
Abstract
The use of museum specimens held in natural history repositories for population and conservation genetic research is increasing in tandem with the use of massively parallel sequencing technologies. Short Tandem Repeats (STRs), or microsatellite loci, are commonly used genetic markers in wildlife and population genetic studies. However, they traditionally suffered from a host of issues including length homoplasy, high costs, low throughput, and difficulties in reproducibility across laboratories. Massively parallel sequencing technologies can address these problems, but the incorporation of museum specimen derived DNA suffers from significant fragmentation and exogenous DNA contamination. Combatting these issues requires extra measures of stringency in the lab and during data analysis, yet there have not been any high-throughput sequencing studies evaluating microsatellite allelic dropout from museum specimen extracted DNA. In this study, we evaluate genotyping errors derived from mammalian museum skin DNA extracts for previously characterized microsatellites across PCR replicates utilizing high-throughput sequencing. We found it useful to classify samples based on DNA concentration, which determined the rate by which genotypes were accurately recovered. Longer microsatellites performed worse in all museum specimens. Allelic dropout rates across loci were dependent on sample quantity, with high concentration museum specimens performing as well and recovering quality metrics nearly as high as the frozen tissue sample. Based on our results, we provide a set of best practices for quality assurance and incorporation of reliable genotypes from museum specimens.
Similar content being viewed by others


Introduction
Natural history repositories contain invaluable specimen collections for scientific use across diverse fields (Lane 1996; Williams 1999; Lister and Group 2011; Blagoderov et al. 2012). Many of these specimens represent populations that no longer exist due to land-use change and anthropogenic landscape alterations over the past century (Smith et al. 2013). Additionally, museum specimens often represent the few or only representatives of endangered or rare species, provide important vouchers for comparison with modern samples, and provide genetic resources for species which may be difficult to sample in wild habitats (Miller et al. 2009; White et al. 2018). Consequently, usage of museum specimens for research incorporating DNA analysis is increasing.
The degraded DNA associated with museum specimens is known to require extra measures of stringency in order to combat issues with exogenous DNA contaminants (Paabo et al. 2004; Rizzi et al. 2012) and highly fragmented endogenous DNA (Campana et al. 2012; Hawkins et al. 2016a; McDonough et al. 2018). Studies which reliably sequence DNA from museum specimens undergo stringent protocols (e.g., processing in appropriate lab spaces) to prevent contamination and to combat the low quantity and fragmented nature of the extracts. Downstream from wet lab procedures, additional bioinformatic steps must be taken to ensure the resulting genetic sequence data represent the target taxa. Truly ancient samples (archaeological and permafrost specimens) have characteristic degradation patterns associated with misincorporation of nucleotides—namely cytosine to uracil deamination—(Hofreiter et al. 2001; Jónsson et al. 2013; Kistler et al. 2017) but degradation patterns are only beginning to be understood and vary by sample type (Shapiro and Hofreiter 2012; Weiß et al. 2016), with museum specimens lacking the characteristic cytosine–uracil deamination (McDonough et al. 2018).
Short tandem repeats (STRs), or microsatellite loci, are useful markers for numerous applications and widely implemented in the field of conservation genetics to evaluate genetic diversity and population structure (e.g., Bilska and Szczecińska 2016; Arbogast et al. 2017). Capillary electrophoresis (CE), a fragment size genotyping technique, was previously standard practice for microsatellite genotyping, but the advent of high-throughput sequencing (HTS) introduced new possibilities (Curto et al. 2019; Tibihika et al. 2019). Degraded DNA present unique obstacles for HTS methodology, yet as more studies incorporate low quality samples, advances in laboratory and bioinformatic processes are making these samples more accessible (Andrews et al. 2018). HTS technology has allowed for rapid identification of microsatellite loci in non-model organisms on a genomic scale (Miller et al. 2013; Silva et al. 2013; Duan et al. 2014; Griffiths et al. 2016), and the simultaneous sequencing of thousands of putative loci as compared to traditional cloning methods (Glenn and Schable 2005). In addition to the cost reduction of microsatellite isolation (as low as $24 USD per locus as detailed in: (Abdelkrim et al. 2009), some of the issues known to occur when genotyping microsatellites via CE can be alleviated using HTS technologies (Vartia et al. 2016). For example, fragment size analysis via CE has been known to provide (albeit sometimes predictably) shifted sizes when samples are run on different machines (Morin et al. 2009), but access to raw sequences from HTS allows precise allele sizing (Darby et al. 2016).
A number of studies have evaluated how to transform raw sequences into microsatellite genotypes (Darby et al. 2016; Vartia et al. 2016; De Barba et al. 2017; Zhan et al. 2017; Barbian et al. 2018; Pimentel et al. 2018; Šarhanová et al. 2018). Each of these genotyping-by-sequencing (GBS) studies has evaluated some aspects of the biases induced when comparing sequences from HTS platforms to CE genotyping. Commonly addressed issues include evaluation of stutter and PCR artifacts (e.g. off-target amplification, PCR product adenylation) as these still occur in HTS-based methods, but can be mitigated using emerging bioinformatic analyses (Barbian et al. 2018; Tibihika et al. 2019). Length homoplasy occurs when alleles of the same length have different nucleotide sequences (Darby et al. 2016), and GBS studies have also shown recovery of additional alleles based on direct sequence analysis, which increases genetic resolution (Curto et al. 2019; Lepais et al. 2020). Although challenges exist for direct comparison of HTS based microsatellite genotypes with those via CE, the ability to generate comparable datasets is paramount in order to build off previous research and inform larger, potentially landscape-based conservation plans.
Despite the wide range of studies already published on GBS, none have specifically evaluated genotyping errors that occur from mammalian study skin sourced DNA (hereafter ‘museum specimen DNA’). Genotyping errors arise when the observed genotype of a sample differs from a consensus genotype (Pompanon et al. 2005), and previous studies have estimated error rates from fecal (De Barba et al. 2017; Barbian et al. 2018), tissue (Vartia et al. 2016; Lepais et al. 2020), and hide and hair samples (Donaldson et al. 2020). Here we invoke HTS GBS to assess genotyping errors derived from Glaucomys oregonensis (Humboldt’s flying squirrel) museum specimen DNA extracts, for five previously characterized microsatellites across PCR replicates of individual samples. More specifically, we analyzed the allelic dropout rate, a type of genotyping error where a true allele fails to amplify in PCR (Broquet and Petit 2004), across three datasets:
-
(1)
Replicate dataset: For every sample, each microsatellite and mitochondrial PCR replicate underwent library preparation.
-
(2)
Pooled dataset: For every sample, all microsatellite and mitochondrial PCR replicates were combined together prior to library preparation.
-
(3)
Bioinformatic dataset: For every sample, bash scripting ‘cat’ commands were used to combine all reads from the replicate dataset prior to genotyping.
The error rates described here will serve as the first for GBS of dry museum study skins, and provide best practices for subsequent studies on museum specimen samples.
Methods
Samples
Total genomic DNA from 147 samples was extracted for a population genetic study on Glaucomys oregonensis (Yuan et al. in prep.). A subset of seven samples was included in this study to establish baseline data for quantifying allelic dropout in degraded source material. From the screened samples, DNA concentrations were measured with a NanoDrop One Spectrophotometer (ThermoScientific) and a Qubit 2.0 (Life Technologies) using a high sensitivity DNA kit. Based on instrument sensitivity, different raw values were used to bin samples as ‘high concentration’ museum specimens (HCMS) or ‘low concentration’ museum specimens (LCMS) (O’Neill et al. 2011). Additional quantification details are provided in the Supplemental Materials. One frozen tissue sourced specimen was also included to observe if similar rates of allelic dropout would be detected from a non-degraded sample (Table 1).
Microsatellite selection
We tested three sample types: tissue, ‘high’, and ‘low’ concentration museum samples to evaluate amplification across microsatellites of varying lengths. Previously published microsatellites from Glaucomys sabrinus (northern flying squirrel)—a species which historically included G. oregonensis- were used in this study (GS-2, GS-4; Zittlau et al. 2000, and GLSA-12, GLSA-22, GLSA-52; Kiesow et al. 2011, Table 2). Two ‘short’ microsatellites were selected (GS-2 and GS-4), which included any marker under 150 bp in length according to published allele sizes, two ‘medium’ (GLSA-12 and GLSA-22, 150–200 bp), and one ‘long’ microsatellite (GLSA-52, > 200 bp).
DNA extraction
Total genomic DNA from museum specimens was isolated using Qiagen QIAamp DNA Mini Kits (Qiagen, Valencia, CA) in a lab designated for degraded DNA, while DNA from tissue samples was processed in a standard DNA facility using Qiagen DNeasy Blood and Tissue Kits following standard animal tissue protocols. DNA was eluted in 100 µl of buffer AE for all museum specimens and 200 µl for tissue samples. The degraded DNA lab protocol included placing a blank extraction control minimally every 12 samples, extensive bleaching, exposing consumables and pipettes to ultraviolet radiation, routine changing of gloves, and processing samples under a PCR workstation (AirClean Systems model AC600). Raw DNA concentrations were evaluated using 1 µl of DNA on a NanoDrop One Spectrophotometer (ThermoScientific).
Polymerase chain reaction
Multiplexed PCR attempts failed repeatedly, so singleplexed PCR was performed in 16 µl reactions containing 2.0 µl of DNA template (~ 0.01–127.4 ng/µl), 4.5 µl of ddH2O, 0.5 µl of each primer, and 8.5 µl of DreamTaq Green PCR Master Mix. PCR negative controls were added minimally every 47 samples with each PCR. When amplifying microsatellites with the GS-2 and GS-4 primers, 0.3 µl of ddH2O was replaced with bovine serum albumin (BSA, New England Biolabs, Inc., 12 mg) to reduce PCR inhibition. For all samples a touchdown PCR profile was used: 95 °C for 1 min; 2 cycles of 95 °C for 15 s, 60 °C for 30 s, 72 °C for 45 s; 2 cycles changing 60 °C to 58 °C; 2 cycles changing 58 °C to 54 °C; 2 cycles changing 54 °C to 52 °C; 35 cycles of 95 °C for 15 s, 50 °C for 30 s, 72 °C for 45 s; 72 °C for 5 min. Successful PCR amplifications were replicated twice in the tissue sample and three times in museum samples. Following amplification, 1.5% agarose gels were run with a 100 bp size standard (Invitrogen) and stained with GelRed (Biotium) or SYBR Green (Invitrogen). To remove residual primers, dNTPs, and nontarget molecules, solid phase reversible immobilization (SPRI) cleaning via magnetic beads was performed (following Rohland and Reich 2012) in a ratio of 1 part PCR product to 1.5X magnetic beads (KAPA beads, Roche). Cleaned PCR products were eluted in 20 µl ddH2O.
A fragment of the mitochondrial cytochrome-b gene was also amplified in PCR and sequenced to ascertain whether levels of mtDNA would be indicative of nDNA. Mitochondrial DNA methods and analysis can be found in the Supplemental Materials.
Library preparation
For the replicate dataset, each microsatellite and mitochondrial PCR replicate underwent library preparation (resulting in 114 libraries). For the pooled dataset, all PCR replicates were combined together prior to library preparation. Two µl of each PCR product for both cytochrome-b and microsatellites were pooled for museum specimens, while 2 µl of each cytochrome-b replicate and 3 µl of each microsatellite replicate were pooled for the tissue sample. For the bioinformatic dataset, we combined all reads from the replicate dataset together using bash scripting ‘cat’ commands prior to genotyping to evaluate if genotypes would vary based on PCR product coverage.
Individual dual iTru style indices (Glenn et al. 2019) were ligated using KAPA Illumina Library Preparation Kits (Roche, # KK8232) following Hawkins et al. (2016b). Libraries were amplified in 25 µl reactions containing 1.25 µl of each iTru adapter, 2.5 µl ddH2O, 7.5 µl of stubby adapter-ligated DNA, and 12.5 µl of KAPA HiFi HotStart ReadyMix. The thermocycler conditions were 98 °C for 45 s; 10 cycles (tissues) or 14 cycles (museum samples) of 98 °C for 15 s, 60 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. A library preparation control (ddH20) was included in all steps. After library prep, an agarose gel was run to ensure successful adapter ligation. Products were purified via SPRI as detailed above.
Sequencing
Sequencing occurred on an Illumina MiSeq using a 2 × 300 PE v.3 kit at the Center for Conservation Genomics, Smithsonian Conservation Biology Institute, Washington DC or using a 2 × 250 PE v.2 Nano kit at the Laboratory of Analytical Biology, National Museum of Natural History, Smithsonian Institution, Washington DC. Reads were demultiplexed and downloaded from the BaseSpace Server (Table S1).
Quality filtering
Samples were run through FastQC v 0.11.9 (Andrews 2010) and CutAdapt v 1.18 (Martin 2011) for quality filtering and adapter removal. Phred scores were required to be ≥ 20 averaged across each read. Prinseq v 0.20.4 (Schmieder and Edwards 2011) was run on each library to determine the proportion of low quality reads (Table S1). Commands are provided in the Supplementary Materials.
Genotyping
CHIIMP v 0.3.1 (Barbian et al. 2018) was used to generate genotypes for all datasets. At each locus, a genotype was called if there were minimally 5 reads (counts.min = 5), sequences were considered alleles only if the read count constituted minimally 5% of the total reads for that locus (fraction.min = 0.05), and all loci were given a 20 bp length buffer. CHIIMP identified the most likely alleles and provided data on whether or not the sample had PCR stutter, PCR artifacts, and more than two possible allele sequences. ConGenR (Lonsinger and Waits 2015) was also run to generate consensus genotypes for the replicate dataset. To determine the ‘final genotypes’ (Table 3) that would be used for population genetic analyses, any genotype with a prinseq quality score < 70% and any called with only one successful PCR replicate were discarded. Then, the genotypes generated from CHIIMP and ConGenR were compared across all datasets, and if there were any incongruent alleles we manually evaluated the sequences to discern the cause, whether it was due to actual differences in repeat motif number, primer site mismatches, or another reason (Online Appendix 1).
Fragment analysis
As the primers used for PCR were not fluorescently labelled for CE, we generated electropherograms for a subset of samples across all loci to evaluate allele size utilizing a TapeStation 2200 (Agilent) with High Sensitivity tapes. PCR products were diluted 1:4–1:10 depending on DNA quantification determined by a Qubit 2.0 using a High Sensitivity kit.
Statistical analyses
Descriptive statistics, a single factor analysis of variance (ANOVA), and a linear regression were calculated in Microsoft Excel v16.16.20 using the data analysis add in. The ANOVA compared the percentage of reads passing prinseq-lite quality filters against sample types. MicroDrop v1.01 (Wang and Rosenberg 2012) was run on the pooled dataset using default parameters to evaluate allelic dropout rates across samples and loci. We did not enforce Hardy–Weinberg Equilibrium on our data due to the low number of alleles and samples. The program was run once on the raw CHIIMP genotypes and once on the final genotypes (Table 3). MicroDrop calculates allelic dropout rates using an expectation–maximization algorithm in a maximum-likelihood method, but it is not designed for replicated PCR samples, so ConGenR (Lonsinger and Waits 2015) was used to evaluate allelic dropout in the replicate dataset across loci and DNA concentration bins. ConGenR calculates allelic dropout rates by comparing all PCR replicate genotypes to a consensus genotype. It is primarily based on Broquet and Petit (2004) but will calculate allelic dropout for homozygous individuals when a false allele is present. Additionally, ConGenR was run once on the raw CHIIMP genotypes and once on the final genotypes.
Results
Effects of sample quality
A total of 387,810 reads were sequenced across all samples. Sample quality was determined by prinseq-lite, which uses standard quality control measures including length distribution, GC content, and ambiguous bases (Table S1). Mean quality across all samples was 85.6%, with a range of 57.43–99.48% (median = 92.6%, mode = 96.82%). Separated by sample type, the mean quality was as follows: 95.99% (SE ± 0.99%), 95.06% (SE ± 0.68%), and 73.84% (SE ± 2.10%) for tissue, HCMS, and LCMS respectively (Table S2). The ANOVA was significant (P = < 0.001) and the regression resulted in an R2 of 0.44 (P = < 0.001), indicating our DNA quantification bins were predictive of sequencing quality.
CHIIMP genotypes were accurate for the tissue sample, especially in the pooled dataset. Mismatched alleles were recovered most frequently in LCMS, which routinely appeared to fail PCR. Mismatches were often associated with PCR stutter, PCR artifacts, or more than two prominent sequences, as identified by CHIIMP (Table 3). Individual samples did not appear to recover specific CHIIMP flags across all replicates, neither did specific microsatellites; however, the locus GS-2 recovered frequent flags for all three metrics (Table 3).
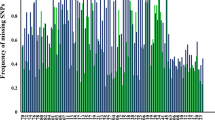
The HCMS recovered consistently high-quality sequences as determined by prinseq-lite. Only a single PCR replicate from UMMZ 79755 had < 85% pass quality metrics. All other replicates were over 85%, and most had over 95% pass quality filters. The quality of LCMS replicates ranged from 69.3 to 81.3%, with a high amount of variation among replicates (Fig. 1). For example, LACM 95619 sequence quality ranged from 67.7 to 92.05% for GS-4. In this instance, three completely different sets of alleles were recovered, providing no confidence in those genotypes despite one replicate recovering a 92.05% quality score.

Scatter plot of average quality of PCR replicates following prinseq-lite. Replicates for each specimen are shown across the x-axis, and the percentage of ‘good’ reads are shown on the y-axis. Samples are sorted by type: tissue, high concentration museum specimens, and low concentration museum specimens. Individuals of the same sample type are separated by a dashed line
Effects of microsatellite length
We recovered genotypes more frequently for shorter microsatellites across all samples. As expected, the tissue sample (HSU 8180) had consistent genotypes called across all replicates, except in one instance where a 2 bp difference was detected between PCR replicates in GS-4 (Table 3). In this case, both the bioinformatic dataset and pooled dataset recovered a homozygous genotype. Based on read depth, it is possible the minor alleles (92 and 94 in replicates 1 and 2 respectively) were sequencing errors or PCR stutter related to high depth of coverage.
The HCMS performed as well and occasionally better than the tissue sample, but often had more than two prominent sequences flagged (Table 3) and still had some missing allele calls (e.g., GS-4 in UMMZ 79755). Overall, HCMS resulting genotypes were reliable, and only appeared to lack confirmation in GLSA-52, the longest microsatellite evaluated.
The LCMS sometimes recovered accurate genotypes, however there was more variation in data quality (Fig. 1), so their genotypes required stringent evaluation. It was clear that as microsatellite length increased, genotype reliability decreased, but even the shortest marker GS-2 lacked four out of nine genotypes, of which two did not match across replicates. GS-4 had one missing and five mismatched genotypes, GLSA-12 had eight missing and one mismatch, GLSA-22 had seven missing, and GLSA-52 was missing all genotypes.
Allelic dropout rates
MicroDrop was run on the raw CHIIMP genotypes called from the pooled dataset and again on the final genotypes. In the pooled dataset, locus specific dropout rates ranged from 0 (GLSA-22) to 48% (GLSA-52) and rates across loci for each sample ranged from 0 (HSU 1836, UMMZ 79760, and LACM 95619) to 40.58% (MVZ 2088) (Table 4). Following manual genotype evaluation, locus specific dropout rates ranged from 0 (GLSA-22 and GS-4) to 36.3% (GLSA-52) and 0 (HSU 1836, UMMZ 79760) to 100% (MVZ 5211) for individual samples. When MVZ 5211 was removed, the highest rate recovered was in LACM 95619 (77.3%). The LCMS had higher dropout rates after manually evaluating genotypes due to the removal of low confidence genotypes.
Allelic dropout rates from ConGenR differed from MicroDrop which was unsurprising given their different algorithmic implementations. In the replicate dataset, locus specific dropout rates ranged from 0 (GLSA-22 and GLSA-52) to 18.75% (GS-2) and stayed the same after manual genotype evaluation (Table 4). The average rate across loci decreased, however, because the rate for GS-4 decreased from 8.33 to 0%. ConGenR also calculated overall allelic dropout rates for HCMS, LCMS, and the tissue sample (Table 4). As expected, dropout rates were highest in LCMS and lowest in the tissue sample.
Electropherogram comparisons
Genotypes from the TapeStation 2200 were either exact matches or 1–20 bp larger than the CHIIMP and ConGenR calls (GS-2, GS-4, and GLSA-52). Some alleles appeared to be larger on the TapeStation (GLSA-12 and GLSA-22) and some replicates also repeatedly failed on the TapeStation and during GBS (MVZ 5211 at GLSA-22 and MVZ 2088 at GLSA-12, Fig. S1).
Discussion
Museum collections are increasingly being used for molecular sequencing, yet comparative studies on the retrieval and reliability of microsatellite genotypes from these data sources are not readily available. Here we show that while museum specimens can recover reliable and important genotypes for elusive species, additional precautions must be made prior to acceptance of HTS generated genotypes, particularly for LCMS.
Best practices
As depicted in Fig. 2, first bin samples based on DNA concentration. Second, we suggest performing minimally three successful PCR replicates per locus prior to genotyping and including mtDNA to ensure endogenous DNA presence. Samples deemed LCMS may require additional replication compared to HCMS. Visualization of PCR products on a TapeStation/Bioanalyzer can inform the need for additional replicates prior to library preparation and sequencing. After successful amplification, perform library preparation on PCR products and sequence to a minimum depth of 1000 reads per sample per microsatellite marker on an Illumina platform with adequate insert length. Samples should be re-sequenced if the minimum read depth is not met.

Best practices flowchart, with emphasis on low concentration samples. By following these practices, we reduced our allelic dropout by about 3% across loci according to MicroDrop v1.01, though we also had to remove many of the LCMS genotypes as we could not confirm authenticity
Next, run CHIIMP and prinseq-lite in parallel to generate genotypes and evaluate sequence quality. Samples with low quality scores from prinseq should be noted as they may be prone to erroneous genotypes. Low concentration samples should be evaluated for mismatched genotypes to determine where the differences occur (e.g., primer region or repeat elements). If the primer sequence varies, manually correct the length as if the entire primer sequence was included, and ignore primer site size mismatches as this is likely an artifact of sequencing or amplification errors. If an allele does not have a priming site error, it is important to evaluate if the size shifts follow evolutionary patterns. For example, if a dinucleotide sequence shifts by two base pairs that makes evolutionary sense. However, stutter sequences are often frame shifted by the repeat motif size, and stutter evaluation in dinucleotide microsatellites is challenging due to the short difference between true alleles and stutter peaks (O’reilly et al. 2000; Barbian et al. 2018). Thus, we also recommend designing tetranucleotide assays whenever possible. CHIIMP calculates the proportion of reads associated with various genotypes, and based on the proportion of sequences with one repeat motif difference it can be flagged as stutter (a modifiable parameter).
Across the replicate dataset we evaluated differences in individual PCR replicate genotypes from samples of varying DNA concentrations. We found HCMS were generally in agreement across the three datasets and provide justification for pooling PCR replicates prior to library preparation, significantly reducing cost. The LCMS were variable, but still often yielded the same allele calls from all three datasets. If a critical sample yields low DNA concentrations, individual library preparation can alleviate dropout concerns in those samples. Otherwise, LCMS individuals should be excluded from population genetic studies.
Microsatellite recovery
Our data separated samples into three categories before genotyping: tissue, HCMS, and LCMS, based on DNA concentration. Genotypes were recovered at a higher rate than expected based on failed PCRs from LCMS. The LCMS recovered a PCR amplification success rate of 21.4% (Table S3) yet recovered 30 genotypes out of 72 possible (42%, Table 3). This did not include removal of problematic genotypes. When only agreeable genotypes were included, this reduced the 30 genotypes to 6 or 8.3%. Despite this low rate of confirmation among the LCMS, this study quantifies rates for genotyping success via GBS on degraded museum specimens for the first time. Alternatively, for HCMS the rate of recovered genotypes was 91.7% (66 out of 72), and 86.1% (62 out of 72) for agreeable genotypes. For the tissue sample, 100% (16 out of 16) of the replicates resulted in a genotype, with 87.5% (14 out of 16) confirmed by the second genotype recovered. This data provides robust support that the rate of disagreement in the HCMS is negligible, only 1.4% lower than the tissue sample, and has shown that samples which do not reliably amplify in PCR are prone to inaccurate genotypes.
Again, the LCMS genotypes recovered required fine scale evaluation to ensure accuracy and repeatability for downstream analyses, as inaccurate allele calls could affect population genetic inferences. Variable genotypes were much more prevalent in LCMS (16 instances in the LCMS versus only two in the HCMS), but may not be specifically due to allelic dropout as in fecal samples (Piggott et al. 2004; Regnaut et al. 2006), since alleles outside the expected bin sizes were recovered (see GS-4 for the LCMS) and only rarely was potential allelic dropout recovered (see GS-2 for LACM 95619). Further optimization of the size buffer setting in CHIIMP may eliminate these alleles, as CE analysis would traditionally ignore peaks outside of the expected size range in programs like GeneMapper™ (Applied Biosystems).
We were not able to compare the final genotypes recovered here to other population genetics studies on G. oregonensis, as this is the first time such a study has occurred in this species. The study performed by Barbian et al. (2018) used GBS to re-genotype chimpanzees with known life-history data and previous CE genotypes, and noted a shift of 1–3 bp in genotype results between their CE and GBS data, which we also recovered with certain loci across the TapeStation electropherograms. We noticed larger fragments (~ 17–45 bp) from the TapeStation than the GBS data at locus GLSA-12 and GLSA-22, which may result from bioinformatic adapter removal steps. Many studies have reported shifted alleles of the same PCR products on different runs of an automated capillary sequencer or with a different size standard (Haberl and Tautz 1999; Ellis et al. 2011).
In concordance with Campana et al. (2012), there was no direct correlation between mtDNA and nDNA amplification success rates in our data. All samples recovered reliable mtDNA signatures, even though many (particularly the LCMS) lacked nDNA at microsatellite loci (Supplemental Materials). Additionally, the HCMS recovered reliable genotypes across all markers, but the longer the microsatellite, the worse the locus amplified. This was especially apparent for the LCMS which failed to recover genotypes for the longest locus. Preferential amplification of short microsatellites was also observed in Rizzi et al. (2012) and Wandeler et al. (2003).
Most genotypes generated by ConGenR matched our final genotypes (Table 3), though in some cases ConGenR indicated uncertainty (denoted with a ‘0’). ConGenR also assigned genotypes to MVZ 5211 at GS-4 and MVZ 2088 at GLSA-12, but we did not as MVZ 5211 had variable allele calls and MVZ 2088 had only one amplified PCR replicate at that locus. For MVZ 2088 at GS-4, the 92/96 genotype ConGenR assigned and the 96/96 genotype we assigned were actually the same sequence with the same number of repeats, but the 96 bp allele contained four more reverse primer base pairs (Online Appendix 1).
The CHIIMP pipeline (Barbian et al. 2018) worked well for our samples, and allowed for optimization and customization of commands for recovering more strict or lenient genotypes. The combination of multiple replicates of PCR, quality filtering, and manual evaluation of CHIIMP results increased our confidence in the genotypes recovered by museum specimens in this study. However, it also highlighted various methods microsatellites could be genotyped which stresses the need to standardize genotyping procedures.
Allelic dropout rates
According to MicroDrop, we recovered high rates of allelic dropout averaging 12.6% in the pooled dataset across all five microsatellite loci. After manual editing, the average rate of allelic dropout decreased to 9.8% across all loci. The average rate of allelic dropout across all loci according to ConGenR, however, was 7.23%, and this rate was reduced to 5.57% after manual evaluation. ConGenR calculates dropout rates based completely on PCR success, so the difference between MicroDrop and ConGenR was within expectations.
MicroDrop allelic dropout rates in the LCMS (average of 19.6%) conformed to rates reported for various studies of avian fecal allelic dropout (mean of 21%, Regnaut et al. 2006) but was much higher than rates reported from chimpanzee fecal samples via GBS (7%, Barbian et al. 2018). This may be due to the fact that Barbian et al. (2018) only genotyped samples at loci that had over 500 reads (counts.min = 500). ConGenR also reported very high LCMS dropout rates (33.3%), but the rate did decrease to 20% after manual evaluation which would conform to rates reported in Regnaut et al. (2006). Results from Donaldson et al. (2020) corroborated our findings, and noted that allelic dropout and false alleles increased while PCR success decreased following sample dilutions.
The HCMS all recovered very low rates of allelic dropout, 2.4% in MicroDrop and 5% in ConGenR, which decreased to < 0.001% and 4.76% respectively after manual evaluation. These three samples performed on par with GBS studies derived from tissue samples (Darby et al. 2016) and provide robust evidence for the utility of museum specimens in recovery of microsatellite genotypes.
In our tissue sample, raw results from CHIIMP generated a high rate of allelic dropout in MicroDrop (14.7%) compared to 0.4% in Darby et al. (2016). Following manual evaluation, the rate was reduced to 8.5%, which was still higher, but this was partially due to one instance of primer trimming (GS-4).
Dropout rates for the pooled dataset separated by length were 7.5% for short, 0.35% for medium, and 48% for long loci. The higher rate observed in short loci is likely attributed to non-specific amplification or higher amounts of stutter and PCR artifacts. After manual evaluation, we recovered a 0.0025%, 6.3%, and 36% dropout rate for short, medium, and long loci respectively.
Number of alleles
We found that the number of alleles recovered by CHIIMP in all datasets was higher than in our final genotypes due to removal of inaccurate alleles from the raw data. The number of alleles per locus remained the same at two loci (GS-2 and GLSA-12, although a different allele was present in the final genotypes for both loci; allele ‘96’ in GS-2 and ‘162’ in GLSA-12), and decreased in the other three loci. Compared to the number of alleles recovered from G. sabrinus, we recovered fewer alleles in GS-2, GS-4, GLSA-22, and GLSA-52, and more alleles in GLSA-12. However, previous research in G. sabrinus included more individuals and a wider geographic distribution. Therefore, we do not expect the alleles recovered here to be exhaustive for G. oregonensis, and our counts seem reasonable for seven individuals.
Furthermore, multiple studies have noted recovery of additional allelic diversity from GBS, traditionally lost in CE, as a result of direct access to allele sequences and the ability to evaluate homoplasy. For example, Darby et al. (2016) found a 44% increase in alleles after accounting for homoplasy in their dataset (164 to 294 alleles). We also recovered homoplastic events in the replicate dataset (Online Appendix 2), and recovered 28 unique alleles based on fragment length but 39 unique alleles based on whole sequence polymorphisms (28.2% increase). The increase in alleles was most prominent in GLSA-52 (3–9 alleles).
Conclusion
GBS is an effective way to generate affordable genotype results for degraded specimens when stringent protocols and deep sequencing is performed. Our costs were under ~ $22 per sample (Online Appendix 3), which is higher than other GBS studies (Darby et al. 2016), however, we only performed singleplex PCR, and if multiplex PCRs were performed the cost could be significantly reduced.
Several bioinformatic pipelines have already been developed to generate microsatellite genotypes from HTS data (De Barba et al. 2017; Barbian et al. 2018; Pimentel et al. 2018; Tibihika et al. 2019), and have screened a variety of starting template types including tissue, hair, and fecal samples. However, this is the first time GBS methods have been applied to evaluate allelic dropout rates from mammalian study skin derived DNA samples. Our results show that when binning samples by DNA concentration, robust genotyping can be recovered from museum specimens, especially for samples deemed HCMS. On the other hand, repeated PCR is necessary for LCMS, and this does not completely eliminate the opportunity for false genotypes to be incorporated into the dataset.
Museum specimens are important as they provide temporal perspective and inclusion of rare species, but appropriate QC measures need to be undertaken to ensure accurate genotypes. Our data demonstrate the ability to reliably incorporate microsatellite genotypes from early twentieth century museum study skins, in combination with modern surveys, to evaluate spatial and temporal shifts in population genetics.
Data availability
Cytochrome-b sequences can be found on GenBank accessions MT498442-MT498448. Raw data from all microsatellite replicates have been uploaded to GenBank SRA under the following Bioproject: PRJNA721448 and Biosamples: SAMN18718222-8. Microsatellite output files from the CHIIMP pipeline can be found at Figshare: 10.25573/data.13491642; 10.25573/data.13491792; 10.25573/data.13492041.
References
Abdelkrim J, Robertson BC, Stanton J-AL, Gemmell NJ (2009) Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. BioTechniques 46:185–192. https://doi.org/10.2144/000113084
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge
Andrews KR, De Barba M, Russello MA, Waits LP (2018) Advances in using non-invasive, archival, and environmental samples for population genomic studies. In: Hohenlohe PA, Rajora OP (eds) Population genomics: wildlife. Springer International Publishing, Cham, pp 63–99
Arbogast BS, Schumacher KI, Kerhoulas NJ et al (2017) Genetic data reveal a cryptic species of New World flying squirrel: Glaucomys oregonensis. J Mammal 98:1027–1041. https://doi.org/10.1093/jmammal/gyx055
Barbian HJ, Connell AJ, Avitto AN et al (2018) CHIIMP: an automated high-throughput microsatellite genotyping platform reveals greater allelic diversity in wild chimpanzees. Ecol Evol 8:7946–7963
Bilska K, Szczecińska M (2016) Comparison of the effectiveness of ISJ and SSR markers and detection of outlier loci in conservation genetics of Pulsatilla patens populations. PeerJ 4:e2504
Blagoderov V, Kitching IJ, Livermore L et al (2012) No specimen left behind: industrial scale digitization of natural history collections. ZooKeys 209:133
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13:3601–3608
Campana MG, Lister DL, Whitten CM et al (2012) Complex relationships between mitochondrial and nuclear DNA preservation in historical DNA extracts. Archaeometry 54:193–202
Curto M, Winter S, Seiter A et al (2019) Application of a SSR-GBS marker system on investigation of European Hedgehog species and their hybrid zone dynamics. Ecol Evol 9:2814–2832. https://doi.org/10.1002/ece3.4960
Darby BJ, Erickson SF, Hervey SD, Ellis-Felege SN (2016) Digital fragment analysis of short tandem repeats by high-throughput amplicon sequencing. Ecol Evol 6:4502–4512
De Barba M, Miquel C, Lobréaux S et al (2017) High-throughput microsatellite genotyping in ecology: improved accuracy, efficiency, standardization and success with low-quantity and degraded DNA. Mol Ecol Resour 17:492–507
Donaldson ME, Jackson K, Rico Y et al (2020) Development of a massively parallel, genotyping-by-sequencing assay in American badger (Taxidea taxus) highlights the need for careful validation when working with low template DNA. Conserv Genet Resour 12:601–610. https://doi.org/10.1007/s12686-020-01146-8
Duan C, Li D, Sun S et al (2014) Rapid development of microsatellite markers for Callosobruchus chinensis using Illumina paired-end sequencing. PloS One 9:e95458
Ellis JS, Gilbey J, Armstrong A et al (2011) Microsatellite standardization and evaluation of genotyping error in a large multi-partner research programme for conservation of Atlantic salmon (Salmo salar L.). Genetica 139:353–367
Glenn TC, Schable NA (2005) Isolating microsatellite DNA loci. Methods Enzymol 395:202–222
Glenn TC, Nilsen RA, Kieran TJ et al (2019) Adapterama I: universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). PeerJ 7:e7755
Griffiths SM, Fox G, Briggs PJ et al (2016) A Galaxy-based bioinformatics pipeline for optimised, streamlined microsatellite development from Illumina next-generation sequencing data. Conserv Genet Resour 8:481–486
Haberl M, Tautz D (1999) Comparative allele sizing can produce inaccurate allele size differences for microsatellites. Mol Ecol 8:1347–1349
Hawkins MT, Hofman CA, Callicrate T et al (2016a) In-solution hybridization for mammalian mitogenome enrichment: pros, cons and challenges associated with multiplexing degraded DNA. Mol Ecol Resour 16:1173–1188
Hawkins MT, Leonard JA, Helgen KM et al (2016b) Evolutionary history of endemic Sulawesi squirrels constructed from UCEs and mitogenomes sequenced from museum specimens. BMC Evol Biol 16:80
Hofreiter M, Serre D, Poinar H et al (2001) Ancient DNA. Nat Rev Genet 2:353–359
Jónsson H, Ginolhac A, Schubert M et al (2013) mapDamage2. 0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29:1682–1684
Kiesow AM, Wallace LE, Britten HB (2011) Characterization and isolation of five microsatellite loci in northern flying squirrels, Glaucomys sabrinus (Sciuridae, Rodentia). West N Am Nat 71:553–556
Kistler L, Ware R, Smith O et al (2017) A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res 45:6310–6320. https://doi.org/10.1093/nar/gkx361
Lane MA (1996) Roles of natural history collections. Ann Mo Bot Gard 83:536–545. https://doi.org/10.2307/2399994
Lepais O, Chancerel E, Boury C et al (2020) Fast sequence-based microsatellite genotyping development workflow. PeerJ 8:e9085. https://doi.org/10.7717/peerj.9085
Lister AM, Group CCR (2011) Natural history collections as sources of long-term datasets. Trends Ecology Evol 26:153–154
Lonsinger RC, Waits LP (2015) ConGenR: rapid determination of consensus genotypes and estimates of genotyping errors from replicated genetic samples. Conserv Genet Resour 7:841–843
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10. https://doi.org/10.14806/ej.17.1.200
McDonough MM, Parker LD, Rotzel McInerney N et al (2018) Performance of commonly requested destructive museum samples for mammalian genomic studies. J Mammal 99:789–802
Miller W, Drautz DI, Janelka JE et al (2009) The mitochondrial genome sequence of the Tasmanian Tiger (Thylacinus cynocephalus). Genome Res 19:213–220
Miller MP, Knaus BJ, Mullins TD, Haig SM (2013) SSR_pipeline: a bioinformatic infrastructure for identifying microsatellites from paired-end Illumina high-throughput DNA sequencing data. J Hered 104:881–885
Morin PA, Manaster C, Mesnick SL, Holland R (2009) Normalization and binning of historical and multi-source microsatellite data: overcoming the problems of allele size shift with allelogram. Mol Ecol Resour 9:1451–1455
O’Neill M, McPartlin J, Arthure K et al (2011) Comparison of the TLDA with the Nanodrop and the reference Qubit system. J Phys Conf Ser 307:012047. https://doi.org/10.1088/1742-6596/307/1/012047
O’reilly PT, Canino MF, Bailey KM, Bentzen P (2000) Isolation of twenty low stutter di-and tetranucleotide microsatellites for population analyses of walleye pollock and other gadoids. J Fish Biol 56:1074–1086
Paabo S, Poinar H, Serre D et al (2004) Genetic analyses from ancient DNA. Annu Rev Genet 38:645–679
Piggott MP, Bellemain E, Taberlet P, Taylor AC (2004) A multiplex pre-amplification method that significantly improves microsatellite amplification and error rates for faecal DNA in limiting conditions. Conserv Genet 5:417–420
Pimentel JS, Carmo AO, Rosse IC et al (2018) High-throughput sequencing strategy for microsatellite genotyping using neotropical fish as a model. Front Genet 9:73
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6:847–859
Regnaut S, Lucas FS, Fumagalli L (2006) DNA degradation in avian faecal samples and feasibility of non-invasive genetic studies of threatened capercaillie populations. Conserv Genet 7:449–453
Rizzi E, Lari M, Gigli E et al (2012) Ancient DNA studies: new perspectives on old samples. Genet Sel Evol 44:21
Rohland N, Reich D (2012) Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. https://doi.org/10.1101/gr.128124.111
Šarhanová P, Pfanzelt S, Brandt R et al (2018) SSR-seq: genotyping of microsatellites using next-generation sequencing reveals higher level of polymorphism as compared to traditional fragment size scoring. Ecol Evol 8:10817–10833
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics (Oxford, England) 27:863–864. https://doi.org/10.1093/bioinformatics/btr026
Shapiro B, Hofreiter M (2012) Ancient DNA: methods and protocols. Springer
Silva PI, Martins AM, Gouvea EG et al (2013) Development and validation of microsatellite markers for Brachiaria ruziziensis obtained by partial genome assembly of Illumina single-end reads. BMC Genomics 14:17
Smith AB, Santos MJ, Koo MS et al (2013) Evaluation of species distribution models by resampling of sites surveyed a century ago by Joseph Grinnell. Ecography 36:1017–1031. https://doi.org/10.1111/j.1600-0587.2013.00107.x
Tibihika PD, Curto M, Dornstauder-Schrammel E et al (2019) Application of microsatellite genotyping by sequencing (SSR-GBS) to measure genetic diversity of the East African Oreochromis niloticus. Conserv Genet 20:357–372
Vartia S, Villanueva-Cañas JL, Finarelli J et al (2016) A novel method of microsatellite genotyping-by-sequencing using individual combinatorial barcoding. R Soc Open Sci 3:150565
Wandeler P, Smith S, Morin PA et al (2003) Patterns of nuclear DNA degeneration over time—a case study in historic teeth samples. Mol Ecol 12:1087–1093
Wang C, Rosenberg NA (2012) MicroDrop: a program for estimating and correcting for allelic dropout in nonreplicated microsatellite genotypes version 1.01. See https://web.stanford.edu/group/rosen/berglab/microdrop.html
Weiß CL, Schuenemann VJ, Devos J et al (2016) Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. R Soc Open Sci 3:160239
White LC, Mitchell KJ, Austin JJ (2018) Ancient mitochondrial genomes reveal the demographic history and phylogeography of the extinct, enigmatic thylacine (Thylacinus cynocephalus). J Biogeogr 45:1–13
Williams SL (1999) Destructive preservation: a review of the effect of standard preservation practices on the future use of natural history collections.Göteborg, Sweden
Zhan L, Paterson IG, Fraser BA et al (2017) megasat: automated inference of microsatellite genotypes from sequence data. Mol Ecol Resour 17:247–256. https://doi.org/10.1111/1755-0998.12561
Zittlau KA, Davis CS, Strobeck C (2000) Characterization of microsatellite loci in northern flying squirrels (Glaucomys sabrinus). Mol Ecol 9:826–827
Acknowledgements
The authors wish to thank Clare O’Connell, Michael Kiso, Jack Lemke, and Evan Miller for providing laboratory assistance; Beatrice Hahn and Jesse Connell for their assistance implementing the CHIIMP pipeline; and Nancy Rotzel and Katie Murphy for performing the sequencing runs at the Center for Conservation Genomics, Smithsonian Conservation Biology Institute, and the Laboratory of Analytical Biology, National Museum of Natural History, Smithsonian Institution, respectively. Several museums allowed for destructive sampling of specimens: MVZ, Chris Conroy, Jim Patton, Eileen Lacey, and Michael Nachman; LACM, Jim Dines, and Kayce Bell; HSU, Alyssa Semerdjian, Nick Kerhoulas, and Allison Bronson; and UMMZ, Cody Thompson.
Funding
This study was funded by MTRH’s discretionary start-up funds, along with an American Society of Mammalogists Grants-in-Aid award, Sigma Xi Grants-in-Aid of Research award (G201903158734905), and Humboldt State University Department of Biology Master’s Student Grant.
Author information
Authors and Affiliations
Contributions
MTRH and SCY conceived of the study and performed laboratory work. SCY and EM performed bioinformatics. MTRH and SCY wrote the manuscript and performed statistical analyses. All authors analyzed the data and approved of the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal research
Not applicable.
Consent to participate
Not applicable.
Consent to publish
All authors are aware and consent to publish.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, S.C., Malekos, E. & Hawkins, M.T.R. Assessing genotyping errors in mammalian museum study skins using high-throughput genotyping-by-sequencing. Conservation Genet Resour 13, 303–317 (2021). https://doi.org/10.1007/s12686-021-01213-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-021-01213-8