
Abstract
Due to the indubitable role of vitamins in maintaining human health, a lot of attention has been paid to the methods and analytical procedures of their determination. Voltammetric methods are of particular interest as they do not involve complex sample preparation, however, close values of the redox potential of some vitamins may result in peak overlapping, hindering the quantitative analysis. This paper addresses the separation of overlapped reduction peak of vitamins B1 and B3 by means of Continuous Wavelet Transform (with appropriately selected mother wavelets) and differentiation of the recorded voltammograms. These numerical algorithms allowed to obtained linear, single-variable calibration functions that comply with the applied in analytical chemistry criteria regarding the correlation coefficients and the limit of detection and quantification. Their relatively simple implementation does not include any manual interpretation step; thus, these algorithms provide impartial and reproducible results. The proposed methodology has been applied in the determination of vitamins B1 and B3 in dietary supplements, providing results consistent with the declaration of the manufacturer.
Graphic abstract

Similar content being viewed by others


Introduction
The term vitamins was introduced in 1912 to encompass the heterogenic group of organic compounds, which are essential for the unerring functioning of the body, but are not synthesized inside the organisms or produced in the amount not sufficient to cover the basic need [1]. For these reasons, they have to be supplemented from external sources, including food and pharmaceuticals. Since both the excess of a particular vitamin (called hypervitaminosis) and its deficiency (hypovitaminosis) or absence (avitaminosis) may result in serious health problems, there is a need for quantitative estimation of vitamins content in their known sources [2]. Particular attention should be paid to dietary supplements, towards which the requirements regarding their chemical composition are not strict enough, thus the declared amounts are often not accurate.
Due to the relatively low costs of the apparatus, short measurement time, and straightforward sample preparation [3], voltammetric methods have been developed for the determination of vitamins in a variety of matrices [2, 4, 5]. Nevertheless, an often encounter situation in electro-analysis is the overlapping of signals resulting from the similar formal potentials of the species present in the analyzed sample [5]. This is notably the case for the multivitamin formulations.
Signal processing allows for extracting useful information from digital and analog signals by means of mathematical methods. The current–potential curve, obtained in every voltammetric experiment, is characterized by high complexity caused by the multiplicity of processes undergoing in the studied system. Therefore, many algorithms for filtration, smoothing, background correction, and separation of the overlapped signals have been developed [6]. The latter group involves the Continuous Wavelet Transform (CWT) and differentiation. Waves represent the distortion of the space and are described by a sinusoid of constant amplitude and frequency or as their superposition. The identification of individual harmonic components is obtained by means of Fourier transform. In comparison, wavelets represent short-time distortion, localized in time, and the application of wavelet analysis leads to the decomposition of the signal into the sum of the scaled and shifted version of the template, known as the mother wavelet Ψ. The modified version Ψab is expressed by Eq. (1) [7]:
where a corresponds to the scale, and b to the shift of Ψ in the time domain. The properly defined mother wavelet has the average value of 0, and the squared norm (the sum of the squared values for each point) equal 1. In CWT, wavelet coefficient CWTx(a, b) for a given scale a and shift b is calculated according to the expression:
where x(t) denotes the transformed signal, and \({\Psi }_{ab}^{*}\) is a conjugate function to Ψab. The obtained coefficients are plotted against the corresponding shift for different scale values. Such graph is known as the scalogram. CWT is a linear transform, meaning that an m-fold increase in the signal causes the CWTx to increase m times (for the same scale). This feature allows performing the calibration with the use of CWTx values in the place of the analytical signal. CWT provides the representation of the signal in the time–frequency domain (parameter b and a, respectively), and thus visualization of the low- and high-frequency signal components. Nonetheless, the CWT performed on the signal x(t), being a function of a single variable, results in the two-variable function CWTx(a, b), so this transform is highly redundant [8]. For the sampled, digital data, the discrete form of Eq. (2) has been proposed [8, 9]:
in which N represents the signal length, 1/Ts denotes the sampling frequency, and the shift b and time t have been expressed as the multiplicity of the sampling period Ts. In voltammetric experiments, the shift parameter can be associated with the applied potential.
Differentiation has been widely applied in spectroscopic measurements [10, 11] and has been gaining more significance in electrochemical measurements [12,13,14]. It allows to account not only for the signal height but also for its width, therefore, the derivative curves store more information. With the increase in derivative order, a higher number of peaks is observed, and their width is lower than for the initial signal [11]. It forms the basis for the signal separation, as high-order derivatives tend to extinguish the wider signals and amplify the narrower ones. Unfortunately, high-order derivatives exhibit an elevated noise level [10], so their practical application is limited. Differentiation is a linear transform, therefore, in the case of voltammetry, the relationship between the peak current and analyte concentration is conserved. It enables to use the value of e.g., the second derivative as the analytical signal.
In this work, the application of the continuous wavelet transform and differentiation of the DP voltammograms to separate the overlapped reduction signal of vitamin B1 (VB1) and B3 (VB3) has been reported. In particular, the mother wavelets in the shape of the theoretical [15] and experimentally recorded DP peak, as well as the second-order derivatives, have been thoroughly investigated. For quantitative analysis, multiple models with one input variable, being the concentration of the individual vitamin, and an output variable in the form of a transformed current value have been proposed. Their evaluation included the determination of the sensitivity, limit of detection (LOD) and quantification (LOQ), and linear range, as well as the comparison of these values with the parameters obtained from the calibration performed in the presence of only one of the studied vitamins. Finally, they have been applied in the determination of VB1 and VB3 in dietary supplements.
Results and discussion
Choice of the working electrode
Vitamins B1 and V3 are reduced at highly negative potentials [5], therefore, for their determination, a working electrode with a high over-potential of hydrogen evolution reaction (HER) is required. In case of the Glassy Carbon Electrode (GCE) and Silver Annular Band Electrode (AgABE), a rapid increase in current was observed as the potential shifted towards more negative values (Fig. 1a). These high background current values will impair the determination of compounds reduced in this potential range, including VB1 and VB3. For those reasons, no peak of VB3 reduction was observed for the three mentioned electrodes. A completely opposite situation was detected in case of the CGMDE, which provided a well-defined peak of VB3 reduction, low background current, and high over-potential of HER. Those unique features, combined with the possibility to precisely control the electrode surface and to renew it before the recording of each subsequent voltammogram, provided high repeatability and reproducibility of the measurements. This was essential to compare the results obtained in separate experiments. Among tested supporting electrolytes, 0.1 mol dm−3 acetate buffer of pH 6.0 provided the best signal-to-noise ratio and the highest sensitivity. Furthermore, due to the high over-potential of the HER of CGMDE in this buffer, it facilitated the interpretation of the VB1 reduction peak.

a Choice of the working electrode based on the signal of 1 mg dm−3 VB3 reduction in 0.1 mol dm−3 acetate buffer of pH 6.0. Inset: magnification of the voltammograms for CGMDE. b DP voltammograms recorded on the CGMDE for concentrations of VB3 and c VB1 reaching from blank up to 2.0 mg dm−3 with the calibration plots in the respective insets
Under previously optimized conditions, quantitative measurements for VB1 and VB3 separately were performed (Fig. 1b, c), based on which the figures of merit were calculated. The obtained values, summarized in Table 1, indicate the superiority of the VB1 determination protocol in terms of the sensitivity, linear range, as well as LOD and LOQ values. In the case of VB3, the deviation from linearity was observed for the concentrations above 1.0 mg dm−3, as depicted in the inset in Fig. 1b. Moreover, the VB3 reduction peak is wider (half-width 73 mV) than the peak of VB1 reduction (40 mV), and the sensitivity ratio of VB1 and VB3 determination protocol is equal to 1.56. These features are going to be crucial at the stage of the separation of overlapped peaks.
Figure 2 presents the DP voltammograms for simultaneous reduction of VB1 and VB3 for the three cases described in the Measurement procedure section. It points out how the presence of a compound with a peak potential close to that of the studied compound can hinder the quantitative evaluation of the recorded voltammograms. Moreover, it demonstrates the issues related to various sensitivities towards the two compounds and differences in their concentrations, that is, the two factors that influence the shape of the resulting voltammograms.

DP voltammograms recorded for the solutions containing: a equal concentrations of VB3 and VB1 ranging from blank to 2.0 mg dm−3 (case 1); b 1.0 mg dm−3 VB3 and VB1 in the range from blank to 2.0 mg dm−3 (case 2); c VB3 in the range from blank to 2.0 mg dm−3 and 1.0 mg dm−3 VB1 (case 3). Supporting electrolyte: 0.1 mol dm−3 acetate buffer (pH 6.0). Experimental conditions: E0 = − 1.0 V, Ek = − 1.55 V, Es = 3 mV, dE = 30 mV, tw = 10 ms, and ts = 10 ms
Continuous wavelet transform
The proposed mother wavelet ΨV, ΨVB1, and ΨVB3 were first normalized and standardized. The obtained curves, depicted in Fig. 3, were used in the signal separation by means of CWT.

Normalized and standardized mother wavelets with the shape of a typical DPV peak (ΨV) and the shape of DPV peak of the reduction of VB1 (ΨVB1) and VB3 (ΨVB3)
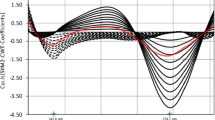
In the next step, the scale of the CWT was selected for each proposed mother wavelet based on the result of the transformation of the DP voltammogram for simultaneous reduction of VB1 and VB3 in the concentration of 1.0 mg dm−3. Among tested scale values, ranging from 10 to 70, the scale 40 for ΨV and the scale 20 in the case of ΨVB1 and ΨVB3 provided the best separation of the signals (Fig. 4). For those scales, there was at least one zero-crossing point in the potential range between the peak potential of VB3 and VB1, thus they were used in the subsequent calculations.

Selection of the scale for continuous wavelet transform with the use of a ΨV, b ΨVB1, and c ΨVB3 mother wavelets for the DP voltammogram of the reduction of 1.0 mg dm−3 VB1 and VB3. Tested scale values ranged from 10 to 70. The transformed signal for the chosen scale is marked in black
The transformed curves exhibited the increase in the wavelet coefficient CWTx following the rise in the concentration of VB1 and VB3 in the first variant of the simultaneous reduction experiment, as depicted in Fig. 5. This particularly applies to the CWTx values at the reduction potential of VB1 and VB3, which plotted against the concentration of the respective vitamin resulted in the linear calibration functions. The figures of merit, summarized in Table 2, fulfilled the defined criteria for the acceptance of devised protocols for quantitative analyses. Noteworthy, despite omitting the background correction step, the intercept did not differ from 0 in every case. The obtained LOD, LOQ, and linear range are comparable with those presented in Table 1. As anticipated, CWT has not improved the range of linear response.

Voltammograms from the Fig. 2a subjected to the CWT with the mother wavelet a ΨV (scale 40), b ΨVB1 (scale 20), and c ΨVB3 (scale 20); inset: corresponding calibration plots
In case no. 2, where the concentration of VB3 was kept constant, hardly any change has been observed in the transformed signals presented in Fig. 6a in the potential range corresponding to the reduction of VB3. Unfortunately, CWT with the ΨV mother wavelet has not provided the correct intercept value, thus failing to comply with the set criteria. The application of ΨVB1 to extract the voltammetric signal of VB1 provided an ideally linear response with the LOD being the lowest among all established. Similar remarks are accurate for case no. 3, characterized by the constant concentration of VB1 (Fig. 6b). The best results have been obtained for the ΨVB3 mother wavelet, as in this specific case all of the established criteria have been fulfilled. These observations proved the legitimacy of using the experimentally based mother wavelets for specific applications.

Mother wavelets ΨVB1 and ΨVB3, based on the experimentally recorded voltammograms, ensure similar slopes for a given vitamin between the investigated cases. Moreover, those mother wavelets offer lower detection and quantification limits. In case of ΨV, which resembles the shape of the theoretical DP peak, the highest sensitivities (slopes) have been observed, however, it failed to provide acceptable results in some of the examined conditions.
Differentiation
For the potential more positive than -1.3 V, hardly any change in current values was observed in the DP voltammogram of the VB1 reduction (Fig. 7a). Therefore, the first and second derivatives of this voltammogram did not significantly differ from 0 in this potential range. This also caused that the DP voltammogram of simultaneous reduction of VB1 and VB3 overlapped with the DP voltammogram of VB3 reduction (in the absence of VB1). The same applies to the first and second derivatives of the recorded voltammograms. Therefore, for potential more positive than -1.3 V, the DP voltammogram and its derivatives depended only on the concentration of VB3. This allowed applying the peak-to-peak approach to establish the value of the analytical signal for VB3 (denoted as hVB3 in Fig. 7a). Similarly, for the potentials more negative than the potential of VB1 reduction, the derivative voltammograms of simultaneous VB1 and VB3 reduction resembled the derivative voltammograms of the VB1 reduction. For this reason, choosing the value for the analytical signal of VB1 (hVB1) in the manner presented in Fig. 7a allowed accounting only for the contribution of VB1. In the potential range between the maximum of the VB1 and VB3 reduction peaks, the derivative voltammograms were influenced by both tested vitamins, therefore, applying this region for calibration purposes would lead to erroneous results. The disparity in the height of VB1 and VB3 analytical signals was caused by the difference in the peak width, since differentiation enhanced the narrower signal of VB1 reduction.

a DP voltammograms for the reduction of VB1, VB3, and simultaneous reduction of VB1 and VB3 with the resulting first (dI/dE) and second (d2I/dE2) derivatives. hVB1 and hVB3 denote the values of analytical signal for VB1 and VB3, respectively. Supporting electrolyte: 0.1 mol dm−3 acetate buffer (pH 6.0). Experimental conditions: E0 = − 1.0 V, Ek = − 1.55 V, Es = 3 mV, dE = 30 mV, tw = 10 ms, and ts = 10 ms. b Second derivatives of the DP voltammograms for simultaneous reduction of VB1 and VB3 in equal concentrations (from blank to 2.0 mg dm−3) with the corresponding calibration curves in the inset
As illustrated in Fig. 7b, the value of the second derivative increased as the concentrations of VB1 (CVB1) and VB3 (CVB3) were raised (case 1). For VB1, the linear relationship was observed in the whole range of tested concentrations; however, the calibration curve for VB3 was linear only for CVB3 lower than 1.0 mg dm−3. These tendencies, as well as the obtained figure of merit summarized in Table 3, resemble those obtained for calibrations performed separately for VB1 and VB3, which is easily understandable considering the linear nature of differentiation.
Cognately, in case no. 2. the hVB1 values were proportional to the concentration of VB1, whereas the hVB3 remained hardly unchanged (Fig. 8a) and vice versa in case 3 (Fig. 8b). The calibration curves and figures of merit obtained for the three cases are comparable to those established in the measurement performed separately for each vitamin. Moreover, the slopes of the calibration curves for a given vitamin are similar between experiment variants, which proves that the chosen analytical signals hVB1 and hVB3 did not depend on the concentration of VB3 and VB1, respectively. For this reason, the necessity to build complex models with multiple input variables can be avoided. The sensitivity ratio of VB1 and VB3 for the calibrations based on the derivative voltammograms is 4.5 times higher than for the calibration performed for VB1 and VB3 individually (Table 1). Therefore, it can be assumed that quantitative analysis of VB1 should be relatively straightforward and far less problematic than in the case of VB3, for which the range of linear response is narrower. The direct comparison of sensitivities obtained based on the DP voltammograms (Table 1) and their transforms (Table 2 and 3) is not possible, since they are expressed in different units.

Real sample analysis
Finally, the proposed methods of signal separation were applied for the determination of VB1 and VB3 in dietary supplements Plusssz Mg and Plusssz Ginseng. In terms of the content of vitamins, they do not differ from other products of this type available on the market. This is justified in the recommended daily dose of vitamins at the level of 1.1 mg VB1 and 16 mg VB3, which can be administered with one supplement tablet. Considering that and the obtained linear ranges of the reported procedures, it is clear that voltammetric determination of VB1 and VB3 has to be performed separately to comply with the accepted standards of quantitative analysis.
Figure 9a presents the DP voltammograms for the determination of VB3 in the Plusssz Mg supplement. The presence of the large peak corresponding to VB3 reduction and a small inflection resulting from the VB1 reduction is an early sign that the above-mentioned contents of VB1 and VB3 may be conserved. The obtained shape of curves in the wavelet domain is similar to that depicted in Fig. 5a because the same mother wavelet (ΨV) has been utilized during the transformation. Based on the calculated wavelet coefficients, the standard additions plots have been drafted, from which vitamin contents have been determined as the intercept with the horizontal axis. The values reported in Table 4 for CWT are the average of the vitamin contents estimated by means of CWT with the use of ΨV, ΨVB1, and ΨVB3 mother wavelets, whereas their standard deviation has been adopted as the determination error. Relating this to the declared content, the recoveries have been calculated.

Determination of VB3 and VB1 in dietary supplements. a DP voltammograms recorded for the Plusssz Mg and for additions of VB3 standard solution (top) with the curves obtained by means of CWT with ΨV mother wavelet (bottom). Inset: corresponding calibration curve. b Second derivatives (bottom) of the DP voltammograms registered for the Plusssz Ginseng and for additions of VB1 standard solution (top). Inset: calibration plot
The recorded voltammograms for every analysis were also subjected to differentiation, and an example of that is presented in Fig. 9b for the determination of VB1 in the Plusssz Ginseng. Based on the obtained second derivatives, the calibration plots have been drafted (inset in Fig. 9b), and the vitamin contents have been calculated (Tab. 4). Moreover, Fig. 9b perfectly illustrates how differentiation facilitates the interpretation of the overlapped peak appearing at the falling edge of another signal even in the case of a very unfavorable ratio of the peak currents.
All of the obtained vitamin contents, summarized in Table 4, are consistent with the declaration of the manufacturer, which proves the accuracy of the developed protocols. Due to the lack of necessity to perform any manual interpretation step, these procedures are more objective and possible to computerize. Thus, it is an important step towards complete automation of the quantitative analysis.
Conclusion
This paper reports two strategies for resolving the problems of overlapping voltammetric peaks with their validation based on the DP voltammograms for simultaneous reduction of vitamins B1 and B3 in various concentration ratios. The first one is Continuous Wavelet Transform (CWT) which searches for a given and appropriately scaled template, known as the mother wavelet, in the recorded signal, allowing for its time–frequency decomposition. By differentiation, not only the signal height but also its width is taken into account, resulting in the amplification of the narrow peaks and the extinction of the broad ones. Better separation is obtained for high-order derivatives; however, they exhibit a high level of noise, deeming the obtained results useless. Preliminary simulations, which utilize the signals recorded for each mixture component separately, are extremely helpful when choosing an appropriate algorithm and the feature used as the analytical signal for calibration purposes.
CWT and differentiation were proved to be suitable for the separation of the reduction peaks of vitamins B1 and B3 with the difference in peak potential of 80 mV. They allowed finding simple relationships between the analytical signal and analyte concentration individually for each vitamin and without the need to reach out to more advanced multivariate calibration strategies. Both algorithms are linear transformations, meaning that, apart from the peak separation, their application results in the scaling of the measured current and change in the unit. Therefore, the relationship between the feature used as analytical signal and analyte concentration resembles the calibration performed in the traditional manner (peak current vs concentration). This leads to the conclusion that CWT and differentiation will not allow to expand the range of linear response of the developed protocols. The proposed methodology has been applied in the determination of VB1 and VB3 in dietary supplements, providing the results consistent with the declaration of the manufacturer.
Experimental
All used chemicals were of analytical grades and used without purification. The acetate buffer of various concentrations and pH values was obtained by mixing the appropriate amount of 1 mol dm−3 acetic acid and its sodium salt (both Sigma Aldrich, ≥ 99%) with fourfold distilled water. The stock solutions of vitamin B1 and vitamin B3 were prepared by dissolving a corresponding amount of thiamine hydrochloride and niacin (DSM Nutritional Products GmbH), respectively, in distilled water. Prior to use, the glassware was cleaned by immersing in 6 mol dm−3 nitric acid (POCh) and subsequent washing with distilled water.
Apparatus and software
All the voltammetric measurements were conducted in a three-electrode system consisting of a Pt wire as an auxiliary electrode, a double-junction silver-silver chloride reference electrode, and a working electrode. As the latter, Glassy Carbon Electrode (BASi, USA, surface area A = 7.1 mm2), Controlled Growth Mercury Drop Electrode (CGMDE, mtm-anko, Poland, A = 1 mm2), and the home-made Silver Annular Band Electrode (AgABE, A = 13.13 mm2 [16]) were tested. For recording the voltammograms, the measurement system comprising the M161 multipurpose electrochemical analyzer, M164 electrode stand, and EAPro Software with the real-time data-acquisition feature was employed. Data presentation and its evaluation by means of CWT and differentiation were performed using the own scripts written in the MATLAB computing environment. Buffer pH value was established using the CPI-505 laboratory pH/ion meter (ELMETRON, Poland). All measurements were performed at room temperature.
Measurement procedures
In the beginning, various experimental conditions, including the working electrode, type, concentration, and pH of the supporting electrolyte, as well as parameters of the Differential Pulse Voltammetry, were tested with the aim to find the most suitable conditions, which will provide repeatable and reproducible results, and high sensitivity of vitamins determination. Unless otherwise stated, differential pulse (DP) voltammograms were recorded on the CGMDE in the 0.1 mol dm−3 acetate buffer (pH 6.0), using the following optimized conditions: start potential E0 = − 1.0 V, end potential Ek = − 1.55 V, step potential Es = 3 mV, pulse amplitude dE = 30 mV, waiting time tw = 10 ms, and sampling time ts = 10 ms. Since the measurements were performed in the range of negative potentials, the solution was deoxygenized by purging argon. In the optimized conditions, the calibration for VB1 and VB3 separately was performed, based on which the range of linear response, the sensitivity of the method, and the limit of detection (LOD) and quantification (LOQ) were established. LOD and LOQ were calculated as the standard deviation of the current for the blank (n = 5) divided by the calibration slope and multiply by 3 and 10, respectively.
In the second part, DP voltammograms for simultaneous reduction of VB1 and VB3 were recorded. In particular, three model cases were considered:
Case 1: the concentrations of VB1 and VB3 were always equal and increased gradually from blank to 2.0 mg dm−3.
Case 2: the concentration of VB3 was equal 1.0 mg dm−3, and that of VB1 increased gradually from blank to 2.0 mg dm−3.
Case 3: the concentration of VB3 increased gradually from blank to 2.0 mg dm−3, and that of VB1 was equal 1.0 mg dm−3.
Case 2 models the situation in which it is hard to determine the beginning of the peak, whereas case 3 represents the difficulty in determining the peak-end, rendering the background correction step impossible to be performed. For every addition of the standard, three subsequent scans were recorded and averaged. Such curves were subjected to the CWT with the use of a mother wavelet in the shape of the ideal DPV peak (ΨV) [8, 15] and the shape of the recorded DPV peak of VB1 (ΨVB1) and VB3 reduction (ΨVB3). For every mother wavelet, the scale of the transformation was selected, so as to provide the best separation of the overlapped peaks. Based on the relationship between the wavelet coefficient CWTx (for a given scale) and vitamin concentration CV, figures of merit of the devised protocols for vitamin determination were calculated.
In the case of differentiation, the averaged voltammograms were additionally preprocessed using the Savitzky–Golay algorithm with the 3rd-degree polynomial and 11-point frames. This step was crucial, since the measurement data have a finished resolution, and its differentiation may introduce further noise. In the end, the calibration curves for the three above-mentioned cases were drawn using the value of the second derivative calculated using the peak-to-peak approach, and the analytical parameters were established.
All the developed calibration functions were deemed suitable and accurate if the correlation coefficient R was greater than 0.995, the intercept value was statistically equal to zero, and the limit of detection and quantification were lower than the concentration corresponding to the first addition of the standard solution.
The developed methods have been employed for the quantification of VB1 and VB3 content in the multivitamin, effervescent dietary supplements Plusssz Mg and Plusssz Ginseng. For each of them, three tablets were weighted, crushed in the mortar to a fine powder. The weight amount corresponding to the average weight of the used tablets was transferred to a 50 cm3 volumetric flask, which was subsequently filled with the distilled water to the mark. Prior to the analysis, the solutions have been centrifuged to accelerate the sedimentation of solid particles, thereby avoiding their introduction to the measurement cell. All determinations have been performed using the standard addition method, in which the DP voltammograms have been registered for the blank, after the introduction of the sample, and following the additions of the standard solutions. For each step, three voltammograms have been recorded.
References
Combs GF (2008) The Vitamins Fundamental Aspect in Nutrition, 3rd edn. Elsevier Academic Press, Amsterdam
Brunetti B (2016) Electroanalysis 28:1930
Madej M, Kochana J, Baś B (2019) Monatsh Chem 150:1655
Huang L, Tian S, Zhao W, Liu K, Guo J (2021) Talanta 222:121645
Lovander MD, Lyon JD, Parr DL, Wang J, Parke B, Leddy J (2018) J Electrochem Soc 165:G18
Jakubowska M (2011) Electroanalysis 23:553
Jakubowska M, Baś B, Kubiak WW (2009) Talanta 79:1398
Jakubowska M (2007) J Electroanal Chem 603:113
Jakubowska M, Piech R (2008) Talanta 77:118
Karpińska J (2004) Talanta 64:801
Cygański A (2009) Metody spektroskopowe w chemii analitycznej, 4th edn. Wydawnictwo Naukowo-Techniczne, Warszawa
Ugliano M (2016) Food Chem 212:837
Hong TK, Rusodimos I, Kim MH (2017) J Electroanal Chem 785:255
Bucur RV (2014) Electrochim Acta 129:76
Britz D (2005) Digital Simulation in Electrochemistry, 3rd edn. Springer, Berlin Heidelberg
Węgiel K, Baś B (2018) J Electrochem Soc 165:H393
Funding
Funding provided by The National Centre for Research and Development (Grant number: POWR.03.02.00-00-1004/16)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Porada, R., Baś, B. Separation of the overlapped vitamin B1 and B3 voltammetric peaks by means of Continuous Wavelet Transform and differentiation. Monatsh Chem 152, 1107–1117 (2021). https://doi.org/10.1007/s00706-021-02788-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-021-02788-6