
A new method for the synthesis of glycyrrhizic acid (GA) conjugates with S-benzyl-L-cysteine using 1-ethyl-3-(3-dimethylaminoproopyl)carbodiimide is proposed. It is established that 3-O-{2-O-[N-(β-D-glucopyranosyluronyl)-L-cysteine-S-benzyl]-N-(β-D-glucopyranosyluronyl)-L-cysteine-S-benzyl}-(3β,20β)-11-oxo-30-(N-carbonyl-L-cysteine-S-benzyl)-30-norolean-12-ene is superior to GA in inhibiting the accumulation of HIV-I virus-specific protein p24 (viral antigen) in MT-4 cell culture (IC50 3 μg/mL, SI 90) and is 50 – 55 times less toxic to cells than azidothymidine.
Similar content being viewed by others
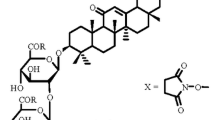

The development of new drugs for treatment and prevention of viral infections of various etiologies is a key problem in chemistry and medicine because of the ubiquitous spread of HIV infections and viral hepatitis B and C and the emergence of new viral infections (A/H1N1, bird and swine flus, Ebola fever, chikungunya, Zika, West Nile, etc.) [1, 2]. Currently, the search for new antiviral agents is associated with the discovery of compounds with new structures and unique mechanisms of action and with chemical modification of already known antiviral compounds to produce derivatives with improved antiviral properties [3]. Use of natural lead compounds with established antiviral activity is a promising approach to the search for new antiviral agents [4, 5]. Glycyrrhizic acid (GA) (1) is such a compound. It is the main triterpene glycoside from licorice roots (Glycyrrhiza glabra L., G. uralensis Fisher) [6,7,8]. Japanese researchers discovered in 1987-1988 the ability of GA to inhibit HIV in vitro and in vivo [9, 10]. GA was successfully employed clinically to treat AIDS patients [11, 12]. The main drawbacks of GA are its low efficacy as an inhibitor of HIV reverse transcriptase and the high doses that are necessary to suppress HIV reproduction [13, 14].
Previously, several GA conjugates with amino acids and dipeptides were shown to possess potent antiviral activity against HIV-1 [15, 16], coronavirus SARS CoV [17], Ep-

stein-Barr virus [18], and A/H1N1/pdm2009 influenza virus [19]. Cysteine-containing GA conjugates turned out to be inhibitors of SARS-associated coronaviruses [17] and immunostimulants [20]. Methods for preparing GA conjugates with various L- and D-amino acid esters via activation of GA carboxylic acids using N-hydroxybenzotriazole (HOBt)–N,N_-dicyclohexylcarbodiimide (DCC) [21], N-hydroxyphthalimide–DCC [22], or N-hydroxysuccinimide (HOSu)–DCC [15, 16] were proposed. Use of DCC to activate GA was accompanied by release of N,N′-dicyclohexylurea, which formed side N-acylureas and contaminated the target products [23]. Water-soluble carbodiimides, in particular 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (DEC), which does not form dicyclohexylurea, are more attractive as condensing agents [24]. The goal of the present work was to develop an improved method using DEC to prepare GA conjugates with S-benzyl-L-cysteine that are interesting for medicine and as antiviral agents.
Experimental Chemical Part
PMR and 13C NMR spectra were recorded with TMS internal standard on a Bruker AM-300 spectrometer at operating frequency 300 and 75.5 MHz. IR spectra were taken from Vaseline-oil mulls on a Prestige-21 IR spectrophotometer (Shimadzu). Optical density was measured in a 1-dm tube at 20 – 22°C (λNa 546 nm) on a PerkinElmer 341 polarimeter.
TLC used Sorbfil plates (Sorbpolimer). Spots of compounds were detected by H2SO4 (5%) in EtOH followed by heating at 110-120°C for 2-3 min. Column chromatography used KSK silica gel (50-150 fraction, Sorbpolimer).
GA (96% pure) prepared as before [25]; 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Sigma-Aldrich, Germany); and S-benzyl-L-cysteine tert-butyl ester hydrochloride (Reanal, Hungary) were used in the work. Et3 N was stored for 1 d over KOH and distilled. Solvents were purified using standard methods [26].
3- O -{2- O -[ N -(β-D-Glucopyranosyluronoyl)-L-cysteine-S-benzyl]-N-(β-D-glucopyranosyluronoyl)-L-cysteine-Sbenzyl}-(3β,20β)-11-oxo-30-(N-carbonyl-L-cysteine-Sbenzyl)-30-norolean-12-ene (III). A solution of GA (0.82 g, 1 mmol) in DMF (20 mL) was treated with HOBt (0.48 g, 3.5 mmol), S-benzyl-L-cysteine tert-butyl ester hydrochloride (1.2 g, 4.0 mmol), DEC (0.67 g, 3.5 mmol), and Et3N (0.8 mL, 8 mmol); stirred at 20 – 22°C for 10 – 12 h, diluted with H2O, and acidified to pH ~ 3 – 4 with citric acid. The precipitate was filtered off, rinsed with H2O, dried, and reprecipitated from aqueous EtOH to afford carboxy-protected conjugate II (1.40 g). IR spectrum, v, cm–1: 3500 – 3200 (OH, NH), 1739 (COOBut), 1662 (C=O), 1621 (C6H5), 1520 (CONH), 1505 (C6H5). Conjugate II (0.68 g, 0.5 mmol) was worked up with CF3COOH solution in CH2Cl2 (2 mL/20 mL) at 20 – 22°C, evaporated to dryness, and chromatographed over a column of silica gel with elution by CHCl3–MeOH–H2O (200:10:1, 100:10:1, 50:10:1, 25:10:1; vol%). Yield 0.35 g (58%) (amorphous compd.); \( {\left[\upalpha \right]}_D^{20} \) +35° (c 0.05, MeOH); lit. [20]: \( {\left[\upalpha \right]}_{20}^D \) +34° (s 0.02; MeOH). IR spectrum, vmax, cm–1: 3600 – 3200 (OH, NH), 1710 (COOH), 1670 (C=O), 1550 (CONH), 1510 (Ph). 13C NMR spectrum (DMF-d7), δ, ppm: 199.1 (C11), 177.2 (C30), 171.3, 170.7 (C6′, C6′), 169.3 (C13), 127.7 (C12), 103.7 (C1′), 102.9 (C1′), 88.1 (C3), 80.1 (C2′), 75.3 (C5″, C5′), 74.8 (C3′), 74.1 (C3′), 73.7 (C3′), 72.6 (C2′), 71.7 (C4′, C4′), 61.0 (C9), 54.3 (C5), 47.8 (C18), 45.7 (C8), 44.6 (C20), 42.8 (C14), 42.5 (C19), 40.4 (C4), 38.8 (C1), 38.4 (C22), 36.1 (C10), 32.2 (C7), 31.9 (C17), 31.1 (C21), 29.5 (C29), 28.6 (C23), 28.3 (C28), 27.8 (C2), 27.3, 27.1 (C15, C16), 22.3 (C27), 17.7 (C26), 16.6 (C6), 15.7 (C24), 15.5 (C25); 3Cys(SBn): 173.0 (COOH), 172.4 (COOH), 172.1 (COOH), 137.8 (C arom.), 137.6 (2C arom.), 130.0 (C arom.), 128.9 (2C arom.), 128.5 (2C arom.), 128.4 (2C arom.), 128.1 (C arom.), 127.9 (2C arom.), 127.8 (C arom.), 127.2 (C arom.), 126.8 (C arom.), 126.4 (2C arom.), 51.3 (CH), 50.8 (CH), 50.5 (CH), 35.7 (CH2), 35.5 (CH2), 35.3 (CH2), 26.8 (CH2), 26.7 (CH2), 26.6 (CH2). Found,%: N 2.8, S 6.7. C57H89N3O19 . Calc.,%: N 3.0, S 6.8. M = 1402.7.
3- O -{2- O -[ N -(β-D-Glucopyranosyluronoyl)-L-cystein e-S-benzyl]-N-(β-D-glucopyranosyluronoyl)-L-cysteine-S-benzyl}-(3β,20β)-11-oxo-30-norolean-12-ene (V). A solution of GA (0.82 g, 1 mmol) in dioxane (20 mL) was treated with HOSu (0.57 g, 5 mmol), DEC (0.57 g, 3 mmol), and Et3N (0.5 mL), stirred at 20 – 22°C for 5 h; treated with S-benzyl-L-cysteine tert-butyl ester hydrochloride (0.76 g, 2.5 mmol) and Et3N (0.3 mL), stored with periodic stirring at 20 – 22°C for 24 h, diluted with cold H2O, and acidified with citric acid to pH ~ 4. The precipitate was filtered off, rinsed with H2O, and dried to afford protected conjugate IV (1.1 g) that was worked up without further purification with CF3COOH in CH2Cl2 as above, evaporated to dryness, and chromatographed over a column of silica gel with elution by CHCl3–MeOH–H2O (200:10:1, 100:10:1, 50:10:1, vol%). Yield 0.72 g (60%); \( {\left[\upalpha \right]}_D^{20} \) +55° (c 0.04; MeOH). Lit. [20]: \( {\left[\upalpha \right]}_D^{20} \) +52° (s 0.02; MeOH). IR spectrum, vmax, cm–1: 3500 – 3200 (OH, NH), 1730 (COOH), 1657 (C=O), 1530 (CONH), 1500 (Ph). 13C NMR spectrum (DMF-d7), δ, ppm: 199.7 (C11), 178.3 (C30), 170.4, 170.3, 170.1 (C6″, C6′, C13), 127.5 (C12), 105.2 (C1′), 104.3 (C1′), 89.0 (C3), 82.4 (C2′), 77.0 (C5′), 76.9 (C5′), 76.0 (C3′), 75.9 (C3″), 74.9 (C2″), 73.4, 73.0 (C4′, C4″), 62.2 (C9), 55.5 (C5), 49.0 (C18), 45.8 (C8), 44.0 (C20), 43.8 (C14), 41.7 (C19), 39.9 (C4), 39.6 (C1), 37.3 (C22), 36.6 (C10), 33.7 (C7), 33.0 (C17), 32.3 (C21), 29.5 (C29), 28.8 (C23), 28.3 (C28), 27.9 (C2), 27.0, 26.0 (C16, C15), 23.5 (C27), 18.9 (C26), 17.8 (C6), 16.8 (C24), 16.7 (C25); 2Cys(SBn): 172.5 (COOH), 172.4 (COOH), 139.2 (C arom.), 138.1 (C arom.), 131.0 (C arom.), 130.0 (C arom.), 129.6 (2C arom.), 129.1 (C arom.), 129.0 (2C arom.), 128.9 (C arom.), 128.4 (C arom.), 128.3 (C arom.), 52.8 (CH), 52.6(CH), 35.8 (CH2), 35.5 (CH2), 26.7 (2CH2). Found,%: N 2.30, S 5.22. C62H84N2O18S2. Calc.,%: N 2.32, S 5.30. M = 1209.42.
Experimental Biological Part
The cytotoxicity and anti-HIV-1 activity of III were studied using the traditional model of primary HIV-infected MT-4 lymphoid cells and HIV-1/EVK strain as described before [13,14,15,16]. The reference drug was GA (96% pure) [25].
Cytotoxicity was determined by dissolving III in DMSO. Aliquots of appropriate dilutions (three for each dilution) were placed into wells of a 96-well plate for cell inoculation. The inoculation concentration was 0.5 106 cells/mL. Cells were cultivated in 96-well plates (Costar, USA) in growth medium [RPMI-1640 with added fetal bovine serum (10%), L-glutamine (0.06%), and gentamycin (100 μg/mL)] at 37°C and 5% CO2 for 4 d. When the incubation was finished, the fraction of viable cells was counted in a Goryaev chamber after staining with trypan blue. A dose-dependent curve was constructed. The compound concentration causing the death of 50% of the cells (CD50) was determined.
The anti-HIV activity of III was assessed by infecting MT-4 cells (concentration 2 ×106 cells/mL) with HIV-1/EVK strain at infection multiple 0.2 – 0.5 infection units per cell for 1 h at 37°C. Infected and control cells (without virus) were diluted with growth medium to an inoculation concentration of 5 (105 cells/mL and placed into wells of 96-well plates. Then, solutions of the tested compound (three wells for each dilution) were placed into the corresponding wells. Then, the plates were cultivated as above. The final concentration of the tested drugs in the cell suspension varied from 0.1 to 100 μg/mL. The inhibitory effect of III was assessed on day 4 of cultivation by measuring the amount of virus antigen p24 using immunoenzyme assay. Furthermore, the fraction of viable cells was determined after staining with trypan blue by counting in a Goryaev chamber. Dose-dependent curves were constructed using the experimental data. The quantitative inhibition characteristics were determined as ID50 , the compound concentration suppressing virus production by 50% or providing 50% protection of cells from death by infection; ID90 , the compound concentration suppressing virus production by 90% or providing 90% protection of cells from death by infection; SI, the selectivity index or ratio of toxic dose CD50 to its effective dose ID50. The standards were azidothymidine (0.1 μg/mL) and GA (100 μg/mL). The quantitative characteristics were determined by processing the results of three measurements using the Excel 2010 program.
Results and Discussion
Carboxy-protected conjugate II was synthesized by condensation of GA with S-benzyl-L-cysteine tert-butyl ester hydrochloride using HOBt–DEC at room temperature (20 – 22°C) in DMF in the presence of an excess of Et3N. Treatment of conjugate II with a solution of CF3COOH inCH2Cl2 at 20 – 22°C produced target product III with three SBnCys moieties in 58% yield after column chromatography (CC) over silica gel (SG). Compound IV with an SBnCysOBut moiety in the carbohydrate part was synthesized using HOSu–DEC in dioxane. The resulting carboxy-protected GA conjugate IV was deprotected without further purification by CF3COOH to afford conjugate V in 60% yield after CC over SG.
Cytotoxicity and antiviral activity of III were studied using the traditional model of primary HIV-infected MT-4 lymphoid cells (HIV-1/EVK strain) as before [13, 15, 16]. The reference drug was GA (96% pure) [25] at a concentration of 100 μg/mL and the well-known anti-HIV drug azidothymidine (AZT) at the therapeutic dose of 0.1 μg/mL [27, 28]. Table 1 presents the quantitative characteristics for inhibition of HIV-1 virus-specific protein p24.
The 50% cytotoxic dose CD50 of the studied compound that caused the death of 50% of the cells was 270 μg/mL. Compound III exhibited pronounced anti-HIV-1 activity, inhibiting with high efficacy the accumulation of virus-specific protein p24 with ID50 = 3 μg/mL or 2.2 μM (compound concentration suppressing virus production by 50%) and ID90 = 100 μg/mL (90% suppression of virus reproduction). The selectivity index (SI, ratio of toxic dose CD50 to effective dose ID50) for inhibition of HIV-1 p24 was 90 for III, which was substantially greater than that for GA, the SI of which varies from 4.4 to 24 depending on the measurement parameters and purity [13, 28]. However, 90% suppression of virus reproduction was attained only with III at high concentration (100 μg/mL). It is noteworthy that III in the studied concentration range did not show noticeable protection from cytopathogenic activity of the virus. Compound III, in contrast to AZT, was nontoxic for HIV-infected cells and was 50 – 55 times less toxic than AZT (CD50 = 3.5 μM) [27, 28].
References
S. I. Hay, A. A. Abajobir, K. H. Abate, et al., Lancet, 390, 1260 – 1344 (2017).
C. A. Daep, J. L. Munoz-Jordan, and E. A. Eugenin, J. NeuroVirol., 20, 539 – 560 (2014).
E. De Clercq and G. Li, Clin. Microbiol. Rev., 29, 695 – 747 (2016).
J. P. Martinez, F. Sasse, M. Bronstrup, et al., Nat. Prod. Rep., 32, 29 – 48 (2015).
A. E. D. Bekhit and A. A. Bekhit, Stud. Nat. Prod. Chem., 42, 195 – 228 (2014).
R. Pompei, S. Laconi, and A. Ingianni, Mini-Rev. Med. Chem., 9, 996 – 1001 (2009).
L. A. Baltina, R. M. Kondratenko, L. A. Baltina, Jr., et al., Khim.-farm. Zh., 43(10), 3 – 12 (2009); Pharm. Chem. J., 43(10), 539 – 548 (2009).
Z.-G. Sun, T.-T. Zhao, N. Lu, et al., Mini-Rev. Med. Chem., 19(10), 826 – 832 (2019).
M. Ito, H. Nakashima, M. Baba, et al., Antiviral Res., 7, 127 – 137 (1987).
M. Ito, A. Sato, K. Hirabayashi, et al., Antiviral Res., 10, 289 – 298 (1988).
A. J. Vlietinck, T. De Bruyne, S. Apers, et al., Planta Med., 64, 97 – 109 (1998).
T. Hattori, S. Ikumatsu, A. Koivo, et al., Antiviral Res., 11, 255 – 261 (1989).
O. A. Plyasunova, I. N. Egoricheva, N. V. Fedyuk, et al., Vopr. Virusol., No. 5 – 6, 235 – 238 (1992).
O. A. Plyasunova, T. V. Il’ina, Ya. Yu. Kiseleva, et al., Vestn. Ross. Akad. Med. Nauk, No. 11, 42 – 46 (2004).
L. A. Baltina, Jr., R. M. Kondratenko, L. A. Baltina, et al., Russ. J. Bioorg. Chem., 35(4), 510 – 517 (2009).
L. A. Baltina, Jr., E. S. Chistoedova, L. A. Baltina, et al., Chem. Nat. Compd., 48(2), 262 – 266 (2012).
G. Hoever, L. A. Baltina, M. Michaelis, et al., J. Med. Chem., 48, 1256 – 1259 (2005).
J. C. Lin, J. M. Cherng, M. S. Hung, et al., Antiviral Res., 79, 6 – 11 (2008).
L. A. Baltina, V. V. Zarubaev, L. A. Baltina, et al., Bioorg. Med. Chem. Lett., 25, 1742 – 1746 (2015).
R. M. Kondratenko, L. A. Baltina, E. V. Vasil’eva, et al., Russ. J. Bioorg. Chem., 30(1), 53 – 50 (2004).
L. A. Baltina, S. A. Ryzhova, E. V. Vasil’eva, et al., Russ. J. Bioorg. Chem., 20(1), 40 – 46 (1994).
L. A. Baltina, Jr., A. I. Fairushina, and L. A. Baltina, Russ. J. Gen. Chem., 85, 2735 – 2738 (2015).
J. H. Jones, M. Bodanszky, D. H. Rich, J. Singh, and D. S. Kemp, in: The Peptides: Analysis, Synthesis, Biology. Vol. 1, Major Methods of Peptide Bond Formation, E. Gross and J. Meienhofer (eds.), Academic Press, New York, London (1979).
L. A. Baltina, Jr., A. I. Fairushina, and L. A. Baltina, Russ. J. Gen. Chem., 86(4), 826 – 829 (2016).
R. M. Kondratenko, L. A. Baltina, S. R. Mustafina, et al., Khim.-farm. Zh., 35(2), 39 – 42 (2001); Pharm. Chem. J., 35(2), 101 – 104 (2001).
A. J. Gordon and R. A. Ford, The Chemist’s Companion, Wiley-Interscience, New York (1972).
E. De Clerq, Trends Pharmacol. Sci, 8, 339 – 345 (1987).
O. A. Plyasunova, Candidate Dissertation in Biological Sciences as a Scientific Article, Kol’tsovo (1992).
L. A. Baltina, Jr., O. V. Stolyarova, R. M. Kondratenko, et al., Khim.-farm. Zh., 48(7), 21 – 25 (2014); Pharm. Chem. J., 48(7), 439 – 443 (2014).
Acknowledgments
The work was performed on State Task Topics AAAAA17-117011910035-6 and AAAA-A20-120012090026-9 and used equipment at the Khimiya CUC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 55, No. 3, pp. 15 – 18, March, 2021.
Rights and permissions
About this article

Cite this article
Baltina, L.A., Kondratenko, R.M., Baltina, L.A. et al. Synthesis of Glycyrrhizic Acid Conjugates with S-Benzyl-L-Cysteine and Their Antiviral Activity. Pharm Chem J 55, 224–227 (2021). https://doi.org/10.1007/s11094-021-02402-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-021-02402-3