
Abstract
The secretory phospholipase A2 (sPLA2) group of secreted enzymes hydrolyze phospholipids and lead to the production of multiple biologically active lipid mediators. sPLA2s and their products (e.g., eicosanoids) play a significant role in the pathophysiology of various inflammatory diseases, including life-threatening lung disorders such as acute lung injury (ALI) and the Acute Respiratory Distress Syndrome (ARDS). The ALI/ARDS spectrum of severe inflammatory conditions is caused by direct (such as bacterial or viral pneumonia) or indirect insults (sepsis) that are associated with high morbidity and mortality. Several sPLA2 isoforms are upregulated in patients with ARDS as well as in multiple ALI preclinical models, and individual sPLA2s exert unique roles in regulating ALI pathophysiology. This brief review will summarize the contributions of specific sPLA2 isoforms as markers and mediators in ALI, supporting a potential therapeutic role for targeting them in ARDS.
Similar content being viewed by others


Introduction
Phospholipase A2 enzymes (PLA2s) are a superfamily of proteins that hydrolyze the sn-2 position of glycerophospholipids to generate free fatty acids and lysophospholipids [1]. PLA2s can be classified into several groups based on their biochemical characteristics, including the cytosolic PLA2 (cPLA2), the secretory PLA2 (sPLA2), and Ca2+-independent PLA2 (iPLA2) groups [1]. These PLA2 enzymes are functionally important due to their capacity to generate lipids that are critical signaling messengers and important regulators of inflammatory processes. PLA2-catalyzed fatty acids, such as arachidonic acid (AA) and its metabolites (especially the eicosanoids), have been implicated in a diversity of inflammatory diseases [2,3,4]. Lysophospholipids, the other major products of PLA2 catalysis, are a large and important family of bioactive lipid molecules with diverse biologic functions [5].
Early studies in the 1980s and 1990s identified PLA2s as potential markers and mediators of acute lung injury (ALI) syndromes. Since that time, there has been extensive investigation into the roles of individual PLA2s in regulating several aspects of pulmonary inflammation [6]. Acute Respiratory Distress Syndrome (ARDS) is a severe form of clinical ALI caused by direct lung injury such as pneumonia, acid aspiration, and mechanical ventilation, and also by indirect injury such as sepsis [7]. In both cases, inflammatory insults, including bacteria (e.g., Staph aureus, Strep pneumoniae), viruses (e.g., SARS-CoV-2, influenza), acid (e.g., due to aspiration), or injurious mechanical forces (e.g., during positive pressure ventilation), lead to the disruption of the alveolo-capillary permeability, dysfunction of pulmonary surfactant, and infiltration of neutrophils; all these processes represent key features of ALI [8]. The resulting protein-rich edema fluid and uncontrolled inflammation (e.g., cytokine storm) cause severe hypoxemia and respiratory failure [8]. Understanding the mechanisms underlying ALI, and identifying molecular targets that can be exploited therapeutically, are the major areas of research focus in the field. This brief review will discuss and summarize our current understanding of the roles of secreted PLA2s in the regulation of lung injury and inflammation. We also will highlight knowledge gaps in the field and potential areas of research for future studies.
Overview of sPLA2 Enzymes
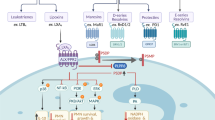
The sPLA2 family consists of low molecular weight (~14–19 kDa) enzymes that are secreted extracellularly. To date, at least 11 mammalian sPLA2s (IB, IIA, IIC, IID, IIE, IIF, III, V, X, XIIA, and XIIB) have been identified, of which XIIB represents the only inactive isoform [9, 10]. For their catalytic activity, sPLA2s require millimolar levels of Ca2+, and their catalytic site contains a histidine-aspartic acid dyad (Fig. 1). In contrast to cPLA2s and iPLA2s, sPLA2s act primarily on phospholipids that are exposed extracellularly, such as the outer plasma membrane of nearby cells, membranes of extracellular vesicles or bacteria, alveolar surfactant, etc. Each sPLA2 member has a distinct biological role attributed to their unique substrate specificity, different cell and tissue distribution, and enzymatic properties [11]. Individual sPLA2s can affect lung function directly by modifying the lipid composition of cell membranes or pulmonary surfactant, or indirectly through the generation of lipid mediators (Fig. 1). In addition to their biological functions, sPLA2s have also emerged as potential biomarkers of ARDS [12], as their expression and secretion are significantly upregulated in patients with ARDS, and in animal and in vitro ALI models (as discussed below).

Mammalian sPLA2 structure and generation of lipid mediators. (Top) Simplified schema representing protein structures for the three mammalian sPLA2 enzymes potentially relevant to acute lung injury syndromes. All active sPLA2s have Ca2+-binding (purple) and catalytic sites (light green), while a C-terminal extension (red) is present in sPLA2-IIA and sPLA2-X. sPLA2-X also contains an N-terminal propeptide (orange), which is cleaved off for enzymatic activation. sPLA2-V has the fewest domains, with no N-terminal propeptide or C-terminal extension, and contains only six disulfides. (Middle) During inflammatory conditions such as ALI/ARDS and sepsis, sPLA2s hydrolyze phospholipids to produce free fatty acids (FFA) and lysophospholipids (LPL). (Bottom) FFAs include oleic acid, linoleic acid, arachidonic acid (AA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA). Through the subsequent actions of cyclooxygenase and 5-lipoxygenase enzymes, AA is converted into prostaglandins, leukotrienes, and other inflammatory eicosanoids. LPL products include the bioactive lipid mediators such as lysophosphatidylcholine (lyso-PC), lysophosphatidyl-ethanolamine (lyso-PE), lysophosphatidylglycerol (lyso-PG), and lysophosphatidic acid (LPA) (color figure online)
Role of sPLA2 Group IIA and IID in ALI
Group II sPLA2 enzymes (IIA, IIC, IID, IIE, IIF) exhibit a membrane binding preference for phosphatidylglycerol (PG), phosphatidylethanolamine (PE), and phosphatidylserine, while they have lower binding capacity for phosphatidylcholine (PC), the main phospholipid in the outer leaflet of mammalian plasma membranes.
Among the sPLA2s, sPLA2 group IIA (also defined as sPLA2-IIA, PLA2G2A, or gIIaPLA2) is the most well-studied sPLA2 and has been associated with a variety of diseases [13]. The circulating levels of sPLA2-IIA are very low in healthy conditions but increase significantly during inflammation or infection [13, 14]. Several studies have highlighted the role of sPLA2-IIA as a biomarker in sepsis and various infections [14,15,16]. sPLA2-IIA is also well-recognized for its antimicrobial role, especially toward gram-positive bacteria since it can directly hydrolyze the membranes of these organisms, which are enriched in PG and PE, the preferred substrates for this specific isoform [17]. In addition, sPLA2-IIA hydrolyzes membrane phospholipids of mitochondria that are released from activated platelets, which serves to promote neutrophil activation and amplify inflammation [18].
A role for sPLA2-IIA in ALI/ARDS is supported by multiple clinical and preclinical studies [11, 19]. Independent investigations have demonstrated increased sPLA2-IIA expression in the bronchoalveolar lavage (BAL) fluid and plasma of patients with ARDS [20,21,22,23], and also both sPLA2-IIA protein and mRNA can be detected in extracellular vesicles in BAL from patients with early stage ARDS [24]. Recently, sPLA2-IIA levels were found to be significantly elevated in the plasma of COVID-19 patients and correlated with disease severity [25]. In vitro, different cell types involved in ALI pathogenesis, such as alveolar macrophages and alveolar epithelial cells, express sPLA2-IIA upon stimulation with inflammatory agents such as endotoxins and TNF-α [20, 26]. sPLA2-IIA levels are also increased in multiple experimental models of ALI in vivo, while direct administration of sPLA2-IIA to the lungs causes respiratory distress and hydrolysis of surfactant PG [27, 28]. This associative data linking sPLA2-IIA to ALI pathophysiology led to studies that explored the therapeutic potential of inhibiting this enzyme. Although preclinical studies demonstrated that specific inhibition of sPLA2-IIA by the small molecule LY315920Na/S-5920 was effective in attenuating lung injury in animal models of ALI and sepsis [29], clinical trials using this same inhibitor failed to provide survival protection in patients with sepsis and organ failure [30, 31]. One potential explanation for these disappointing results is that sPLA2-IIA also has strong bactericidal effects, as mentioned earlier, and exerts protective roles against infections [14]. Therefore, although it could represent a useful marker of lung injury, sPLA2-IIA inhibition might impair the host defense against bacterial infections, and thus worsen ALI caused by these organisms. Despite extensive research into sPLA2-IIA, it is evident that new studies are needed to further explore the differential roles of sPLA2-IIA in regulating ALI and understand the integrated effects of its pro- and anti-inflammatory roles.
Recently, another isoform in the sPLA2-II group, sPLA2-IID (or PLA2G2D), was identified as a potential important mediator in respiratory infections. sPLA2-IID expression is increased in aged CD11c+ cells (alveolar macrophage and respiratory dendritic cells), while middle-aged mice lacking sPLA2-IID are protected against SARS-CoV (i.e., SARS) infection [32]. In the absence of sPLA2-IID, there was enhanced dendritic cell migration in the lymph nodes, augmented antiviral T-cell responses, reduced lung damage, and increased survival following viral infection. Increased sPLA2-IID expression in lung CD11c+ cells resulted in upregulation of several eicosanoids that might be involved in the initial immune response [32]. Based on these findings, it will be intriguing to further explore the role of this specific sPLA2 isoform in COVID-19 pathogenesis. Another in vivo study has demonstrated that sPLA2-IID expression is increased in the lungs of LPS-treated mice, further implicating this specific isoform in ALI pathogenesis [33].
Role of sPLA2-V in ALI
sPLA2 group V (defined as sPLA2-V, gVPLA2, or PLA2G5) is less well-studied compared to sPLA2-IIA; however, research in the last decade has provided strong evidence that it is involved in several inflammatory disease processes [34]. In contrast to sPLA2-II, sPLA2-V has a high capacity to bind PC, a major phospholipid in the outer leaflet of the mammalian plasma membrane and pulmonary surfactant. Some reports suggest that sPLA2-V preferentially releases fatty acids with a low degree of unsaturation, such as oleic and linoleic acids [11]; however, a large body of evidence also implicates sPLA2-V in AA mobilization and eicosanoid generation [11, 34,35,36].
One of the major effects of sPLA2-V in the lung is the hydrolysis of lung surfactant. Surfactant, a complex mixture of phospholipids and proteins secreted by alveolar epithelial type II cells, functions to reduce the surface tension at the air–liquid interface in the lung [37]. Dysfunctional surfactant results in alveolar injury and is a major hallmark of ALI. sPLA2-V contributes to disruption of surfactant activity by hydrolyzing PC, one of its major phospholipid components. This is evident in neonate mice overexpressing sPLA2-V that die within 8 h from birth due to increased surfactant hydrolysis and resulting severe lung damage [38].
In addition to directly affecting lung function by altering the surfactant activity, preclinical work from our group and others has demonstrated additional mechanisms by which sPLA2-V may participate in ALI pathogenesis (summarized in Fig. 2). In a direct model of LPS-induced ALI in mice, sPLA2-V protein and mRNA expression were increased in lung tissues [39]. sPLA2-V genetic deletion (i.e., knockout mice) or inhibition (after treatment with MCL-3G1, a specific sPLA2-V antibody) resulted in significant decreases in LPS-induced lung edema and inflammation [39]. In ALI, lung endothelial barrier disruption is a key event toward edema formation and facilitation of inflammatory cell accumulation into the alveolar space [40, 41]. Given the role of sPLA2-V in regulating lung edema in vivo, our group further explored the contribution of sPLA2-V in regulating lung endothelial cell (EC) function. These in vitro studies demonstrated that sPLA2-V expression is constitutively expressed in unstimulated lung EC and is upregulated upon treatment with LPS [42]. LPS is a potent inflammatory stimulus that induces actin cytoskeletal remodeling and disruption of EC junctions, leading to gap formation and increased permeability. Specific blockade of sPLA2-V by MCL-3G1 attenuates these cytoskeletal rearrangements and disruption of EC barrier by LPS [42]. Consistent with these findings, another group reported that increased mechanical forces (i.e., stretch) that occur during mechanical ventilation in patients with ARDS (i.e., ventilator-induced lung injury or VILI) also upregulate sPLA2-V in lung EC [43]. Similar to LPS, high levels of mechanical stretch cause EC barrier dysfunction and induce pro-inflammatory signaling. Genetic deficiency or inhibition of sPLA2-V (by the MCL-3G1) protects against lung vascular leak and inflammation in vivo in mice subjected to VILI [43]. In response to mechanical stretch, sPLA2-V upregulates lung EC ICAM-1 expression and cytokine production, as well as increased neutrophil adhesion to activated EC [43]. These studies from our group and others provide strong evidence that sPLA2-V is a critical mediator of lung EC barrier dysfunction in ALI. However, this prior work employed relatively simplistic ALI models using only one stimulus that does not fully recapitulate the human disease. To advance these observations further, our laboratory recently has developed a clinically relevant model of bacterial infection-induced ALI, which employs the live pathogen methicillin-resistant Staph aureus (MRSA). MRSA-induced pneumonia is a common cause for sepsis and ARDS, and recent studies have described the injurious effects of Staph aureus on lung EC [44, 45]. In our preliminary studies, MRSA induces the expression of sPLA2-V in lung EC, and its blockade or deficiency inhibits MRSA-induced EC barrier disruption in vitro and ALI in vivo (unpublished data). This work is ongoing.

sPLA2-V mediates acute lung injury pathophysiology. In response to multiple ALI-relevant stimuli, sPLA2-V expression (green hexagons) is increased in lung endothelium and directly hydrolyzes phospholipids to promote inflammatory processes (1). These include neutrophil activation, adhesion to lung EC, and migration to the interstitium and alveolar space to further drive inflammation (2). sPLA2-V also directly causes lung EC dysfunction and leads to gap formation and increased vascular permeability (3). During ALI, sPLA2-V is increased in the alveolar space where it hydrolyzes lung surfactant and contributes to alveolar injury, hyaline membrane formation, accumulation of edema fluid, and inflammatory cell recruitment (4, 5). In addition, sPLA2-V may alter lung macrophage function, including phagocytosis (6) (color figure online)
Although these findings have established a functional role for sPLA2-V in modulating endothelial injury and inflammation during ALI, the mechanisms underlying these effects are less well-understood. sPLA2-V induction in lung EC has been associated with increased cell surface expression [42, 43], suggesting that inflammatory stimuli induce its translocation to the outer membrane, where it can directly modulate barrier function. Indeed, exogenous administration of recombinant sPLA2-V causes significant disruption of the lung EC barrier [42]. Intracellular pathways activated by sPLA2-V include F-actin rearrangement (cytoskeleton remodeling), stress fiber formation, and adherens junction disruption. These changes were inhibited after blocking sPLA2-V activity using the MCL-3G1 antibody [42]. Notably, administration of the related sPLA2-IIA enzyme has no effect on EC barrier function [42].
There are several possible mechanisms by which sPLA2-V mediates EC barrier disruption, involving direct outer plasma membrane hydrolysis by sPLA2-V, and/or indirect disruption caused by the sPLA2-V-induced mediators/signaling [46]. As described above, sPLA2-V activity on the outer membrane of cells generates multiple bioactive lipid mediators (Fig. 1). Our group explored the effects of these products on EC permeability induced by sPLA2-V. Lysophosphatidyl-choline (lyso-PC), lysophosphatidyl-glycerol (lyso-PG), lysophosphatidic acid, and arachidonic acid all failed to cause significant barrier disruption in cultured lung EC [46]. Future studies may examine the role of other specific sPLA2-V hydrolysis products on EC permeability, such as oleic and linoleic acids, which can mediate EC dysfunction [47,48,49]. Another mechanism by which sPLA2-V may disrupt EC barrier function is by activating specific downstream intracellular signaling pathways. sPLA2-V activates members of the MAP kinase family and cPLA2 (group IV) in lung EC [46]. MAPK enzymes are known to regulate multiple EC functions [50], while cPLA2 is an inflammatory mediator that can be activated by both ERK and sPLA2-V enzymes [51, 52]. However, specific inhibition of these pathways did not attenuate the effects of sPLA2-V on EC permeability, suggesting that their activation is not required for sPLA2-V-mediated effects [46]. Whether other downstream signaling pathways activated by sPLA2-V contribute to EC permeability remains to be determined. At a whole-organism level, EC disruption may be mediated by the effects of sPLA2-V activity on neighboring cells. As one example, a recent study demonstrated that sPLA2-V activates neutrophils to release VEGF, a potent endothelial barrier disrupting agent [53]. EC–neutrophil interactions play a critical role in the progression of ALI, and sPLA2-V may mediate this cellular cross-talk to contribute to lung EC barrier failure.
To understand how sPLA2-V mediates its responses, it is also important to characterize its interactions with the plasma membrane. sPLA2-V binds to cells by interacting with cell surface heparan sulfate proteoglycans or by binding to phospholipids [54]. We demonstrated that heparinase treatment that results in degradation of the cell surface heparin sulfate moieties did not affect EC barrier disruption by sPLA2-V; however, in this study, we did not assess whether heparinase efficiently blocks sPLA2-V binding [46]. It is likely that binding of sPLA2-V to heparan sulfate proteoglycans is an important mechanism for sPLA2-V internalization and degradation, as discussed below. Whether sPLA2-V binds to other cell surface molecules, such as receptors, to activate downstream signaling that mediates EC barrier disruption should also be considered. Indeed, a recent report suggested that sPLA2-V mediates its effects in neutrophils by interacting with integrins or a PLA2 receptor (PLAR1) [53].
The maintenance of lung endothelial function and its protection from barrier disrupting stimuli are critical for the prevention of ALI. Understanding these processes may also facilitate the development of endothelium-targeted therapeutics for ARDS. As sPLA2-V appears to be an important regulator of endothelial barrier function and a promising therapeutic target, it is pivotal to understand not only the mechanisms by which this enzyme exerts its effects, but also how its expression, secretion, and clearance are regulated. Early work demonstrated that sPLA2-V binding to cell surface heparan sulfate proteoglycans mediates its internalization and degradation in human neutrophils [54]. More recent studies from our laboratory have identified a new clearance mechanism, in which sPLA2-V upon internalization into lung ECs associates with autophagosomes and is eliminated through a lysosome-related pathway [55]. Conditions in which this pathway is dysregulated could result in impaired clearance of this inflammatory enzyme and exaggeration of injurious effects during ALI.
In addition to the central role of sPLA2-V in regulating the lung endothelial barrier, other studies have implicated this enzyme in host immune responses. Human macrophages activated by IL-4 (M2 phenotype) express increased levels of sPLA2-V and exhibit increased phagocytosis of yeast-derived zymosan and bacteria, a process mediated by lysophosphatidyl-ethanolamine (LPE), an sPLA2-V product [56]. Macrophages lacking sPLA2-V have impaired phagocytosis, which can be restored after the exogenous addition of LPE [56]. Alternatively, sPLA2-V regulates phagocytosis by translocating to the phagosomes to mediate maturation of phagolysosomes [57]. Similar to macrophages, neutrophils are another component of the immune system that play a key role in phagocytosis and pathogen clearance. In a model of E.coli pneumonia, sPLA2-V-deficient mice have higher bacterial loads in the alveolar space compared to wild-type mice [58]. Although the exact mechanisms by which sPLA2-V affects bacterial loads remain unclear, this study suggested that the absence of sPLA2-V may result in diminished accumulation of neutrophils in the alveolar space [58]. Notably, although sPLA2-V is stored in neutrophil granules and may be liberated upon their activation to directly target bacteria, sPLA2-V has no direct effect on E.coli viability [58]. Other studies have demonstrated that sPLA2-V plays an important role in the generation of eicosanoids in macrophages and neutrophils [35, 54, 59]. Based on these prior investigations, future studies should explore the specific roles of sPLA2-V in modulating immune cell functions and signaling in ALI.
Although there is strong evidence that sPLA2-V modulates key functions of several cell types involved in ALI pathogenesis, in contrast to sPLA2-IIA there is a paucity of data regarding the potential role of sPLA2-V as a biomarker. One published study detected sPLA2-V in the BAL of infants with ARDS [22], while we have observed significant amounts of the enzyme in BAL of mice treated with LPS, MRSA, or high-tidal volume mechanical ventilation (unpublished data). These observations suggest that sPLA2-V may also have an important role as an ALI biomarker that warrants further study.
Role of sPLA2-X in ALI
Group X sPLA2 (defined as sPLA2-X, gXPLA2, PLA2G10, or GX-sPLA2), which is structurally related to both group-I and -II sPLA2, exhibits the most potent hydrolytic activity toward PC and induces the release of AA more efficiently than other sPLA2s [13, 60]. sPLA2-X is synthesized as a zymogen and requires proteolytic cleavage for activation [13]. Despite the high affinity of sPLA2-X for PC, the major surfactant phospholipid, mice overexpressing sPLA2-X display minimal abnormality of the respiratory tract function and have normal alveolar architecture and unaltered surfactant [38].This observation may be due to the fact that the sPLA2-X remains inactive under physiologic conditions and only becomes activated after proteolysis triggered by inflammatory stimuli [38].
Several studies have investigated the role of sPLA2-X as a potent regulator of airway inflammation underlying asthma (reviewed in refs. [13, 61]); however, its involvement in ALI is less well-understood. One study suggested that sPLA2-X may be involved in acute inflammatory processes induced by influenza infection [62]. Following H1N1 infection, sPLA2-X lung expression is increased, while targeted deletion of sPLA2-X significantly increased survival. Mechanistically, sPLA2-X produces several inflammatory lipid mediators at the early phase of inflammation and dysregulates the adaptive immune system [62]. It remains to be determined if inhibition of sPLA2-X activity during bacterial or viral infections has therapeutic potential in ALI.
Other sPLA2s and ALI
Knowledge is limited about other sPLA2s and their potential involvement in ALI. sPLA2-IB, similar to sPLA2-X, exists as an inactive proenzyme that can be cleaved to become active. In vitro studies have demonstrated that sPLA2-IB is released from alveolar epithelial cells upon Pseudomonas aeruginosa infection (a common cause of ALI) and that sPLA2-IB causes PC efflux, a process mediated by ABCTA1, a lipid-export pump [63]. This and other studies that have demonstrated increased levels of sPLA2-IB in ALI [22, 64] suggest that this isoform may play a functional role in ALI pathogenesis, but additional studies are needed.
Future Directions and Conclusion
Despite strong evidence that members of the sPLA2 family are critical mediators of ALI, there remains a lack of effective therapeutic strategies to target these enzymes. This is due in part to existing knowledge gaps regarding sPLA2 biology and its roles in regulating disease pathogenesis. In this review, we have summarized key findings about the regulation and distinct roles of individual sPLA2 isoforms in ALI and discussed some specific research areas that would benefit from future studies. In addition to those mentioned above, some other suggested future directions are the following: (1) although sPLA2s are secreted enzymes, they can also act intracellularly, through mechanisms that need to be elucidated. (2) In addition to their extracellular enzymatic activity, sPLA2s also interact with receptors or other molecules on the cell surface (e.g., PLA2 receptors, proteoglycans, integrins). New studies are needed to identify potential interacting or binding molecules for these enzymes and characterize the resultant downstream signaling. (3) Understanding the mechanisms underlying sPLA2 expression, secretion, and clearance under normal and pathological conditions would contribute to the development of new strategies to target the sPLA2s. (4) Several mutations in sPLA2 enzymes have been associated with diseases such as atherosclerosis, cardiovascular disease, and asthma [65]. Recently a rare mutation (R123H) was also identified in sPLA2-IIA in two infants with ARDS [66]. New studies are needed to identify additional mutations or SNPs associated with ARDS, and to understand how these genomic alterations affect sPLA2 function. (5) There is evidence that PLA2 isoforms interact with each other, but a potential role for PLA2 cross-talk in ALI is understudied. Finally, (6) improved understanding is needed regarding the effects of the lipid mediators produced by sPLA2 activity on lung cells.
In summary, there is significant evidence demonstrating that in association with ALI pathophysiology, sPLA2s are upregulated in different cell types in the lung compartment, their extracellular levels are highly elevated, and individual isoforms exert specialized biologic functions contributing to pro- or anti-inflammatory signaling. Due to their biological roles and extracellular presence during lung inflammation, sPLA2s represent attractive targets for therapeutic and/or diagnostic biomarker development.
References
Murakami, M., Sato, H., & Taketomi, Y. (2020). Updating phospholipase A(2) biology. Biomolecules 10(10), 1457.
Dennis, E. A., & Norris, P. C. (2015). Eicosanoid storm in infection and inflammation. Nat Rev Immunol, 15, 511–523.
Astudillo, A. M., Balboa, M. A., & Balsinde, J. (2019). Selectivity of phospholipid hydrolysis by phospholipase A(2) enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim Biophys Acta Mol Cell Biol Lipids, 1864, 772–783.
Mouchlis, V. D., & Dennis, E. A. (2019). Phospholipase A(2) catalysis and lipid mediator lipidomics. Biochim Biophys Acta Mol Cell Biol Lipids, 1864, 766–771.
Tan, S. T., Ramesh, T., Toh, X. R., & Nguyen, L. N. (2020). Emerging roles of lysophospholipids in health and disease. Prog Lipid Res, 80, 101068.
Hurley, B. P., & McCormick, B. A. (2008). Multiple roles of phospholipase A2 during lung infection and inflammation. Infect Immun, 76, 2259–2272.
Shaver, C. M., & Bastarache, J. A. (2014). Clinical and biological heterogeneity in acute respiratory distress syndrome: direct versus indirect lung injury. Clin Chest Med, 35, 639–653.
Thompson, B. T., Chambers, R. C., & Liu, K. D. (2017). Acute respiratory distress syndrome. N Engl J Med, 377, 562–572.
Murakami, M., Taketomi, Y., Miki, Y., Sato, H., Yamamoto, K., & Lambeau, G. (2014). Emerging roles of secreted phospholipase A2 enzymes: the 3rd edition. Biochimie, 107(Pt A), 105–113.
Murakami, M. (2019). Novel functions of phospholipase A(2)s: overview. Biochim Biophys Acta Mol Cell Biol Lipids, 1864, 763–765.
Murakami, M., Sato, H., Miki, Y., Yamamoto, K., & Taketomi, Y. (2015). A new era of secreted phospholipase A(2). J Lipid Res, 56, 1248–1261.
Wang, Y., Wang, H., Zhang, C., Zhang, C., Yang, H., Gao, R., & Tong, Z. (2019). Lung fluid biomarkers for acute respiratory distress syndrome: a systematic review and meta-analysis. Crit Care, 23, 43.
Murakami, M., Yamamoto, K., Miki, Y., Murase, R., Sato, H., & Taketomi, Y. (2016). The roles of the secreted phospholipase A(2) gene family in immunology. Adv Immunol, 132, 91–134.
van Hensbergen, V. P., Wu, Y., van Sorge, N. M., & Touqui, L. (2020). Type IIA secreted phospholipase A2 in host defense against bacterial infections. Trends Immunol, 41, 313–326.
Tan, T. L., Ahmad, N. S., Nasuruddin, D. N., Ithnin, A., Tajul Arifin, K., Zaini, I. Z., & Wan Ngah, W. Z. (2016). CD64 and group II secretory phospholipase A2 (sPLA2-IIA) as biomarkers for distinguishing adult sepsis and bacterial infections in the emergency department. PLoS ONE 11, e0152065.
Ahmad, N. S., Tan, T. L., Arifin, K. T., Ngah, W. Z. W., & Yusof, Y. A. M. (2020). High sPLA2-IIA level is associated with eicosanoid metabolism in patients with bacterial sepsis syndrome. PLoS ONE, 15, e0230285.
Dore, E., & Boilard, E. (2019). Roles of secreted phospholipase A(2) group IIA in inflammation and host defense. Biochim Biophys Acta Mol Cell Biol Lipids, 1864, 789–802.
Boudreau, L. H., Duchez, A. C., Cloutier, N., Soulet, D., Martin, N., Bollinger, J., Paré, A., Rousseau, M., Naika, G. S., Lévesque, T., Laflamme, C., Marcoux, G., Lambeau, G., Farndale, R. W., Pouliot, M., Hamzeh-Cognasse, H., Cognasse, F., Garraud, O., Nigrovic, P. A., Guderley, H., Lacroix, S., Thibault, L., Semple, J. W., Gelb, M. H., & Boilard, E. (2014). Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood, 124, 2173–2183.
Kitsiouli, E., Nakos, G., & Lekka, M. E. (2009). Phospholipase A2 subclasses in acute respiratory distress syndrome. Biochim Biophys Acta, 1792, 941–953.
Kitsiouli, E., Antoniou, G., Gotzou, H., Karagiannopoulos, M., Basagiannis, D., Christoforidis, S., Nakos, G., & Lekka, M. E. (2015). Effect of azithromycin on the LPS-induced production and secretion of phospholipase A2 in lung cells. Biochim Biophys Acta, 1852, 1288–1297.
Nakos, G., Kitsiouli, E., Hatzidaki, E., Koulouras, V., Touqui, L., & Lekka, M. E. (2005). Phospholipases A2 and platelet-activating-factor acetylhydrolase in patients with acute respiratory distress syndrome. Crit Care Med, 33, 772–779.
De Luca, D., Lopez-Rodriguez, E., Minucci, A., Vendittelli, F., Gentile, L., Stival, E., Conti, G., Piastra, M., Antonelli, M., Echaide, M., Perez-Gil, J., & Capoluongo, E. D. (2013). Clinical and biological role of secretory phospholipase A2 in acute respiratory distress syndrome infants. Crit Care, 17, R163.
Seeds, M. C., Grier, B. L., Suckling, B. N., Safta, A. M., Long, D. L., Waite, B. M., Morris, P. E., & Hite, R. D. (2012). Secretory phospholipase A2-mediated depletion of phosphatidylglycerol in early acute respiratory distress syndrome. Am J Med Sci, 343, 446–451.
Papadopoulos, S., Kazepidou, E., Antonelou, M. H., Leondaritis, G., Tsapinou, A., Koulouras, V. P., Avgeropoulos, A., Nakos, G., & Lekka, M. E. (2020). Secretory phospholipase A(2)-IIA protein and mRNA pools in extracellular vesicles of bronchoalveolar lavage fluid from patients with early acute respiratory distress syndrome: a new perception in the dissemination of inflammation? Pharmaceuticals 13(11), 415.
Snider, J. M., You, J. K., Wang, X., Snider, A. J., Hallmark, B., Seeds, M. C., Sergeant, S., Johnstone, L., Wang, Q., Sprissler, R., Zhang, H. H., Luberto, C., Kew, R. R., Hannun, Y. A., McCall, C. E., Yao, G., Del Poeta, M., & Chilton, F. H. (2021). Group IIA secreted phospholipase A (2) plays a central role in the pathobiology of COVID-19. medRxiv. 02.22.21252237.
Touqui, L., & Arbibe, L. (1999). A role for phospholipase A2 in ARDS pathogenesis. Mol Med Today, 5, 244–249.
Chabot, S., Koumanov, K., Lambeau, G., Gelb, M. H., Balloy, V., Chignard, M., Whitsett, J. A., & Touqui, L. (2003). Inhibitory effects of surfactant protein A on surfactant phospholipid hydrolysis by secreted phospholipases A2. J Immunol, 171, 995–1000.
Attalah, H. L., Wu, Y., Alaoui-El-Azher, M., Thouron, F., Koumanov, K., Wolf, C., Brochard, L., Harf, A., Delclaux, C., & Touqui, L. (2003). Induction of type-IIA secretory phospholipase A2 in animal models of acute lung injury. Eur Respir J, 21, 1040–1045.
Furue, S., Mikawa, K., Nishina, K., Shiga, M., Ueno, M., Tomita, Y., Kuwabara, K., Teshirogi, I., Ono, T., Hori, Y., Matsukawa, A., Yoshinaga, M., & Obara, H. (2001). Therapeutic time-window of a group IIA phospholipase A2 inhibitor in rabbit acute lung injury: correlation with lung surfactant protection. Crit Care Med, 29, 719–727.
Abraham, E., Naum, C., Bandi, V., Gervich, D., Lowry, S. F., Wunderink, R., Schein, R. M., Macias, W., Skerjanec, S., Dmitrienko, A., Farid, N., Forgue, S. T., & Jiang, F. (2003). Efficacy and safety of LY315920Na/S-5920, a selective inhibitor of 14-kDa group IIA secretory phospholipase A2, in patients with suspected sepsis and organ failure. Crit Care Med, 31, 718–728.
Zeiher, B. G., Steingrub, J., Laterre, P. F., Dmitrienko, A., Fukiishi, Y., Abraham, E., & Group, E. S. (2005). LY315920NA/S-5920, a selective inhibitor of group IIA secretory phospholipase A2, fails to improve clinical outcome for patients with severe sepsis. Crit Care Med, 33, 1741–1748.
Vijay, R., Hua, X., Meyerholz, D. K., Miki, Y., Yamamoto, K., Gelb, M., Murakami, M., & Perlman, S. (2015). Critical role of phospholipase A2 group IID in age-related susceptibility to severe acute respiratory syndrome-CoV infection. J Exp Med, 212, 1851–1868.
Hamaguchi, K., Kuwata, H., Yoshihara, K., Masuda, S., Shimbara, S., Oh-ishi, S., Murakami, M., & Kudo, I. (2003). Induction of distinct sets of secretory phospholipase A(2) in rodents during inflammation. Biochim Biophys Acta, 1635, 37–47.
Samuchiwal, S. K., & Balestrieri, B. (2019). Harmful and protective roles of group V phospholipase A(2): Current perspectives and future directions. Biochim Biophys Acta Mol Cell Biol Lipids, 1864, 819–826.
Ruipérez, V., Casas, J., Balboa, M. A., & Balsinde, J. (2007). Group V phospholipase A2-derived lysophosphatidylcholine mediates cyclooxygenase-2 induction in lipopolysaccharide-stimulated macrophages. J Immunol, 179, 631–638.
Muñoz, N. M., Kim, Y. J., Meliton, A. Y., Kim, K. P., Han, S. K., Boetticher, E., O’Leary, E., Myou, S., Zhu, X., Bonventre, J. V., Leff, A. R., & Cho, W. (2003). Human group V phospholipase A2 induces group IVA phospholipase A2-independent cysteinyl leukotriene synthesis in human eosinophils. J Biol Chem, 278, 38813–38820.
Agudelo, C. W., Samaha, G., & Garcia-Arcos, I. (2020). Alveolar lipids in pulmonary disease. A review. Lipids Health Dis, 19, 122.
Ohtsuki, M., Taketomi, Y., Arata, S., Masuda, S., Ishikawa, Y., Ishii, T., Takanezawa, Y., Aoki, J., Arai, H., Yamamoto, K., Kudo, I., & Murakami, M. (2006). Transgenic expression of group V, but not group X, secreted phospholipase A2 in mice leads to neonatal lethality because of lung dysfunction. J Biol Chem, 281, 36420–36433.
Munoz, N. M., Meliton, A. Y., Meliton, L. N., Dudek, S. M., & Leff, A. R. (2009). Secretory group V phospholipase A2 regulates acute lung injury and neutrophilic inflammation caused by LPS in mice. Am J Physiol Lung Cell Mol Physiol, 296, L879–L887.
Belvitch, P., Htwe, Y. M., Brown, M. E., & Dudek, S. (2018). Cortical actin dynamics in endothelial permeability. Curr Top Membr, 82, 141–195.
Dudek, S. M., & Garcia, J. G. (2001). Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985), 91, 1487–1500.
Dudek, S. M., Munoz, N. M., Desai, A., Osan, C. M., Meliton, A. Y., & Leff, A. R. (2011). Group V phospholipase A2 mediates barrier disruption of human pulmonary endothelial cells caused by LPS in vitro. Am J Respir Cell Mol Biol, 44, 361–368.
Meliton, A. Y., Munoz, N. M., Meliton, L. N., Birukova, A. A., Leff, A. R. & & Birukov, K. G. (2013). Mechanical induction of group V phospholipase A(2) causes lung inflammation and acute lung injury. Am J Physiol Lung Cell Mol Physiol, 304, L689–L700.
Meliton, A. Y., Meng, F., Tian, Y., Sarich, N., Mutlu, G. M., Birukova, A. A. & & Birukov, K. G. (2015). Oxidized phospholipids protect against lung injury and endothelial barrier dysfunction caused by heat-inactivated Staphylococcus aureus. Am J Physiol Lung Cell Mol Physiol, 308, L550–L562.
Becker, K. A., Fahsel, B., Kemper, H., Mayeres, J., Li C., Wilker, B., Keitsch, S., Soddemann, M., Sehl, C., Kohnen, M., Edwards, M. J., Grassmé, H., Caldwell, C. C., Seitz, A., Fraunholz, M., Gulbins, E. (2018). Staphylococcus aureus alpha-toxin disrupts endothelial-cell tight junctions via acid sphingomyelinase and ceramide. Infect Immun 86(1), e00606–17.
Munoz, N. M., Desai, A., Meliton, L. N., Meliton, A. Y., Zhou, T., Leff, A. R., & Dudek, S. M. (2012). Group V phospholipase A(2) increases pulmonary endothelial permeability through direct hydrolysis of the cell membrane. Pulm Circ, 2, 182–192.
Beilman, G. (1995). Pathogenesis of oleic acid-induced lung injury in the rat: distribution of oleic acid during injury and early endothelial cell changes. Lipids, 30, 817–823.
Gremmels, H., Bevers, L. M., Fledderus, J. O., Braam, B., van Zonneveld, A. J., Verhaar, M. C., & Joles, J. A. (2015). Oleic acid increases mitochondrial reactive oxygen species production and decreases endothelial nitric oxide synthase activity in cultured endothelial cells. Eur J Pharmacol, 751, 67–72.
Ghosh, A., Gao, L., Thakur, A., Siu, P. M., & Lai, C. W. K. (2017). Role of free fatty acids in endothelial dysfunction. J Biomed Sci, 24, 50.
Barabutis, N., Verin, A., & Catravas, J. D. (2016). Regulation of pulmonary endothelial barrier function by kinases. Am J Physiol Lung Cell Mol Physiol, 311, L832–L845.
Letsiou, E., Kitsiouli, E., Nakos, G., & Lekka, M. E. (2011). Mild stretch activates cPLA2 in alveolar type II epithelial cells independently through the MEK/ERK and PI3K pathways. Biochim Biophys Acta, 1811, 370–376.
Balestrieri, B., & Arm, J. P. (2006). Group V sPLA2: classical and novel functions. Biochim Biophys Acta, 1761, 1280–1288.
Loffredo, S., Borriello, F., Iannone, R., Ferrara, A. L., Galdiero, M. R., Gigantino, V., Esposito, P., Varricchi, G., Lambeau, G., Cassatella, M. A., Granata, F., & Marone, G. (2017). Group V secreted phospholipase A(2) induces the release of proangiogenic and antiangiogenic factors by human neutrophils. Front Immunol, 8, 443.
Kim, K. P., Rafter, J. D., Bittova, L., Han, S. K., Snitko, Y., Munoz, N. M., Leff, A. R., & Cho, W. (2001). Mechanism of human group V phospholipase A2 (PLA2)-induced leukotriene biosynthesis in human neutrophils. A potential role of heparan sulfate binding in PLA2 internalization and degradation. J Biol Chem, 276, 11126–11134.
Meliton, L. N., Zhu, X., Brown, M., Epshtein, Y., Kawasaki, T., Letsiou, E., & Dudek, S. M. (2020). Degradation of group V secretory phospholipase A(2) in lung endothelium is mediated by autophagy. Microvasc Res 129,103954.
Rubio, J. M., Rodríguez, J. P., Gil-de-Gómez, L., Guijas, C., Balboa, M. A., & Balsinde, J. (2015). Group V secreted phospholipase A2 is upregulated by IL-4 in human macrophages and mediates phagocytosis via hydrolysis of ethanolamine phospholipids. J Immunol, 194, 3327–3339.
Balestrieri, B., Maekawa, A., Xing, W., Gelb, M. H., Katz, H. R., & Arm, J. P. (2009). Group V secretory phospholipase A2 modulates phagosome maturation and regulates the innate immune response against Candida albicans. J Immunol, 182, 4891–4898.
Degousee, N., Kelvin, D. J., Geisslinger, G., Hwang, D. M., Stefanski, E., Wang, X. H., Danesh, A., Angioni, C., Schmidt, H., Lindsay, T. F., Gelb, M. H., Bollinger, J., Payré, C., Lambeau, G., Arm, J. P., Keating, A., & Rubin, B. B. (2011). Group V phospholipase A2 in bone marrow-derived myeloid cells and bronchial epithelial cells promotes bacterial clearance after Escherichia coli pneumonia. J Biol Chem, 286, 35650–35662.
Han, S. K., Kim, K. P., Koduri, R., Bittova, L., Munoz, N. M., Leff, A. R., Wilton, D. C., Gelb, M. H., & Cho, W. (1999). Roles of Trp31 in high membrane binding and proinflammatory activity of human group V phospholipase A2. J Biol Chem, 274, 11881–11888.
Hanasaki, K., Ono, T., Saiga, A., Morioka, Y., Ikeda, M., Kawamoto, K., Higashino, K., Nakano, K., Yamada, K., Ishizaki, J., & Arita, H. (1999). Purified group X secretory phospholipase A(2) induced prominent release of arachidonic acid from human myeloid leukemia cells. J Biol Chem, 274, 34203–34211.
Nolin, J. D., Murphy, R. C., Gelb, M. H., Altemeier, W. A., Henderson, Jr., W. R., & Hallstrand, T. S. (2019). Function of secreted phospholipase A(2) group-X in asthma and allergic disease. Biochim Biophys Acta Mol Cell Biol Lipids, 1864, 827–837.
Kelvin, A. A., Degousee, N., Banner, D., Stefanski, E., Leόn, A. J., Angoulvant, D., Paquette, S. G., Huang, S. S., Danesh, A., Robbins, C. S., Noyan, H., Husain, M., Lambeau, G., Gelb, M., Kelvin, D. J., & Rubin, B. B. (2014). Lack of group X secreted phospholipase A2 increases survival following pandemic H1N1 influenza infection. Virology, 454-455, 78–92.
Agassandian, M., Miakotina, O. L., Andrews, M., Mathur, S. N., & Mallampalli, R. K. (2007). Pseudomonas aeruginosa and sPLA2 IB stimulate ABCA1-mediated phospholipid efflux via ERK-activation of PPARalpha-RXR. Biochem J, 403, 409–420.
Rae, D., Porter, J., Beechey-Newman, N., Sumar, N., Bennett, D., & Hermon-Taylor, J. (1994). Type 1 prophospholipase A2 propeptide in acute lung injury. Lancet, 344, 1472–1473.
Khan, M. I., & Hariprasad, G. (2020). Human secretary phospholipase A2 mutations and their clinical implications. J Inflamm Res, 13, 551–561.
Righino, B., Minucci, A., Pirolli, D., Capoluongo, E., Conti, G., De Luca, D., De, & Rosa, M. C. (2018). In silico investigation of the molecular effects caused by R123H variant in secretory phospholipase A2-IIA associated with ARDS. J Mol Graph Model, 81, 68–76.
Acknowledgements
We dedicate this article to our esteemed colleague, mentor, and friend, Dr Viswanathan Natarajan. His impactful scientific achievements have greatly advanced our understanding of how bioactive lipids participate in multiple physiologic and pathophysiologic processes. In addition, he has provided outstanding mentorship for many junior investigators, including the authors of this review. The entire field owes Dr Natarajan a great debt.
Funding
This work was supported by AHA 14GRNT20460186 (S.M.D.), NIH R01 HL133059 (S.M.D.), and T32 HL082547 (S.M.D.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Letsiou, E., Htwe, Y.M. & Dudek, S.M. Secretory Phospholipase A2 Enzymes in Acute Lung Injury. Cell Biochem Biophys 79, 609–617 (2021). https://doi.org/10.1007/s12013-021-01003-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-021-01003-x