse strains poses a challenge to establish suitable nomenclature [27]. Complete sequencing of the Ig gene loci of the different strains will be required to determine how much structural variation there is and how much of the strain difference can be ascribed to true allelic variation.
Genetic variation in Ig heavy loci can affect functional outcomes of immune responses. Early studies of immune responses to structurally simple haptens revealed that the hapten-specific antibodies were highly similar and utilised the same V gene combinations [28]. Structural studies revealed that the key residues in the Ig V genes that facilitated the hapten recognition in mice were germline-encoded [29]. More recent studies have indicated that genetic restriction in response also applies to complex antigens. In a study of the B-cell response to phycoerythrin, a fluorescent protein here used as an immunising antigen, it was observed that different inbred strains make qualitatively different responses [30]. The two strains of mice used in that study only differ in their Ig heavy chain loci, while the rest of their genes are identical. C57BL/6 and C.B-17 (Ighb) mice had larger numbers of phycoerythrin-specific naive B cells and generated smaller germinal centre responses and larger numbers of IgM memory cells and plasmablasts than BALB/c (Igha) mice. BALB/c mice, on the contrary, elicited more affinity-matured switched Ig memory B cells. The properties of phycoerythrin-specific B cells in C57BL/6 mice correlated with the usage of a single VH that in the germline form of C57BL/6 but not in BALB/c mice afforded high-affinity phycoerythrin binding. These results suggest that some individuals may be genetically predisposed to generate non-canonical memory B-cell responses to certain antigens because of avid antigen binding via germline-encoded VH elements.
Genetic variation in murine Ig loci can also affect disease susceptibility as shown for autoimmune diseases [31]. In a study of collagen-induced arthritis, an experimental mouse model for human rheumatoid arthritis, it was found that a pathogenic antibody response to collagen II is genetically controlled by a strong influence of the Ig heavy chain locus. Antibodies targeting a major epitope of collagen II mainly utilised the same VH gene (mouse IGHV1-4). Comparing Ig V genes of the susceptible strain with that of the non-susceptible strain, the authors noted differences at two germline positions, which they considered to be allelic variants. Site-directed mutagenesis and X-ray crystallography revealed that these two residues were critical for recognition of the epitope thereby providing an explanation why the non-disease susceptible mouse strain would not produce antibodies against this major collagen epitope.
Polymorphic variation within the human Ig loci
Early studies
Before repertoire sequencing became widely available, early studies on Ig polymorphisms in disease association were performed by restriction fragment length polymorphism (RFLP) technique or detection of alleles by gene-specific polymerase-chain-reaction (PCR). One such study suggested that a polymorphism in IGHV1-69 was linked to susceptibility to rheumatoid arthritis in a specific population that lacked the HLA-DRB1 epitope [32], and several studies revealed a possible role of the IGHV locus in multiple sclerosis [33], particularly the IGHV2 gene family [34, 35]. Possible disease associations from older studies with less detailed locus characterisation (reviewed by Watson & Breden [36]) are difficult to interpret in the light of current knowledge of the Ig loci.
Studies of germline Ig variation by repertoire analysis
For now, high-throughput repertoire studies have been the most prevalent and accessible way to explore germline Ig variation. Such studies rely on rearranged Ig mRNA transcripts that are used to generate cDNA for the preparation of amplicon sequencing libraries. Repertoire studies have led to the inference of several novel Ig V alleles [13, 14, 37,38,39,40,41], however, new alleles must be validated from genomic DNA in order to be added to the IMGT/GENE-DB, which currently serves as the reference germline database. Since it is not always possible to obtain additional samples from the same individuals, the Inferred Allele Review sub-Committee (IARC) [42] has been set up to review previously unreported inferred Ig V alleles. The IARC and IMGT have been working closely to include inferred variants for which there is sufficient evidence from multiple studies in the IMGT/GENE-DB. IARC assesses novel allele candidates from inferred genotypes, which are deposited to the Open Germline Receptor Database (OGRDB) [18]. Currently, 16 inferred heavy chain and 2 light chain IgV alleles have been approved and added to the IMGT/GENE-DB [20] reference database following IARC assessment and recommendation.
Despite a large number of known germline V alleles (Table 1), more are still being discovered [37, 43]. This suggests that the amount of germline polymorphisms in Ig V genes is likely underestimated, especially since most of the known Ig V alleles are only from individuals of European ancestry [44]. Most studies have only investigated the coding region of V genes and left the non-coding regions unexplored. Although haplotypes and potential gene deletions can be inferred using specialised software [45], the non-coding regions of the loci as well as D, J or C variation still remain underexplored compared to V genes.
Complete sequencing of human Ig haplotypes
Current knowledge of the non-coding Ig regions stems mostly from the studies that assembled full-length germline DNA sequences of the Ig loci using cosmid or BAC clones and Sanger sequencing [11, 46]. There has been a lack of high-throughput methods suitable for exploring the Ig loci from gDNA. Although Ig haplotyping from repertoire data is possible, it has its limitations as repertoire studies that utilise rearranged Ig transcripts can only explore expressed genes. Furthermore, haplotype inference from such Ig repertoire data can only determine which genes are on the same chromosome, but it cannot determine their order, and neither can it tell us anything about the non-coding regions. Whole-genome studies are not suitable either, since the Ig genes are highly similar and frequent duplications make the assembly of Ig loci from short reads extremely difficult. Recently, a new method has been presented, which utilises long-read SMRT sequencing (Pacific Biosciences) to sequence the Ig loci [47]. For analysis of such data, the authors have also developed a custom software, IGenotyper, which allows exploration of Ig haplotype diversity from genomic DNA by generating high-quality assemblies. Apart from enabling complete haplotype phasing thanks to the long reads, this method also retains accuracy when multiplexing, allowing to sequence multiple samples at the same time and making it high-throughput [47]. It provides a much-needed alternative to Ig repertoire sequencing (short-read, high-throughput) or cosmid/BAC clone Sanger-seq-based methods (long-read, low-throughput).
This new method has been used to analyse Ig haplotypes from family trios and this has led to characterisation of novel structural variants and V alleles in the heavy chain locus [47]. Although the number of explored full-length Ig loci is still very limited, the fact that nearly all Ig haplotypes studied so far had unique features demonstrates that the Ig haplotype diversity is large. Since Ig genes in their germline configuration have affinity to certain antigens (Fig. 3) and can facilitate efficient immune response, the absence of certain genes or their duplication can affect the resulting Ig repertoire. Thus, haplotype variation can influence the immune response and could potentially have a profound impact on one’s ability to fight infections [48].

A Antibody specificity is determined by the amino-acid composition of the variable region. B Polymorphisms at residues which are critical for the antibody-antigen interaction can result in loss of binding capacity [23, 65, 71]. Not all antibody residues are essential for antigen recognition (C) and Ig polymorphisms at positions that are not critical for binding may have little to no effect on the antibody binding capacity. D In addition to amino-acid changes, post-translational modifications in the variable domain may also affect the binding capacity of an antibody by either enhancing or abolishing it [83].
Convergent antibody responses to pathogens influenced by Ig polymorphisms
As pathogens display a multitude of antigens, the antibody responses generated during infection are typically very diverse. Nevertheless, finding highly similar antibodies that are shared between different individuals in response to pathogens is not uncommon, and some examples of infections where such convergent responses have been observed are influenza [49], dengue [50], ebola [51] and SARS-CoV-2 [52, 53]. Investigating responses to individual epitopes, for instance, when studying antibodies that have neutralising activity to viruses, it is common to find convergent and stereotyped antibody responses [54]. This suggests that antibody fitness for a defined epitope governs the selection of BCRs engaged in the response and that the rules for selection are similar across individuals. Importantly, the phenomenon of convergent antibody responses provides the foundation for genetic effects to play out. Biases in the V gene usage as observed in antigen-specific/disease-specific BCR repertoires suggest that germline variation in these genes might have a functional effect (Fig. 4). It also suggests that to see genetic effects on recognition of antigen, studies will need to focus on recognition of single and defined epitopes [55]. Examples of conserved germline-encoded residues that are crucial for recognition of defined epitopes are shown in Table 2. We expect many more such examples to be uncovered in the future. In the following, we will review selected antibody responses to particular antigens where such convergence has been observed and where Ig polymorphisms have been demonstrated to affect the antibody response.

A Ig V genes exist in many allelic forms, which differ in one or more nucleotides. Non-synonymous polymorphisms that change the amino-acid sequence may also alter the antigen-binding capacity of the resulting antibody. B For some V genes, the expression levels of alleles in heterozygous individuals are uneven, with one allele more frequently used in the repertoire than the other allele [37, 38, 106]. C There exist differences in the distribution of alleles among different population groups. As a result, vaccines relying on a specific Ig allele may not elicit equally efficient responses across ethnicities [23, 71].
Broadly neutralising antibodies against HIV
For vaccination against HIV, broadly neutralising antibodies targeting the HIV envelope are considered particularly useful. The VRC01-class of antibodies targeting the CD4-binding site on HIV-1 glycoprotein 120 [56] (gp120) are among the broadest neutralising antibodies [57, 58]. These antibodies share common features including the highly conserved use of the IGHV1-2 gene paired with a light chain with a short complementarity determining region 3 of 5 amino acids [58, 59]. The light chain of VRC01 antibodies is frequently encoded by kappa genes, although some combinations with lambda have also been reported [58, 60]. Structural studies revealed that Trp50, Asn58, Arg71 and Trp100B in the IGHV1-2*02-encoded heavy chain form contact with conserved residues of the gp120 epitope via hydrogen bonds (Trp50-Asn280gp120; Asn58-Arg456gp120; Trp100B-Asn279gp120) and salt bridges (Arg71-Asp368gp120) [61, 62].
Due to differences between IGHV1-2 alleles, the ability to produce VRC01-class BCR depends on the alleles in an individual’s genotype [23, 60]. Allele IGHV1-2*05 was found incompatible with producing VRC01-class BCR, likely due to a difference in the amino-acid residue at position 50, where IGHV1-2*05 contains Arg50 instead of Trp50 [23, 60]. In a study by Lee and colleagues, the authors showed that although IGHV1-2*04 contains the critical residue Trp50, individuals heterozygous for IGHV1-2*04 had a lower amount of VRC01-precursor naive B cells [23]. The reason for this is unclear because the only difference between alleles *02 and *04 is located in the framework region 3 at position 66 (Arg in *02 and Trp in *04). Since IGHV1-2 alleles that decrease or hamper the chance of producing VRC01-class antibodies are relatively common in the population [23], not all individuals are equally able to produce potent neutralising antibodies to target the HIV-1 gp120 epitope.
Antibodies against Plasmodium falciparum circumsporozoite protein
For vaccination against malaria, elicitation of antibodies to the Plasmodium falciparum circumsporozoite protein (PfCSP) appears to provide protection in animal models [63]. These antibodies, which target a linear repeat region of the PfCSP antigen, show a striking bias in usage of IGHV3-33/IGKV1-5 both when elicited in malaria-exposed individuals [64] or in malaria-vaccinated individuals [65,66,67]. A key residue for recognition of the linear PfCSP repeat epitope, as also demonstrated by X-ray crystallography, is the germline-encoded tryptophan (Trp) at position 52 in the CDRH2 [65]. Genes that are highly similar to IGHV3-33, namely IGHV3-30, IGHV3-30-3 and IGHV3-30-5; differ at position 52, where their sequence encodes serine (Ser) instead of Trp. On mutating high-affinity antibodies to encode Ser52 instead of Trp52, the reactivity of such mutants to PfCSP was reduced. Interestingly, the study of malaria-exposed individuals of Tanzania revealed a putative novel variant of IGHV3-30 encoding Trp52 instead of Ser52 [64].
Antibody response against the seasonal flu vaccine
Exploration of antibody repertoires in individuals vaccinated against seasonal flu has revealed a stereotyped response against the hemagglutinin stem of the influenza virus. Neutralising antibodies against the influenza A hemagglutinin stem have a biased usage of IGHV1-69 in their heavy chain [68, 69]. Structural and site-directed mutagenesis studies have shown that a hydrophobic residue at position 53 and phenylalanine (Phe) at position 54 in IGHV1-69 are critical for binding [70]. Of note, amino-acid position 54 in IGHV1-69 is polymorphic containing either Phe or leucine (Leu). In a study of antibody repertoires in individuals vaccinated with seasonal flu vaccine individuals, it was observed that individuals who carried only Leu54 variants in their genome did not make a proper response. Interestingly, there was a difference in the frequency of these variants among ethnicities. The Leu54 variants were more prevalent among Europeans and nearly absent among Africans [71].
Antibody neutralisation of virulence factors of commensal bacteria
Staphylococcus aureus (S.aureus) is commonly found on the skin and in the upper respiratory tract of healthy individuals, but it can also become an opportunistic pathogen. The bacteria require iron to become pathogenic, and S.aureus obtains this critical factor by using haem iron from the host [72]. This process involves an iron surface determinant (Isd) system, particularly the IsdB surface receptor, which interacts with the haem-binding part of human haemoglobin [73]. By studying the antibody response against IsdB in healthy individuals, it was observed that antibodies that bind two domains of IsdB (NEAT1 and NEAT2) have a biased V gene usage [74]. NEAT1 was mostly neutralised by antibodies utilising germline-encoded IGHV4-39, and NEAT2 was neutralised by antibodies utilising the IGHV1-69 gene. Interestingly, IGHV1-69 alleles that encoded Arg at position 50 instead of Gly or Ala led to complete abolishment of the binding, thus losing the capacity for neutralisation. Individuals with different germline variants of IGHV1-69 could therefore have different susceptibility to infection by S.aureus.
Convergent antibodies in coeliac disease
Coeliac disease is caused by a harmful immune response to cereal gluten proteins. Unlike other autoimmune diseases with strong HLA associations, the antigens recognised by disease-specific T cells and B cells are known [75]. Coeliac patients have CD4 + T cells that recognise deamidated gluten peptides in the context of disease-associated HLA-DQ allotypes. In addition, these patients have B cells/plasma cells, which are specific to the autoantigen transglutaminase 2 and deamidated gluten peptides. Both types of antibodies have a biased usage of VH and VL pairs that is observed across individuals. Transglutaminase 2 specific plasma cells present in the coeliac disease gut lesion have a particularly prominent bias for the IGHV5-51:IGKV1-5 pair [76,77,78], but IGHV3-48:IGLV5-45 and IGHV4-34:IGKV1-39 are also frequently used [78]. The VH:VL usage of BCR/antibody depends on the epitope of transglutaminase 2 that is being recognised [79]. Similarly to transglutaminase specific cells, plasma cells with BCR specific to deamidated gluten peptides utilise stereotypical pairings of IGHV3-15:IGKV4-1; IGHV3-23:IGLV4-69 and IGHV3-74:IGKV4-1 [80, 81].
An Arg residue at position 55 was found to be critical for binding of the stereotyped IGHV3-15:IGKV4-1 antibody to deamidated gluten peptides [82]. This residue was also critical for binding in IGHV3-74:IGKV4-1 antibodies [81]. Interestingly, one plasma cell-specific for deamidated gluten peptide isolated from the lesion of a coeliac disease patient carried the IGHV4-4:IGKV4-1 pair, and its heavy chain was encoded by the IGHV4-4*07 allele. This allele carries Arg at position 55 in contrast to the IGHV4-4*01 allele that carries Glu. Upon mutating the antibody to carry Glu at position 55, the antibody binding was lost [81]. This finding of one antibody from one patient does not say much about the role of allele IGHV4-4*07 in coeliac disease susceptibility, but it demonstrates that polymorphic variation in Ig genes influences the antibody response to deamidated gluten peptides. Further studies are required to understand how Ig polymorphisms shape the B-cell response to gluten in coeliac disease.
Changes in affinity due to Fab glycosylation at germline-encoded residues
Ig molecules, like other proteins, can be subject to post-translational modifications, such as N-/O-linked glycosylation [83,84,85,86] or tyrosine sulfation [87]. Glycosylation is probably the most widely studied post-translational modification of antibodies, and it can occur both at the variable domain (antigen-binding fragment, Fab) or the constant region (crystalisable fragment, Fc). N-glycosylation usually occurs at specific motifs called sequons that are made of Asn-X-Ser/Thr, where asparagine (Asn) becomes glycosylated. The residues that are targeted for O-glycosylation are serine (Ser) or threonine (Thr) [88]. Modification of an amino-acid residue within the antigen-binding domain (Fab) of an antibody or a BCR can have an effect on its binding properties (Fig. 3D). Glycosylation status can be altered in response to various stimuli including infection, aging, smoking, etc [89].
The number of residues in the Fab domain that can be glycosylated depends on the germline gene used to make that Ig molecule since the variable region can be coded by many different genes. Glycosylation of the variable region is more frequent for antibodies encoded by the IGHV4 family and occurs less frequently in those encoded by IGHV1 or IGHV3 family [90]. The IGHV4-34*01 allele, in particular, contains a germline-encoded Asn-X-Ser/Thr sequon in its CDR2 region, which promotes N-linked glycosylation. Other genes with germline-encoded sequon include for example IGHV1-8 or IGHV5-10-1 [90]. Additional sequons within the variable region can be created via somatic hypermutation [86].
Glycosylation in the V region might affect the antigen-binding capacity of antibodies by either increasing [84, 85] or reducing their binding capacity [83]. In some cases, N-linked glycosylation of the variable domain can decrease the self-reactivity of an antibody [83]. A study conducted in mice has shown that an antibody produced by self-reactive B cells had reduced capacity to bind its self-antigen after undergoing a mutation of Ser to Asn at position 52; a change that can enable N-linked glycosylation [83]. When the Asn52 antibody was expressed in bacteria, where glycosylation does not occur, the self-reactivity of this antibody was restored, suggesting an impact of glycosylation on autoantigen binding. This study highlights the important role of post-translational modifications occurring in the Ig variable domain.
Constant region function and germline gene variation
While the interaction of an antigen occurs at the Ig variable region, the constant region of an Ig molecule is responsible for interaction with other components of the immune system [91]. The Ig constant region determines the isotype and subclass of an Ig molecule, and it can be recognised and bound by various Fc receptors (FcR), through which antibodies can exert their effector functions [92, 93]. Binding of the constant region to FcR or the polymeric Ig receptor (pIgR) is important for transport across epithelial surfaces [94]. Antibody function may be further modified by post-translational modification. Glycosylation of a conserved Asn residue at position 279 in the constant region [95] by different sugar moieties can be used to alter the function of an antibody, as is often seen during infection [91].
Antibody effector functions can be also influenced by Ig allotypes [96], which are polymorphic variations identified in the constant regions of immunoglobulins. Allotypes are described for IgG1, IgG2, IgG3, IgA2, and Ig kappa. Due to linkage of the human Ig heavy constant genes (ordered IGHG3, IGHG1, IGHA1, IGHG2, IGHG4, IGHE and IGHA2) Gm-Am haplotypes can be defined [97]. Differences in Ig allotypes seem to affect binding to Fc receptors and consequently, this can have a further effect on antibody-mediated immune responses [98, 99]. Allotypes may also play a role in the immune response to infections and in autoimmunity. Exploration of frequency of IgG1 allotypes in human cytomegalovirus (HCMV) infection revealed that individuals with allotype G1m17 had higher levels of HCMV-specific IgG1 antibodies as well as higher amounts of total HCMV-specific Ig when compared to individuals homozygous for the G1m3 allotype [100, 101]. Similarly, subclass composition of IgG antibodies against bacterial antigens was also found to correlate with different allotypes [102]. In multiple sclerosis, G1m1 allotype was dominant among intrathecal antibody-producing B cells of G1m1/G1m3 heterozygous patients, and these cells also displayed preferential usage of the IGHV4 paired with IGKV1 [103]. The link of IgG constant region polymorphisms to stereotyped V gene usage could possibly reflect recognition of particular antigen by disease-related B cells in multiple sclerosis.
The constant regions of different Ig subclasses are encoded by different genes and alleles. It is important to note that the same allotype, characterised by a defined amino-acid residue at a specific position, can be encoded by different Ig constant alleles. Currently, there is a lack of studies describing the germline variation of the constant Ig genes. A relatively recent study from Brazil identified 28 novel IGHG alleles among 357 individuals from diverse population groups, suggesting there is a quite extensive germline variation in the constant that remains unexplored [104]. Some of the identified polymorphisms represented amino-acid changes, although the majority were synonymous mutations. Polymorphisms affecting residues important for glycosylation or FcR binding could have potential implications on the function of the immune system.
Effect of non-coding polymorphisms on the Ig repertoire
In addition to coding polymorphisms, the expression levels of different alleles in heterozygous individuals might also play a role in the ability to develop a sufficient immune response. Different expression levels of Ig V alleles in heterozygous individuals have been reported in multiple studies (Fig. 4B) [23, 37, 38], but the reason for such uneven expression remains unclear. It is likely that non-coding regions might play a role in the regulation of Ig expression, but this has been very little explored [105]. Although the non-coding regions have received slightly more attention in the past years [37, 106], polymorphisms in these regions remain poorly characterised.
Promoters
Each Ig V gene has its own promoter that regulates its transcription. Ig promoters are composed of conserved elements that include, but are not limited to, a TATA-box, an octamer/decamer, and various other conserved motifs (Fig. 5) [107, 108] Elements of the Ig promoter serve as binding sites for transcription factors that regulate the expression of various genes [109]. Promoters of the heavy, kappa, and lambda V genes are quite different from each other, and there are also differences among the different V gene families within the same Ig locus. An interesting feature of some Ig promoters is their directionality. The IGHV6-1 promoter, which has a TATA-box upstream and downstream of the octamer, can function in both directions [110]. Two putative TATA-boxes were also observed in the promoter of IGHV5-51 and IGHV3 genes by analysing sequences listed in the IMGT/GENE-DB [20, 37]. The functional significance of this phenomenon and its possible impact on antibody production is not clear.

Each V gene has a promoter that contains various conserved elements, including the TATA-box. The transcription initiation site is located a few nucleotides downstream of the TATA-box and it is followed by the 5′ untranslated region (5′UTR). The leader sequence serves as a signalling sequence for the translated peptide and is later cleaved off. It is encoded by L-PART1 and L-PART2, which are separated by an intron in the germline DNA. The start of L-PART1 corresponds with the start codon for translation (ATG). The V-REGION is the part of a V gene that is translated and directly contributes to the Ig paratope. At the 3′ end of a V gene lies the V recombination signal sequence (V-RS), which, as its name suggests, is essential for the recombination of a V gene with a D or J gene.
5′UTRs and leader sequences
Germline polymorphisms in the 5′UTR and the leader sequences have been largely overlooked until recently [37]. Since high-throughput sequencing platforms have limitations in terms of the length of sequences, many library preparation protocols produce amplicons that only capture the V-REGION and the junction. Nevertheless, the 5′UTRs and leader sequences of Ig V genes also contain polymorphisms [37, 106]. The role of such upstream polymorphisms is unknown for now, but mutating the sequence downstream of the transcription initiation site (corresponding to the 5′UTR) was shown to decrease transcription efficiency [111]. The leader sequence is located downstream of the 5′UTR (Fig. 5) and is encoded by L-PART1 and L-PART2, which are spliced together in a functional Ig transcript. The leader sequence has an important role in in vitro antibody production since it is translated and serves as a peptide signal sequence before being cleaved off. Experimental studies aiming to optimise antibody production have shown that differences in the leader sequences had an influence on the efficiency of antibody expression in vitro [112, 113].
Intron
In the germline DNA, the leader sequences of an Ig V gene are separated by an intron, which is spliced out in the mature mRNA (Fig. 5). Since the intron is absent in Ig repertoire studies, germline variation in this region is unexplored. Introns might also play a role in shaping the Ig repertoire. Different splicing patterns of introns in kappa genes using cryptic splice acceptor sites were found to alter the mRNA levels of the same genes [114]. Recently, partial intron retention was observed in the Ig light chain transcripts from naive BCR repertoires, particularly in genes with low expression levels [106]. Intron retention in other genes often introduces a premature stop codon in the transcript, which makes the mRNA more susceptible to degradation. It is possible that alternative splicing could be a way to regulate the expression of light chain V genes.
Recombination signal sequence
The V recombination signal sequence (V-RS, also sometimes abbreviated as RSS) is crucial for the recombination of a V gene with a D or J gene, and producing a functional Ig transcript. Polymorphisms in this sequence can affect the recombination efficiency, as was observed in the case of IGKV2D-29 (previously known as A2 or VA2). Antibodies produced in response to vaccination by Haemophilus influenzae type B polysaccharide (Hib PS) were observed to preferentially utilise the IGKV2D-29 light chain gene [115]. It was demonstrated that a polymorphism at the V-RS sequence of this gene correlates with lower recombination frequency [116, 117]. Navajos have a 10-fold higher incidence of Haemophilus influenzae type B infection compared with control populations, and as this polymorphism is frequent among Navajos, it was hypothesised that this may be the underlying reason for higher disease susceptibility in this population [116, 117].
Despite the importance of the V-RS region, there has been very little research on V-RS polymorphisms in the past few years. Since the V-RS is spliced out and no longer present in the rearranged Ig mRNA, it is not possible to study this region from Ig repertoire data. Therefore, little is known about the variation in V-RS and the functional effects of polymorphisms.
Enhancers and their role in regulating Ig expression
The IgH loci contain three main enhancer clusters near the C genes (Fig. 1). These can be bound by important transcription factors such as NF-kB [118]. There is a lack of studies describing polymorphisms in these enhancers and their potential effect. Existing studies on Ig enhancer polymorphisms were done on a very small scale and there are conflicting conclusions about their potential role in diseases [119, 120]. However, mutations in Ig heavy enhancer have been previously implicated in B-cell lymphomas [121]. The 3′ regulatory region and the switch regions of human and murine Ig loci have been shown to contain hotspots for oestrogen receptor binding [122, 123]. A study in mice that investigated the role of Ig enhancers found differences between male and female BCR repertoire [123]. These in-vitro experiments showed that the addition of oestrogen to purified mouse splenic B cells increased the amount of Ig heavy chain transcripts. Additionally, the presence of oestrogen receptors bound to the switch regions was identified by chromatin immunoprecipitation. The authors suggested that such hormonal regulation could affect the expression of different Ig isotypes. These findings could also provide a potential explanation for the differences in Ig repertoires between males and females, and perhaps it could shed light on factors involved in the increased frequency of some autoimmune diseases among females. It is however unclear how frequent polymorphisms in this region of the locus are and what the functional effect of such polymorphisms would be.
The impact of BCR genes on susceptibility for autoimmune disease: The jury is still out
Knowing that MHC genes with their central role in adaptive immunity by far are the chief determinant for autoimmunity, it is conceivable that other genes feeding into adaptive immune pathways are susceptibility genes for autoimmune diseases. For diseases with MHC associations, primary associations with MHC class I allotypes are seen with seronegative diseases while primary associations with MHC class II allotypes are seen in diseases with autoantibodies or where there is evidence for the involvement of B cells, such as multiple sclerosis [124]. As twin studies have demonstrated that a person’s BCR repertoire is affected by genes [125, 126], finding BCR genes among susceptibility genes for seropositive autoimmune diseases would be expected. In fact, very few studies have reported finding in keeping with this notion. A study of the Oceanic population reported that the IGHV4-61*02 allele is associated with a higher risk of rheumatic heart disease [127]. Further, a susceptibility locus for Kawasaki disease was identified among Ig genes [128], and in a later study, researchers identified a particular SNP within the IGHV3-66 gene as a risk factor for this disease [129]. However, the scarcity of results does not imply that BCR genes are not susceptibility genes. The coverage of genetic markers in the chips used for typing in genome-wide association analysis (GWAS) for the BCR (i.e., IGH, IGK and IGL) as well as TCR (TRA and TRB) loci is scarce and much less than that for MHC (HLA) (Table 3). Hence, these loci have yet to be scrutinised in extensive association analysis. This will only become possible when the genomic structures, knowledge of allelic variation, and knowledge of linkage disequilibrium in these loci have become more complete.
Concluding remarks and future perspectives
Genomic variation in the Ig loci shapes a person’s Ig repertoire [48] and it can have a profound effect on the ability to develop a sufficient immune response to a specific antigen [130], as described above. Due to the large complexity of the Ig loci, the options to study these genomic regions have been somehow limited. High-throughput Ig repertoire studies have been central in characterising germline variants of Ig V genes (and recently also IGHD [131] and TCRβ [132] genes), however, such studies have their limitations. Since repertoire studies utilise mRNA (transcribed to cDNA) and not genomic DNA, they only provide information about the coding parts of the Ig loci. Yet, promoters, introns, V-RS and upstream sequences of Ig V genes also seem to play a role in regulating Ig expression. There is a need for studies that would characterise genetic variation in the non-coding regions of Ig loci and explore their functional implications. This could potentially help explain the uneven expression of Ig alleles in heterozygous individuals. The main issue in characterising non-coding genetic variation in Ig loci has been a lack of suitable high-throughput methods that would allow amplification and high-throughput sequencing of larger sections of the Ig loci. Recently, a novel method has been published, which utilises long-read high-throughput sequencing to explore germline Ig loci [47, 133]. This will hopefully enable researchers to study genomic Ig variation more effectively.
Another limitation of repertoire studies is their technical aspect, particularly the accuracy of sequencing methods and the software used for analysis. Although high-throughput methods are widely used for characterisation of polymorphisms in the genome, each method has its own biases and limitations that can affect the data interpretation [134,135,136,137]. Currently, there is a lack of benchmarking and proof-of-concept studies that would demonstrate the accuracy of polymorphism inference in a synthetic dataset containing defined sequences. Nevertheless, Ig repertoire studies remain a valuable tool that has played a key role in characterising genomic variation in Ig loci.
We expect that many more examples of functional impact of Ig polymorphisms will be described in the future, particularly in studies focusing on defined antigenic epitopes where there is usage of convergent VH/VL. In the relatively near future, we should also learn whether Ig polymorphisms have an impact on risk and development of autoimmune disease.
References
Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–81.
Safonova Y, Pevzner PA. V(DD)J recombination is an important and evolutionarily conserved mechanism for generating antibodies with unusually long CDR3s. Genome Res. 2020;30:1547–58.
McBride OW, Battey J, Hollis GF, Swan DC, Siebenlist U, Leder P. Localization of human variable and constant region immunoglobulin heavy chain genes on subtelomeric band q32 of chromosome 14. Nucleic Acids Res. 1982;10:8155–70.
McBride OW, Heiter PA, Hollis GF, Swan D, Otey MC, Leder P. Chromosomal location of human kappa and lambda immunoglobulin light chain constant region genes. J Exp Med. 1982;155:1480–90.
Lötscher E, Zimmer FJ, Klopstock T, Grzeschik KH, Jaenichen R, Straubinger B, et al. Localization, analysis and evolution of transposed human immunoglobulin V kappa genes. Gene. 1988;69:215–23.
Lefranc M-P, Lefranc G. The Immunoglobulin FactsBook. Academic Press; 2001.
Tan J, Pieper K, Piccoli L, Abdi A, Perez MF, Geiger R, et al. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature. 2016;529:105–9.
Pieper K, Tan J, Piccoli L, Foglierini M, Barbieri S, Chen Y, et al. Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature. 2017;548:597–601.
Koning MT, Vletter EM, Rademaker R, Vergroesen RD, Trollmann IJM, Parren P, et al. Templated insertions at VD and DJ junctions create unique B-cell receptors in the healthy B-cell repertoire. Eur J Immunol. 2020;50:2099–101.
Chen Y, Xu K, Piccoli L, Foglierini M, Tan J, Jin W, et al. Structural basis of malaria RIFIN binding by LILRB1-containing antibodies. Nature. 2021;592:639–643.
Matsuda F, Ishii K, Bourvagnet P, Kuma K-I, Hayashida H, Miyata T, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med. 1998;188:2151–62.
Pallarès N, Lefebvre S, Contet V, Matsuda F, Lefranc M-P. The human immunoglobulin heavy variable genes. Exp Clin Immunogenet. 1999;16:36–60.
Gadala-Maria D, Yaari G, Uduman M, Kleinstein SH. Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles. Proc Natl Acad Sci USA. 2015;112:E862–E870.
Gadala-Maria D, Gidoni M, Marquez S, Vander Heiden JA, Kos JT, Watson CT, et al. Identification of subject-specific immunoglobulin alleles from expressed repertoire sequencing data. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.00129.
Corcoran MM, Phad GE, Bernat NV, Stahl-Hennig C, Sumida N, Persson MAA, et al. Production of individualized V gene databases reveals high levels of immunoglobulin genetic diversity. Nat Commun. 2016;7:13642.
Ralph DK, Matsen 4th FA. Per-sample immunoglobulin germline inference from B cell receptor deep sequencing data. PLoS Comput Biol. 2019;15:e1007133
Vázquez Bernat N, Corcoran M, Nowak I, Kaduk M, Castro Dopico X, Narang S, et al. Rhesus and cynomolgus macaque immunoglobulin heavy-chain genotyping yields comprehensive databases of germline VDJ alleles. Immunity. 2021;54:355–366.e4.
Lees W, Busse CE, Corcoran M, Ohlin M, Scheepers C, Matsen FA, et al. OGRDB: a reference database of inferred immune receptor genes. Nucleic Acids Res. 2020;48:D964–D970.
IgPdb. The IgPdb Database. https://cgi.cse.unsw.edu.au/~ihmmune/IgPdb/information.php.
Giudicelli V, Chaume D, Lefranc M-P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33:D256–D261.
Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu Rev Immunol. 2003. https://doi.org/10.1146/annurev.iy.08.040190.004013.
Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. 2015;15:185–9.
Lee JH, Toy L, Kos JT, Safonova Y, Schief WR, Watson CT, et al. Vaccine genetics of IGHV1-2 VRC01-class broadly neutralizing antibody precursor naïve human B cells. bioRxiv. 2021. 10.1101/2021.03.01.433480.
Collins AM, Wang Y, Roskin KM, Marquis CP, Jackson KJL. The mouse antibody heavy chain repertoire is germline-focused and highly variable between inbred strains. Philos Trans R Soc Lond B Biol Sci 2015;370:20140236. https://doi.org/10.1098/rstb.2014.0236.
Watson CT, Kos JT, Gibson WS, Newman L, Deikus G, Busse CE, et al. A comparison of immunoglobulin IGHV, IGHD and IGHJ genes in wild-derived and classical inbred mouse strains. Immunol Cell Biol. 2019;97:888–901.
Johnston CM, Wood AL, Bolland DJ, Corcoran AE. Complete sequence assembly and characterization of the C57BL/6 mouse Ig heavy chain V region. J Immunol. 2006;176:4221–34.
Busse CE, Jackson KJL, Watson CT, Collins AM. A proposed new nomenclature for the immunoglobulin genes of Mus musculus. Front Immunol. 2019;10:2961.
Wysocki LJ, Gridley T, Huang S, Grandea AG 3rd, Gefter ML. Single germline VH and V kappa genes encode predominating antibody variable regions elicited in strain A mice by immunization with p-azophenylarsonate. J Exp Med. 1987;166:1–11.
Parhami-Seren B, Kussie PH, Strong RK, Margolies MN. Conservation of binding site geometry among p-azophenylarsonate-specific antibodies. J Immunol. 1993;150:1829–37.
Pape KA, Maul RW, Dileepan T, Paustian AS, Gearhart PJ, Jenkins MK. Naive B cells with high-avidity germline-encoded antigen receptors produce persistent IgM+ and transient IgG+ memory B cells. Immunity. 2018;48:1135–1143.e4.
Raposo B, Dobritzsch D, Ge C, Ekman D, Xu B, Lindh I, et al. Epitope-specific antibody response is controlled by immunoglobulin VH polymorphisms. J Exp Med. 2014;211:405–11.
Vencovský J, Zd’árský E, Moyes SP, Hajeer A, Ruzicková Š, Cimburek Z, et al. Polymorphism in the immunoglobulin VH gene V1‐69 affects susceptibility to rheumatoid arthritis in subjects lacking the HLA‐DRB1 shared epitope. Rheumatology. 2002;41:401–10.
Walter MA, Gibson WT, Ebers GC, Cox DW. Susceptibility to multiple sclerosis is associated with the proximal immunoglobulin heavy chain variable region. J Clin Invest. 1991;87:1266–73.
Hashimoto LL, Walter MA, Cox DW, Ebers GC. Immunoglobulin heavy chain variable region polymorphisms and multiple sclerosis susceptibility. J Neuroimmunol. 1993;44:77–83.
Wood NW, Sawcer SJ, Kellar-Wood HF, Holmans P, Clayton D, Robertson N, et al. Susceptibility to multiple sclerosis and the immunoglobulin heavy chain variable region. J Neurol. 1995;242:677–82.
Watson CT, Breden F. The immunoglobulin heavy chain locus: genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13:363–73.
Mikocziova I, Gidoni M, Lindeman I, Peres A, Snir O, Yaari G, et al. Polymorphisms in human immunoglobulin heavy chain variable genes and their upstream regions. Nucleic Acids Res. 2020;48:5499–510.
Gidoni M, Snir O, Peres A, Polak P, Lindeman I, Mikocziova I, et al. Mosaic deletion patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. Nat Commun. 2019;10:628.
Romo-González T, Morales-Montor J, Rodríguez-Dorantes M, Vargas-Madrazo E. Novel substitution polymorphisms of human immunoglobulin VH genes in Mexicans. Hum Immunol. 2005;66:731–9.
Scheepers C, Shrestha RK, Lambson BE, Jackson KJL, Wright IA, Naicker D, et al. Ability to develop broadly neutralizing HIV-1 antibodies is not restricted by the germline Ig gene repertoire. J Immunol. 2015;194:4371–8.
Boyd SD, Gaëta BA, Jackson KJ, Fire AZ, Marshall EL, Merker JD, et al. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J Immunol. 2010;184:6986–92.
Ohlin M, Scheepers C, Corcoran M, Lees WD, Busse CE, Bagnara D, et al. Inferred allelic variants of immunoglobulin receptor genes: a system for their evaluation, documentation, and naming. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.00435.
Vázquez Bernat N, Corcoran M, Hardt U, Kaduk M, Phad GE, Martin M, et al. High-quality library preparation for NGS-based immunoglobulin germline gene inference and repertoire expression analysis. Front Immunol. 2019;10:660.
Peng K, Safonova Y, Shugay M, Popejoy AB, Rodriguez OL, Breden F, et al. Diversity in immunogenomics: the value and the challenge. Nat Methods. 2021. https://doi.org/10.1038/s41592-021-01169-5.
Peres A, Gidoni M, Polak P, Yaari G. RAbHIT: R antibody haplotype inference tool. Bioinformatics. 2019;35:4840–2.
Watson CT, Steinberg KM, Huddleston J, Warren RL, Malig M, Schein J, et al. Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet. 2013;92:530–46.
Rodriguez OL, Gibson WS, Parks T, Emery M, Powell J, Strahl M, et al. A novel framework for characterizing genomic haplotype diversity in the human immunoglobulin heavy chain locus. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.02136.
Collins AM, Yaari G, Shepherd AJ, Lees W, Watson CT. Germline immunoglobulin genes: disease susceptibility genes hidden in plain sight? Curr Opin Syst Biol. 2020. https://doi.org/10.1016/j.coisb.2020.10.011.
Jackson KJL, Liu Y, Roskin KM, Glanville J, Hoh RA, Seo K, et al. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe. 2014;16:105–14.
Parameswaran P, Liu Y, Roskin KM, Jackson KKL, Dixit VP, Lee J-Y, et al. Convergent antibody signatures in human dengue. Cell Host Microbe. 2013;13:691–700.
Davis CW, Jackson KJL, McElroy AK, Halfmann P, Huang J, Chennareddy C, et al. Longitudinal analysis of the human B cell response to Ebola virus infection. Cell. 2019;177:1566–1582.e17.
Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584:437–42.
Nielsen SCA, Yang F, Jackson KJL, Hoh RA, Röltgen K, Jean GH, et al. Human B cell clonal expansion and convergent antibody responses to SARS-CoV-2. Cell Host Microbe. 2020;28:516–525.e5.
Dunand CJH, Wilson PC. Restricted, canonical, stereotyped and convergent immunoglobulin responses. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140238.
Akbar R, Robert PA, Pavlović M, Jeliazkov JR, Snapkov I, Slabodkin A, et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021;34:108856.
Zhou T, Georgiev I, Wu X, Yang Z-Y, Dai K, Finzi A, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–7.
Li Y, O’Dell S, Walker LM, Wu X, Guenaga J, Feng Y, et al. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J Virol. 2011;85:8954–67.
Zhou T, Lynch RM, Chen L, Acharya P, Wu X, Doria-Rose NA, et al. Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell. 2015;161:1280–92.
West AP Jr, Diskin R, Nussenzweig MC, Bjorkman PJ. Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc Natl Acad Sci USA. 2012;109:E2083–90.
Yacoob C, Pancera M, Vigdorovich V, Oliver BG, Glenn JA, Feng J, et al. Differences in allelic frequency and CDRH3 region limit the engagement of HIV Env immunogens by putative VRC01 neutralizing antibody precursors. Cell Rep. 2016;17:1560–70.
Scharf L, West AP Jr, Gao H, Lee T, Scheid JF, Nussenzweig MC, et al. Structural basis for HIV-1 gp120 recognition by a germ-line version of a broadly neutralizing antibody. Proc Natl Acad Sci USA. 2013;110:6049–54.
Scharf L, West AP, Sievers SA, Chen C, Jiang S, Gao H, et al. Structural basis for germline antibody recognition of HIV-1 immunogens. Elife. 2016. https://doi.org/10.7554/eLife.13783.
Foquet L, Hermsen CC, van Gemert G-J, Van Braeckel E, Weening KE, Sauerwein R, et al. Vaccine-induced monoclonal antibodies targeting circumsporozoite protein prevent Plasmodium falciparum infection. J Clin Invest. 2014;124:140–4.
Tan J, Sack BK, Oyen D, Zenklusen I, Piccoli L, Barbieri S, et al. A public antibody lineage that potently inhibits malaria infection through dual binding to the circumsporozoite protein. Nat Med. 2018;24:401–7.
Imkeller K, Scally SW, Bosch A, Martí GP, Costa G, Triller G, et al. Antihomotypic affinity maturation improves human B cell responses against a repetitive epitope. Science. 2018;360:1358–62.
Murugan R, Buchauer L, Triller G, Kreschel C, Costa G, Pidelaserra Martí G, et al. Clonal selection drives protective memory B cell responses in controlled human malaria infection. Sci Immunol. 2018. 10.1126/sciimmunol.aap8029.
Oyen D, Torres JL, Wille-Reece U, Ockenhouse CF, Emerling D, Glanville J, et al. Structural basis for antibody recognition of the NANP repeats in Plasmodium falciparum circumsporozoite protein. Proc Natl Acad Sci USA. 2017;114:E10438–E10445.
Wheatley AK, Whittle JRR, Lingwood D, Kanekiyo M, Yassine HM, Ma SS, et al. H5N1 vaccine-elicited memory B cells are genetically constrained by the IGHV locus in the recognition of a neutralizing epitope in the hemagglutinin stem. J Immunol. 2015;195:602–10.
Pappas L, Foglierini M, Piccoli L, Kallewaard NL, Turrini F, Silacci C, et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature. 2014;516:418–22.
Avnir Y, Tallarico AS, Zhu Q, Bennett AS, Connelly G, Sheehan J, et al. Molecular signatures of hemagglutinin stem-directed heterosubtypic human neutralizing antibodies against influenza A viruses. PLoS Pathog. 2014;10:e1004103.
Avnir Y, Watson CT, Glanville J, Peterson EC, Tallarico AS, Bennett AS, et al. IGHV1-69 polymorphism modulates anti-influenza antibody repertoires, correlates with IGHV utilization shifts and varies by ethnicity. Sci Rep. 2016;6:20842.
Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol. 2006;188:8421–9.
Bowden CFM, Chan ACK, Li EJW, Arrieta AL, Eltis LD, Murphy MEP. Structure-function analyses reveal key features in Staphylococcus aureus IsdB-associated unfolding of the heme-binding pocket of human hemoglobin. J Biol Chem. 2018;293:177–90.
Yeung YA, Foletti D, Deng X, Abdiche Y, Strop P, Glanville J, et al. Germline-encoded neutralization of a Staphylococcus aureus virulence factor by the human antibody repertoire. Nat Commun. 2016;7:13376.
Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol. 2013;13:294–302.
Marzari R, Sblattero D, Florian F, Tongiorgi E, Not T, Tommasini A, et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J Immunol. 2001;166:4170–6.
Di Niro R, Mesin L, Zheng N-Y, Stamnaes J, Morrissey M, Lee J-H, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012;18:441–5.
Roy B, Neumann RS, Snir O, Iversen R, Sandve GK, Lundin KEA, et al. High-throughput single-cell analysis of B cell receptor usage among autoantigen-specific plasma cells in celiac disease. J Immunol. 2017;199:782–91.
Iversen R, Di Niro R, Stamnaes J, Lundin KEA, Wilson PC, Sollid LM. Transglutaminase 2-specific autoantibodies in celiac disease target clustered, N-terminal epitopes not displayed on the surface of cells. J Immunol. 2013;190:5981–91.
Steinsbo O, Henry Dunand CJ, Huang M, Mesin L, Salgado-Ferrer M, Lundin KEA. et al. Restricted VH/VL usage and limited mutations in gluten-specific IgA of coeliac disease lesion plasma cells. Nat Commun. 2014;5:4041
Lindeman I, Zhou C, Eggesbø LM, Miao Z, Polak J, Lundin KEA, et al. Longevity, clonal relationship, and transcriptional program of celiac disease-specific plasma cells. J Exp Med. 2021. https://doi.org/10.1084/jem.20200852.
Snir O, Chen X, Gidoni M, Pré MF du, Zhao Y, Steinsbø Ø, et al. Stereotyped antibody responses target posttranslationally modified gluten in celiac disease. JCI Insight. 2017. https://doi.org/10.1172/jci.insight.93961.
Sabouri Z, Schofield P, Horikawa K, Spierings E, Kipling D, Randall KL, et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc Natl Acad Sci USA. 2014;111:E2567–75.
Wallick SC, Kabat EA, Morrison SL. Glycosylation of a VH residue of a monoclonal antibody against alpha (1-6) dextran increases its affinity for antigen. J Exp Med. 1988;168:1099–109.
Leibiger H, Wüstner D, Stigler RD, Marx U. Variable domain-linked oligosaccharides of a human monoclonal IgG: structure and influence on antigen binding. Biochem J. 1999;338(Pt 2):529–38.
Dunn-Walters D, Boursier L, Spencer J. Effect of somatic hypermutation on potential N-glycosylation sites in human immunoglobulin heavy chain variable regions. Mol Immunol. 2000;37:107–13.
Choe H, Li W, Wright PL, Vasilieva N, Venturi M, Huang C-C, et al. Tyrosine sulfation of human antibodies contributes to recognition of the CCR5 binding region of HIV-1 gp120. Cell. 2003;114:161–70.
de Haan N, Falck D, Wuhrer M. Monitoring of immunoglobulin N- and O-glycosylation in health and disease. Glycobiology. 2020;30:226–40.
Gudelj I, Lauc G, Pezer M. Immunoglobulin G glycosylation in aging and diseases. Cell Immunol. 2018;333:65–79.
van de Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, van Schie KA, Kruithof S, Berkowska MA, et al. Adaptive antibody diversification through N-linked glycosylation of the immunoglobulin variable region. Proc Natl Acad Sci USA. 2018;115:1901–6.
Irvine EB, Alter G. Understanding the role of antibody glycosylation through the lens of severe viral and bacterial diseases. Glycobiology. 2020;30:241–53.
Torres M, Casadevall A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 2008;29:91–97.
DiLillo DJ, Ravetch JV. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res. 2015;3:704–13.
Rojas R, Apodaca G. Immunoglobulin transport across polarized epithelial cells. Nat Rev Mol Cell Biol. 2002;3:944–55.
Huber R, Deisenhofer J, Colman PM, Matsushima M, Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature. 1976;264:415–20.
de Taeye SW, Bentlage AEH, Mebius MM, Meesters JI, Lissenberg-Thunnissen S, Falck D, et al. FcγR Binding and ADCC Activity of Human IgG Allotypes. Front Immunol. 2020;11:740.
Lefranc M-P, Lefranc G. Human Gm, Km, and Am allotypes and their molecular characterization: a remarkable demonstration of polymorphism. In: Immunogenetics. Totowa, NJ: Humana Press; 2012, p. 635–80.
Atherton A, Armour KL, Bell S, Minson AC, Clark MR. The herpes simplex virus type 1 Fc receptor discriminates between IgG1 allotypes. Eur J Immunol. 2000;30:2540–7.
Kratochvil S, McKay PF, Chung AW, Kent SJ, Gilmour J, Shattock RJ. Immunoglobulin G1 allotype influences antibody subclass distribution in response to HIV gp140 vaccination. Front Immunol. 2017;8:1883.
Pandey JP, Kistner-Griffin E, Radwan FF, Kaur N, Namboodiri AM, Black L, et al. Immunoglobulin genes influence the magnitude of humoral immunity to cytomegalovirus glycoprotein B. J Infect Dis. 2014;210:1823–6.
Simon B, Weseslindtner L, Görzer I, Pollak K, Jaksch P, Klepetko W, et al. Subclass-specific antibody responses to human cytomegalovirus in lung transplant recipients and their association with constant heavy immunoglobulin G chain polymorphism and virus replication. J Heart Lung Transpl. 2016;35:370–7.
Carson RT, McDonald DF, Kehoe MA, Calvert JE. Influence of Gm allotype on the IgG subclass response to streptococcal M protein and outer membrane proteins of Moraxella catarrhalis. Immunology. 1994;83:107–13.
Lindeman I, Polak J, Qiao S-W, Holmøy T, Høglund RA, Vartdal F, et al. Stereotyped B-cell responses are linked to IgG constant region polymorphisms in multiple sclerosis. bioRxiv. 2021. 2021.04.23.441098.
Calonga-Solís V, Malheiros D, Beltrame MH, Vargas L de B, Dourado RM, et al. Unveiling the diversity of immunoglobulin heavy constant gamma (IGHG) gene segments in Brazilian populations reveals 28 novel alleles and evidence of gene conversion and natural selection. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.01161.
Kenter AL, Watson CT, Spille J-H. Igh locus polymorphism may dictate topological chromatin conformation and V gene usage in the Ig repertoire. Front Immunol. 2021;12:1724.
Mikocziova I, Peres A, Gidoni M, Greiff V, Yaari G, Sollid LM. Alternative splice variants and germline polymorphisms in human immunoglobulin light chain genes. bioRxiv. 2021. 2021.02.05.429934.
Falkner FG, Zachau HG. Correct transcription of an immunoglobulin κ gene requires an upstream fragment containing conserved sequence elements. Nature. 1984;310:71–74.
Bemark M, Liberg D, Leanderson T. Conserved sequence elements in K promoters from mice and humans: implications for transcriptional regulation and repertoire expression. Immunogenetics. 1998;47:183–95.
Vázquez-Arreguín K, Tantin D. The Oct1 transcription factor and epithelial malignancies: Old protein learns new tricks. Biochim Biophys Acta. 2016;1859:792–804.
Sun Z, Kitchingman GR. Bidirectional transcription from the human immunoglobulin VH6 gene promoter. Nucleic Acids Res. 1994;22:861–8.
Pelletier MR, Hatada EN, Scholz G, Scheidereit C. Efficient transcription of an immunoglobulin κ promoter requires specific sequence elements overlapping with and downstream of the transcriptional start site. Nucleic Acids Res. 1997;25:3995–4003.
Haryadi R, Ho S, Kok YJ, Pu HX, Zheng L, Pereira NA, et al. Optimization of heavy chain and light chain signal peptides for high level expression of therapeutic antibodies in CHO cells. PLoS One. 2015;10:e0116878.
Gibson SJ, Bond NJ, Milne S, Lewis A, Sheriff A, Pettman G, et al. N-terminal or signal peptide sequence engineering prevents truncation of human monoclonal antibody light chains. Biotechnol Bioeng. 2017;114:1970–7.
Chou CL, Morrison SL. Intron sequences determine the expression of kappa light chain genes. Mol Immunol. 1994;31:99–107.
Lucas AH, Langley RJ, Granoff DM, Nahm MH, Kitamura MY, Scott MG. An idiotypic marker associated with a germ-line encoded kappa light chain variable region that predominates the vaccine-induced human antibody response to the Haemophilus influenzae b polysaccharide. J Clin Invest. 1991;88:1811–8.
Feeney AJ, Atkinson MJ, Cowan MJ, Escuro G, Lugo G. A defective Vkappa A2 allele in Navajos which may play a role in increased susceptibility to haemophilus influenzae type b disease. J Clin Invest. 1996;97:2277–82.
Nadel B, Tang A, Escuro G, Lugo G, Feeney AJ. Sequence of the spacer in the recombination signal sequence affects V(D)J rearrangement frequency and correlates with nonrandom Vκ usage In vivo. J Exp Med. 1998;187:1495–503.
Lefranc G, Lefranc MP. Regulation of the immunoglobulin gene transcription. Biochimie. 1990;72:7–17.
Frezza D, Giambra V, Cianci R, Fruscalzo A, Giufrè M, Cammarota G, et al. Increased frequency of the immunoglobulin enhancer HS1,2 allele 2 in coeliac disease. Scand J Gastroenterol. 2004;39:1083–7.
Liu J, Law RA, Koles PG, Saxe JC, Bottomley M, Sulentic CEW. Allelic frequencies of the hs1.2 enhancer within the immunoglobulin heavy chain region in Dayton, Ohio patients screened for celiac disease with duodenal biopsy. Dig Liver Dis. 2017;49:887–92.
Ghazzaui N, Issaoui H, Ferrad M, Carrion C, Cook-Moreau J, Denizot Y, et al. Eμ and 3’RR transcriptional enhancers of the IgH locus cooperate to promote c-myc-induced mature B-cell lymphomas. Blood Adv. 2020;4:28–39.
Hurwitz JL, Jones BG, Sealy RE, Xu B, Fan Y, Partridge JF, et al. Hotspots for hormone response elements in Sα switch regions of immunoglobulin heavy chain loci; how estrogen may influence class switch recombination (CSR) and IgA/IgG isotype expression by activated B cells. J Immunol. 2016;196:198.3–198.3.
Jones BG, Sealy RE, Penkert RR, Surman SL, Maul RW, Neale G, et al. Complex sex-biased antibody responses: estrogen receptors bind estrogen response elements centered within immunoglobulin heavy chain gene enhancers. Int Immunol. 2019;31:141–56.
Sollid LM, Pos W, Wucherpfennig KW. Molecular mechanisms for contribution of MHC molecules to autoimmune diseases. Curr Opin Immunol. 2014;31:24–30.
Glanville J, Kuo TC, von Büdingen H-C, Guey L, Berka J, Sundar PD, et al. Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc Natl Acad Sci USA. 2011;108:20066–71.
Rubelt F, Bolen CR, McGuire HM, Heiden JAV, Gadala-Maria D, Levin M, et al. Individual heritable differences result in unique cell lymphocyte receptor repertoires of naïve and antigen-experienced cells. Nat Commun. 2016;7:11112.
Parks T, Mirabel MM, Kado J, Auckland K, Nowak J, Rautanen A, et al. Association between a common immunoglobulin heavy chain allele and rheumatic heart disease risk in Oceania. Nat Commun. 2017;8:14946.
Tsai F-J, Lee Y-C, Chang J-S, Huang L-M, Huang F-Y, Chiu N-C, et al. Identification of novel susceptibility loci for Kawasaki disease in a Han Chinese population by a genome-wide association study. PLoS ONE. 2011;6:e16853.
Johnson TA, Mashimo Y, Wu J-Y, Yoon D, Hata A, Kubo M, et al. Association of an IGHV3-66 gene variant with Kawasaki disease. J Hum Genet. 2020: 1–15.
Slabodkin A, Chernigovskaya M, Mikocziova I, Akbar R, Scheffer L, Pavlović M, et al. Individualized VDJ recombination predisposes the available Ig sequence space. bioRxiv. 2021. https://doi.org/10.1101/2021.04.19.440409.
Bhardwaj V, Franceschetti M, Rao R, Pevzner PA, Safonova Y. Automated analysis of immunosequencing datasets reveals novel immunoglobulin D genes across diverse species. PLoS Comput Biol. 2020;16:e1007837.
Omer A, Peres A, Rodriguez OL, Watson CT, Lees W, Polak P, et al. T cell Receptor Beta (TRB) germline variability is revealed by inference from repertoire data. bioRxiv. 2021. https://doi.org/10.1101/2021.05.17.444409.
Ford M, Haghshenas E, Watson CT, Sahinalp SC. Genotyping and copy number analysis of immunoglobin heavy chain variable genes using long reads. iScience. 2020;23:100883.
Smakaj E, Babrak L, Ohlin M, Shugay M, Briney B, Tosoni D, et al. Benchmarking immunoinformatic tools for the analysis of antibody repertoire sequences. Bioinformatics. 2020;36:1731–9.
Khan TA, Friedensohn S, Gorter de Vries AR, Straszewski J, Ruscheweyh H-J, Reddy ST. Accurate and predictive antibody repertoire profiling by molecular amplification fingerprinting. Sci Adv. 2016;2:e1501371.
Menzel U, Greiff V, Khan TA, Haessler U, Hellmann I, Friedensohn S, et al. Comprehensive evaluation and optimization of amplicon library preparation methods for high-throughput antibody sequencing. PLoS ONE. 2014;9:e96727.
Barennes P, Quiniou V, Shugay M, Egorov ES, Davydov AN, Chudakov DM, et al. Benchmarking of T cell receptor repertoire profiling methods reveals large systematic biases. Nat Biotechnol. 2021;39:236–45.
Acknowledgements
We thank Benedicte A. Lie and Marte K. Viken (University of Oslo) for providing the numbers displayed in Table 3. We would also like to thank Frode Vartdal (University of Oslo) for a helpful discussion about Ig allotypes. All figures were created with BioRender.com.
Funding
We acknowledge generous support by UiO World-Leading Research Community to VG and LMS, UiO:LifeScience Convergence Environment Immunolingo to VG, EU Horizon 2020 iReceptorplus (#825821) to VG and LMS, a Research Council of Norway FRIPRO project (#300740) to VG, South-Eastern Norway Regional Health Authority (#201611) to LMS and Stiftelsen Kristian Gerhard Jebsen (SKGJ-MED-017, K.G. Jebsen Coeliac Disease Research Centre) to LMS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPETING INTERESTS
VG declares advisory board positions in aiNET GmbH and Enpicom B.V. VG is a consultant for Roche/Genentech.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mikocziova, I., Greiff, V. & Sollid, L.M. Immunoglobulin germline gene variation and its impact on human disease. Genes Immun 22, 205–217 (2021). https://doi.org/10.1038/s41435-021-00145-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41435-021-00145-5
This article is cited by
-
Genetic background of juniper (Juniperus spp.) consumption predicted by fecal near-infrared spectroscopy in divergently selected goats raised in harsh rangeland environments
BMC Genomics (2024)
-
Adaptive immune receptor repertoire analysis
Nature Reviews Methods Primers (2024)
-
Genetic variation in the immunoglobulin heavy chain locus shapes the human antibody repertoire
Nature Communications (2023)
-
The dynamic interface of genetics and immunity: toward future horizons in health & disease
Genes & Immunity (2023)
-
Enhancer-instructed epigenetic landscape and chromatin compartmentalization dictate a primary antibody repertoire protective against specific bacterial pathogens
Nature Immunology (2023)
