
Abstract
Synthesis and structural characterization of two heterodinuclear ZnII-LnIII complexes with the formula [ZnLn(HL)(µ-OAc)(NO3)2(H2O)x(MeOH)1-x]NO3 · n H2O · n MeOH [Ln = Pr (1), Nd (2)] and the crystal and molecular structure of [ZnNd(HL)(µ-OAc)(NO3)2(H2O)] [ZnNd(HL)(OAc)(NO3)2(H2O)](NO3)2 · n H2O · n MeOH (3) are reported. The asymmetrical compartmental ligand (E)-2-(1-(2-((2-hydroxy-3-methoxybenzylidene)amino)-ethyl)imidazolidin-2-yl)-6-methoxyphenol (H2L) is formed from N1,N3-bis(3-methoxysalicylidene)diethylenetriamine (H2valdien) through intramolecular aminal formation, resulting in a peripheral imidazoline ring. The structures of 1–3 were revealed by X-ray crystallography. The smaller ZnII ion occupies the inner N2O2 compartment of the ligand, whereas the larger and more oxophilic LnIII ions are found in the outer O2O2’ site.
Graphic Abstract
Synthesis and structural characterization of two heterodinuclear ZnII-LnIII complexes (Ln = Pr, Nd) bearing an asymmetrical compartmental ligand formed in situ from N1,N3-bis(3-methoxysalicylidene)diethylenetriamine (H2valdien) through intramolecular aminal formation are reported.

Similar content being viewed by others


Introduction
Acyclic and macrocyclic Schiff base ligands are among the most extensively used ligands in coordination chemistry [1]. In general, Schiff bases can be readily prepared in good yields through condensation of primary amines with aldehyde or ketones. Owing to the ease of their synthesis, their versatility and ability to form stable complexes with almost all transition metals, Schiff base ligands have enormously contributed to the development of coordination chemistry and their transition metal complexes have been particularly important in bioinorganic chemistry, magnetochemistry, catalysis [2,3,4] and biomedical and related applications [5].
Polydentate Schiff base chelate ligands derived from condensation of 2-hydroxy-3-methoxybenzaldehyde (o-vanillin) or derivatives as aldehyde component with polyamines have been exploited for the synthesis of homo- and heterodinuclear complexes. These have attracted research attention because of interesting properties such as magnetism and luminescence [6,7,8,9,10,11,12,13,14,15,16,17,18]. N1,N3-bis(3-methoxysalicylidene)diethylenetriamine (H2valdien), which is obtained via Schiff base condensation of two equivalents of o-vanillin with diethylenetriamine, belongs to this class of ligands. After deprotonation of the phenol moieties, the resulting valdien2− anion represents a compartmental ligand [19, 20], providing an inner binding site with an N3O2 donor set and an outer binding site with an outer O2O2’ donor set. Whereas the inner compartment is expected to accommodate metal ions from the 3d row, the outer site may bind the more oxophilic lanthanide ions [6, 7, 21, 22]. Nevertheless, the inner compartment can also host lanthanide ions [8, 23,24,25,26]. Moreover, the H2valdien ligand has also been encountered in a chelate-spacer-chelate bridging mode in dinuclear complexes with the aliphatic secondary amine linkages remaining unbound to the metal ions [27,28,29,30].
Herein, we report on the synthesis and structural characterization of two heterodinuclear ZnII-LnIII complexes with the general formula [ZnLn(HL)(µ-OAc)(NO3)2(H2O)x(MeOH)1-x]NO3 · n H2O · n MeOH [Ln = Pr (1), Nd (2)], containing bridging acetate ions and the asymmetrical compartmental ligand (E)-2-(1-(2-((2-hydroxy-3-methoxybenzylidene)amino)-ethyl)imidazolidin-2-yl)-6-methoxy-phenol (H2L) formed in situ from H2valdien through isomerization by intramolecular aminal formation, resulting in a peripheral imidazoline ring [27]. Such rearrangements are due to the reversibility of the Schiff base (imine) formation [31]. The rearranged ligand H2L thus formed likewise represents a compartmental ligand but exhibits lower symmetry and a smaller inner compartment than the parent H2valdien ligand. In addition, the structure of the serendipitously discovered compound [ZnNd(HL)(µ-OAc)(NO3)2(H2O)] [ZnNd(HL)(OAc)(NO3)2(H2O)](NO3)2 · n H2O · n MeOH (3) is described.
Experimental Section
General
The H2valdien ligand was synthesised as described in the literature [23]. Zn(OAc)2 · 2 H2O (Fischer Scientific), Pr(NO3)3 · 6 H2O and Nd(NO3)3 · 6 H2O (Sigma Aldrich) were purchased and used as received. Methanol was of reagent grade. CHN microanalysis was carried out by Mikroanalytisches Labor Kolbe (Mülheim, Germany).
Synthesis of 1 and 2
Zn(OAc)2 · 2 H2O (0.220 g, 1.0 mmol) dissolved in 10 mL of methanol was added to H2valdien (0.371 g, 1.0 mmol) dissolved in 10 mL of acetonitrile and the mixture was stirred under reflux at 40 °C for 1 h. Subsequently, the yellow precipitate so formed was added to Pr(NO3)3 · 6 H2O (0.435 g, 1.0 mmol) for 1 or Nd(NO3)3 · 6 H2O (0.438 g, 1.0 mmol) for 2 in 40 mL of methanol and the reaction mixture was refluxed for a further 3 h. The solution was then filtered and the filtrate was set aside undisturbed at ambient temperature. Yellow–brown crystals of 1 and yellow crystals of 2 suitable for single-crystal X-ray diffraction were obtained after several days. Analytical data for the compounds are given below.
[ZnPr(HL)(µ-OAc)(NO3)2(H2O)0.35(MeOH)0.65]NO3 · 2 MeOH · H2O (1; L2− = C20H23N3O42−): Yield: 0.580 g (0.62 mmol, 62%). Anal. calcd. for C24.65H40.30N6O19PrZn (M = 931.00 g mol−1): C 31.8, H 4.4, N 9.0%; found: C 31.9, H 4.3, N 9.2%. MS(ESI+): m/z [H3L]+ calcd. for C20H26N3O4+ 372.2, found 372.2, [Zn(HL)]+ calcd. for C20H24N3O4Zn+ 434.1, found 434.1; IR(ATR): 1640 cm–1 (C = N stretch).
[ZnNd(HL)(µ-OAc)(NO3)2(H2O)0.75(MeOH)0.25]NO3 · 3 MeOH (2; L2− = C20H23N3O42−): Yield: 0.570 g (0.60 mmol, 60%). Anal. calcd. for C25.24H41.50N6NdO19Zn (M = 942.64 g mol−1): C 32.2, H 4.4, N 8.9%; found: C 32.1, H 4.3 N 9.2%. MS(ESI+): m/z [H3L]+ calcd. for C20H26N3O4+ 372.2, found 372.2, [Zn(HL)]+ calcd. for C20H24N3O4Zn+ 434.1, found 434.1; IR(ATR): 1640 cm–1 (C = N stretch).
Physical Methods
Energy-dispersive X-ray spectroscopy (EDX) was undertaken on a Hitachi S3500N scanning electron microscope using a Si(Li) Pentafet Plus detector from Oxford Instruments GmbH with a 25 kV excitation voltage, 600 s measuring time and 100 × magnification from a fine powder sample sprinkled on a self-adhesive carbon guide tap. IR spectra were measured in the range 4000–400 cm–1 with a Bruker ALPHA Platinum-ATR FT-IR spectrometer. ESI mass spectra were recorded on a Q ExactiveTM Plus Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany).
X-ray Crystallography
The X-ray intensity data were collected on a Bruker AXS Kappa Mach3 APEXII diffractometer at T = 100(2) K, using Mo-Kα radiation (λ = 0.71073 Å) from an Incoatec IµS microfocus X-ray source with Helios mirrors. The data were processed with SAINT [32] and absorption corrections were carried out with SADABS [33]. The crystal structures were solved with SHELXT [34] and refined with SHELXL-2018/3 [35]. Disordered parts of the structures were refined with appropriate geometrical restraints and using free variables for the occupancies (see supplementary crystallographic data). Carbon-bound hydrogen atoms were placed in geometrically calculated positions and refined using the appropriate riding model. Hydrogen atoms attached to nitrogen and oxygen were treated by semi-free refinement using appropriate distance restraints. Some solvate methanol and water hydrogen atoms could not be located in the final difference Fourier map and were therefore excluded from the structure refinement. The structure of 3 was refined as an inversion twin, resulting in a Flack x parameter of 0.440(19) [36].
Crystal data and refinement details for 1: C25H39.68N6O18.34PrZn, Mr = 924.03, monoclinic, P21/n, Z = 4, a = 11.3498(15) Å, b = 14.8170(19) Å, c = 21.488(3) Å, β = 101.864(2)°, V = 3536.5(8) Å3, F(000) = 1870, crystal size 0.223 × 0.066 × 0.031 mm, ρcalcd = 1.735 g cm−3, μ = 2.125 mm−1, 2θmax = 62.22°, reflections collected/unique 104,376 / 11,339 (Rint = 0.0522), parameters/restraints 512/16, R1 [I > 2σ(I)] = 0.0246, wR2 (all data) = 0.0592, S = 1.031, Δρmax/Δρmin = 0.65/ − 0.46 e Å−3.
Crystal data and refinement details for 2: C23.94H35.26N6NdO18.90Zn, Mr = 919.13, monoclinic, P21/n, Z = 4, a = 11.3439(13) Å, b = 14.7501(18) Å, c = 21.454(3) Å, β = 101.914(2)°, V = 3512.4(7) Å3, F(000) = 1848, crystal size 0.086 × 0.044 × 0.024 mm, ρcalcd = 1.736 g cm−3, μ = 2.231 mm−1, 2θmax = 62.14°, reflections collected/unique 153,810/11,217 (Rint = 0.1000), parameters/restraints 529/85, R1 [I > 2σ(I)] = 0.0384, wR2 (all data) = 0.0942, S = 1.048, Δρmax/Δρmin = 1.48/ − 1.30 e Å−3.
Crystal data and refinement details for 3: C22H27N6NdO18.05Zn, Mr = 873.87, monoclinic, Pn, Z = 4, a = 11.2800(2) Å, b = 15.749(3) Å, c = 18.5316(8) Å, β = 102.908(2)°, V = 3209.0(6) Å3, F(000) = 1742, crystal size 0.130 × 0.060 × 0.030 mm, ρcalcd = 1.809 g cm−3, μ = 2.435 mm−1, 2θmax = 66.18°, reflections collected/unique 88,652/24,240 (Rint = 0.1126), parameters/restraints 869/2, R1 [I > 2σ(I)] = 0.0705, wR2 (all data) = 0.1700, S = 1.025, Δρmax/Δρmin = 2.08/ − 1.48 e Å−3.
Results and Discussion
The H2valdien ligand was prepared through Schiff base condensation of o-vanillin and diethylenetriamine in a 2:1 molar ratio [23]. Reaction with Zn(OAc)2 · 2 H2O and, subsequently, with Ln(NO)3 · 6 H2O (Ln = Pr or Nd) in methanol under reflux conditions afforded the heterodinuclear ZnII-LnIII complexes 1 (Ln = Pr) and 2 (Ln = Nd), as depicted in Scheme 1. X-ray crystallography revealed that the H2valdien compartmental ligand underwent an isomerization through an intramolecular aminal formation during the complexation reaction, resulting in an imidazolidine ring in the periphery. This phenomenon has been observed previously for the H2valdien ligand and it was suggested that ring contraction optimizes binding of the ZnII ion [27]. Some minor discrepancies between the sum formulae derived from elemental analysis of the bulk material as synthesized and those obtained from X-ray crystallography are ascribed to partial loss of co-crystallized solvents on drying before analysis. The presence of Zn and respectively, Pr and Nd in 1 and 2 was confirmed by EDX analysis (Fig. S1 in the supplementary material). The IR band at 1640 cm−1 observed for both 1 and 2 (Figs. S2 and S3 in the supplementary material) is assigned to the imine C = N stretching vibration and agrees well with that reported for a heterodinuclear ZnII-LaIII complex having the reported formula “[ZnLa(HL)(NO3)(S)](NO3)”, where S = H2O or C2H5OH, (CSD refcode: XODFOM) [27] and [Zn4Dy2(L)2(L’)2(N3)2]Cl2 · 2 H2O (BIRXEI), [Zn4Tb2(L)2(L’)2(Cl)2][ZnN3Cl3] · 2 H2O (BIRXOS), and [Zn4Gd2(L)2(L’)2(Cl)2][ZnN3Cl3] · 2 H2O (BIRZAG) [37], containing the same rearranged compartmental ligand (herein abbreviated H2L) and in the case of the latter three complexes the carbamate ligand L’, derived from L by the absorption of CO2 from the air in the presence of ZnII. In the ESI+ mass spectra of 1 and 2 (Figs. S4-S7 in the supplementary material), the peaks at m/z 372.2 and 434.1 can be assigned to the fragment ions [H3L]+ and [Zn(HL)]+, respectively.

Synthesis of 1 and 2. The coordination site on Ln occupied by water or methanol in the solid-state is represented by water only in the diagram for the sake of clarity. Co-crystallized solvent molecules are not shown
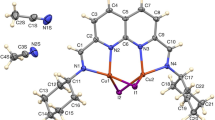
Figure 1 depicts the molecular structures of the cationic complexes in 1 and 2 in the solid-state, as determined by X-ray crystallography. The structures of 1 and 2 were found to be isostructural. The ZnII ion occupies the inner N2O2 compartment of the rearranged ligand, whereas the LnIII ion is situated in the outer O2O2 compartment. The intramolecular distance between the two metal ions is ca. 3.5 Å. The coordination sphere of the five-coordinate ZnII ion is best described as square-pyramidal with the imine (N1) and aminal nitrogen (N2) atoms and the bridging phenolate oxygen atoms of the chelate ligand in the basal plane and an acetate oxygen atom in the apical position. The geometry index τ5 is 0.31 for 1 and 0.32 for 2 [38], indicating that the coordination geometry lies between square-pyramidal and trigonal–bipyramidal but closer to square-pyramidal (C4v symmetry). The LnIII ion is ten-coordinate with the two bridging phenolate and the two methoxy oxygen atoms of the chelate ligand occupying four coordination sites. The remaining positions are filled by two nitrate ions in a symmetrically bidentate coordination mode [39, 40], a water or alternatively a methanol oxygen atom (site of O13), and an oxygen atom of the µ-acetato-κO,O’ ligand. The coordination geometry of the LnIII ion can be best described as an approximate sphenocorona (C2v symmetry), as determined by comparison with ideal polyhedra using continuous shape measures [41, 42]. As structural consequence of the intramolecular aminal formation, the H2L compartmental ligand adopts a bent conformation with the mean planes of the two aromatic rings being almost perpendicular (dihedral angle ca. 80°). A similar conformation of the ligand was found for XODFOM. In the crystal, the aminal nitrogen atom N3 is protonated, making the complex cationic, and forms N − H···O hydrogen bonds to a methanol molecule of crystallisation and a nitrate counter ion, which balances the charge.

Molecular structures of 1 and 2 in the crystal. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms (except for those attached to nitrogen), counter ions and solvent molecules are omitted for clarity. The site of O13 is occupied by water or methanol (not shown here for the sake of clarity) in the crystal
Serendipitously, we found a crystal in one crystallization batch of 2, representing an unknown methanol solvate hydrate of a co-crystal (3) of [ZnNd(HL)(µ-OAc)(NO3)2(H2O)]NO3 and a structural isomer [ZnNd(HL)(OAc)(NO3)2(H2O)]NO3 (Fig. 2). In the latter, a water molecule occupies the apical position at ZnII and the acetato ligand binds solely to NdIII in a symmetrical bidentate fashion. The coordination geometries of NdIII and ZnII are retained in the two isomers. In 3, the geometry index τ5 is 0.14 for Zn1 in [ZnNd(HL)(µ-OAc)(NO3)2(H2O)]+ and 0.43 for Zn1 in [ZnNd(HL)(OAc)(NO3)2(H2O)]+.

Molecular structures of the two isomeric cations in the crystal structure of [ZnNd(HL)(µ-OAc)(NO3)2(H2O)] [ZnNd(HL)(OAc)(NO3)2(H2O)](NO3)2 ⋅ n H2O ⋅ n MeOH (3). Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms (except for those attached to nitrogen), counter ions and solvent molecules are omitted for clarity
The structures of 1 and 2 appear to be isostructural with the above-mentioned XODFOM, which has the reported molecular composition “[ZnLa(HL)(NO3)3(S)](NO3)” (S = H2O or C2H5OH) [27], in which water and ethanol alternatively occupy one coordination site at LaIII. However, whereas a µ-acetato-κO,O’ ligand bridges the ZnII and LnIII in 1 and 2, in XODFOM a bridging nitrate ion the ZnII and LaIII ions is reported. The reported N−O bond of the non-coordinating oxygen atom of the bridging nitrate ion in XODFOM is unusually long at 1.429(13) Å [39, 40], and the corresponding atomic displacement parameters are rather large, which may be a warning sign for incorrect atom type assignment [43,44,45]. Taking the reported synthetic route into account, the presence of a bridging acetate ligand in XODFOM is a possibility, since the precursor complex described as “[Zn(valdien)] · 1.5 CH3OH”, which was not structurally characterized by X-ray crystallography, was prepared from H2valdien and Zn(OAc)2 · 2 H2O. Considering previous work by Naskar et al. [29], the constitution of the precursor complex might have been rather [Zn2(H2valdien)2(OAc)2]. Elemental analysis calcd. for [Zn2(H2valdien)2(OAc)2] (C 53.40, H 5.50, N 8.49%) differs little from that reported for “[Zn(valdien)] · 1.5 CH3OH” (C 53.10, H 5.80, N 8.30%) by Benetollo et al. [27]. We should note that the crystal structure of [Zn(valdien)] · CH3OH was published very recently [46], but an additional base (LiOH) was used in the synthesis contrary to the synthesis of “[Zn(valdien)] · 1.5 CH3OH” reported by Benetollo et al. The syn-syn bidentate bridging mode of the nitrate ion is known for inorganic nitrates [39, 47], but is rather unusual for organic or organometallic nitrato complexes [40]. In this connection, we note that the external N−O distances in the crystal structure with the CSD refcode ADURAV, reportedly containing two syn-syn bridging bidentate nitrato ligands between LaIII and ZnII, at 1.506(18) and 1.52(2) Å are also suspiciously long [48]. This coordination mode is, in contrast, well known for carboxylate ions [49]. The comparable C21−C22 bond lengths of 1.499(3) and 1.495(6) Å in 1 and 2, respectively, and the corresponding atomic displacement parameters clearly support the presence of acetate ions at this site in XODFOM.
Conclusions
We have synthesized the heterodinuclear ZnII-LnIII complexes 1 and 2 by successive treatment of the H2valdien compartmental ligand with Zn(OAc)2 · 2 H2O and, respectively, Pr(NO3)3 · 6 H2O and Nd(NO)3 · 6 H2O, affording the asymmetrical, ring-contracted isomerized compartmental ligand H2L from H2valdien in situ. Such a rearrangement of the H2valdien ligand, which has been described the literature, is enabled through the reversibility of the Schiff base condensation. Its occurrence in the formation of 1 and 2 can be ascribed to a better accommodation of the smaller ZnII ion in the inner N2O2 binding site instead of the inner N3O2 site of the parent H2valdien. As anticipated, the LnIII ions are found in the outer O2O2’ compartment with counter ions and solvent molecules filling the remaining coordination sites of the ten-fold coordinated ions. Bond lengths, atomic displacement parameters and electron density maps resulting from the X-ray structural analysis provide clear evidence that the ZnII and LnIII ions in 1 and 2 are additionally linked by acetate anions in a syn-syn bridging mode rather than by nitrate anions, as has been proposed for similar structures. Detection of the [Zn(HL)]+ ion but no LnIII adducts by ESI mass spectrometry suggests that the binding of ZnII to the inner pocket of the ligand is, as expected, more stable than that of the LnIII ions in the outer compartment. The crystal structure of 3 reveals that structural isomers of 2 occur.
Data Availability
CCDC 2025504-2025506 contain the supplementary crystallographic data for 1-3. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Code Availability
Not applicable.
References
Hernández-Molina R, Mederos A (2003) 1.19—Acyclic and macrocyclic Schiff base ligands. In: McCleverty JA, Meyer TJ (eds) Comprehensive coordination chemistry II. Pergamon, Oxford, pp 411–446
Abu-Dief AM, Mohamed IMA (2015) A review on versatile applications of transition metal complexes incorporating Schiff bases. Beni-Suef Univ J Basic Appl Sci 4(2):119–133. https://doi.org/10.1016/j.bjbas.2015.05.004
Dalia SA, Afsan F, Hossain MS, Khan M, Zakaria C, Zahan M, Ali M (2018) A short review on chemistry of schiff base metal complexes and their catalytic application. Int J Chem Stud 6(3):2859–2866
Al Zoubi W, Ko YG (2017) Schiff base complexes and their versatile applications as catalysts in oxidation of organic compounds: part I. Appl Organomet Chem 31(3):e3574. https://doi.org/10.1002/aoc.3574
More MS, Joshi PG, Mishra YK, Khanna PK (2019) Metal complexes driven from Schiff bases and semicarbazones for biomedical and allied applications: a review. Mater Today Chem 14:100195. https://doi.org/10.1016/j.mtchem.2019.100195
Andruh M (2015) The exceptionally rich coordination chemistry generated by Schiff-base ligands derived from o-vanillin. Dalton Trans 44(38):16633–16653. https://doi.org/10.1039/C5DT02661J
Pasatoiu TD, Tiseanu C, Madalan AM, Jurca B, Duhayon C, Sutter JP, Andruh M (2011) Study of the luminescent and magnetic properties of a series of heterodinuclear [ZnIILnIII] complexes. Inorg Chem 50(13):5879–5889. https://doi.org/10.1021/ic200426w
Long J, Habib F, Lin P-H, Korobkov I, Enright G, Ungur L, Wernsdorfer W, Chibotaru LF, Murugesu M (2011) Single-molecule magnet behavior for an antiferromagnetically superexchange-coupled dinuclear dysprosium(III) complex. J Am Chem Soc 133(14):5319–5328. https://doi.org/10.1021/ja109706y
Oyarzabal I, Artetxe B, Rodriguez-Dieguez A, Garcia J, Seco JM, Colacio E (2016) A family of acetato-diphenoxo triply bridged dimetallic ZnIILnIII complexes: SMM behavior and luminescent properties. Dalton Trans 45(23):9712–9726. https://doi.org/10.1039/c6dt01327a
Liu C-M, Zhang D-Q, Su J-B, Zhang Y-Q, Zhu D-B (2018) Single-molecule magnet behavior of 1D coordination polymers based on DyZn2(salen)2 units and Pyridin-N-Oxide-4-Carboxylate: structural divergence and magnetic regulation. Inorg Chem 57(17):11077–11086. https://doi.org/10.1021/acs.inorgchem.8b01653
Wong W-K, Liang H, Wong W-Y, Cai Z, Li K-F, Cheah K-W (2002) Synthesis and near-infrared luminescence of 3d–4f bi-metallic Schiff base complexes. New J Chem 26(3):275–278. https://doi.org/10.1039/b104175b
Echenique-Errandonea E, Zabala-Lekuona A, Cepeda J, Rodriguez-Dieguez A, Seco JM, Oyarzabal I, Colacio E (2018) Effect of the change of the ancillary carboxylate bridging ligand on the SMM and luminescence properties of a series of carboxylate-diphenoxido triply bridged dinuclear ZnLn and tetranuclear Zn2Ln2 complexes (Ln = Dy, Er). Dalton Trans 48(1):190–201. https://doi.org/10.1039/c8dt03800g
Fondo M, Corredoira-Vazquez J, Garcia-Deibe AM, Sanmartin-Matalobos J, Herrera JM, Colacio E (2017) Designing ligands to isolate ZnLn and Zn2Ln complexes: field-induced single-ion magnet behavior of the ZnDy, Zn2Dy, and Zn2Er analogues. Inorg Chem 56(10):5646–5656. https://doi.org/10.1021/acs.inorgchem.7b00165
Zhao S, Liu X, Wong W-Y, Lü X, Wong W-K (2014) Near infrared luminescent hexanuclear zinc–lanthanide prisms: synthesis, structure and luminescent properties. Inorg Chim Acta 414:160–164. https://doi.org/10.1016/j.ica.2014.02.003
Liu C-M, Zhang DQ, Hao X, Zhu DB (2020) Zn2Ln2 complexes with carbonate bridges formed by the fixation of carbon dioxide in the atmosphere: single-molecule magnet behaviour and magnetocaloric effect. Dalton Trans 49(7):2121–2128. https://doi.org/10.1039/c9dt04480a
Maeda M, Hino S, Yamashita K, Kataoka Y, Nakano M, Yamamura T, Kajiwara T (2012) Correlation between slow magnetic relaxation and the coordination structures of a family of linear trinuclear Zn(II)-Ln(III)-Zn(II) complexes (Ln = Tb, Dy, Ho, Er, Tm and Yb). Dalton Trans 41(44):13640–13648. https://doi.org/10.1039/c2dt31399e
Long J, Ivanov MS, Khomchenko VA, Mamontova E, Thibaud J-M, Rouquette J, Beaudhuin M, Granier D, Ferreira RAS, Carlos LD, Donnadieu B, Henriques MSC, Paixão JA, Guari Y, Larionova J (2020) Room temperature magnetoelectric coupling in a molecular ferroelectric ytterbium(III) complex. Science 367(6478):671. https://doi.org/10.1126/science.aaz2795
Miroslaw B, Cristóvão B, Hnatejko Z (2018) Heterometallic ZnII–LnIII–ZnII Schiff base complexes with linear or bent conformation—synthesis, crystal structures. Luminescent and magnetic characterization. Molecules 23(7):1761. https://doi.org/10.3390/molecules23071761
Vigato PA, Tamburini S (2008) Advances in acyclic compartmental ligands and related complexes. Coord Chem Rev 252(18–20):1871–1995. https://doi.org/10.1016/j.ccr.2007.10.030
Vigato PA, Peruzzo V, Tamburini S (2012) Acyclic and cyclic compartmental ligands: recent results and perspectives. Coord Chem Rev 256(11–12):953–1114. https://doi.org/10.1016/j.ccr.2012.01.009
Liu C-M, Zhang D-Q, Hao X, Zhu D-B (2014) Trinuclear [CoIII2–LnIII] (Ln=Tb, Dy) single-ion magnets with mixed 6-Chloro-2-Hydroxypyridine and Schiff base ligands. Chem Asian J 9(7):1847–1853. https://doi.org/10.1002/asia.201402001
Zhao L, Wu J, Xue S, Tang J (2012) A linear 3d–4f tetranuclear CoIII2DyIII2 single-molecule magnet: synthesis, structure, and magnetic properties. Chem Asian J 7(10):2419–2423. https://doi.org/10.1002/asia.201200548
Brunet G, Habib F, Korobkov I, Murugesu M (2015) Slow magnetic relaxation observed in dysprosium compounds containing unsupported near-linear hydroxo- and fluoro-bridges. Inorg Chem 54(13):6195–6202. https://doi.org/10.1021/acs.inorgchem.5b00343
Bag P, Chakraborty A, Rouzières M, Clérac R, Butcher RJ, Chandrasekhar V (2014) Oxalato-bridged neutral octanuclear heterometallic complexes [Ln4K4(L)4(μ-H2O)4(NO3)2(μ-Ox)] (Ln = Dy(III), Gd(III), Tb(III), Ho(III); LH3 = N[CH2CH2N═CH-C6H3-2-OH-3-OMe]3; Ox = (C2O4)2–): synthesis, structure, magnetic and luminescent properties. Cryst Growth Des 14(9):4583–4592. https://doi.org/10.1021/cg500677t
Habib F, Brunet G, Vieru V, Korobkov I, Chibotaru LF, Murugesu M (2013) Significant enhancement of energy barriers in dinuclear dysprosium single-molecule magnets through electron-withdrawing effects. J Am Chem Soc 135(36):13242–13245. https://doi.org/10.1021/ja404846s
Habib F, Lin P-H, Long J, Korobkov I, Wernsdorfer W, Murugesu M (2011) The use of magnetic dilution to elucidate the slow magnetic relaxation effects of a Dy2 single-molecule magnet. J Am Chem Soc 133(23):8830–8833. https://doi.org/10.1021/ja2017009
Benetollo F, Di Bernardo P, Tamburini S, Vigato PA, Zanonato P (2008) Mononuclear and polynuclear complexes with a side-off compartmental Schiff base. Inorg Chem Commun 11(3):246–251. https://doi.org/10.1016/j.inoche.2007.11.022
Usman M, Arjmand F, Khan RA, Alsalme A, Ahmad M, Tabassum S (2017) Biological evaluation of dinuclear copper complex/dichloroacetic acid cocrystal against human breast cancer: design, synthesis, characterization, DFT studies and cytotoxicity assays. RSC Adv 7(76):47920–47932. https://doi.org/10.1039/C7RA08262B
Naskar B, Modak R, Maiti DK, Drew MGB, Bauzá A, Frontera A, Das Mukhopadhyay C, Mishra S, Das Saha K, Goswami S (2017) A Schiff base platform: structures, sensing of Zn(II) and PPi in aqueous medium and anticancer activity. Dalton Trans 46(29):9498–9510. https://doi.org/10.1039/C7DT01932G
Noor S, Goddard R, Kumar S, Ahmad N, Sabir S, Mitra P, Seidel RW (2018) On the chiral Z′ = 2 crystal structure of [Cu2(H2valdien)2](NO3)2 [H2valdien = N1, N3-bis(3-methoxysalicylidene)diethylenetriamine]. J Chem Crystallogr 48(4):164–169. https://doi.org/10.1007/s10870-018-0724-4
Belowich ME, Stoddart JF (2012) Dynamic imine chemistry. Chem Soc Rev 41(6):2003–2024. https://doi.org/10.1039/C2CS15305J
SAINT (2012). Bruker AXS Inc., Madison, Wisconsin, USA
SADABS (2012). Bruker AXS Inc., Madison, Wisconsin, USA
Sheldrick GM (2015) SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr A Found Adv 71(Pt 1):3–8. https://doi.org/10.1107/S2053273314026370
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr C Struct Chem 71(Pt 1):3–8. https://doi.org/10.1107/S2053229614024218
Flack H (1983) On enantiomorph-polarity estimation. Acta Crystallogr A 39(6):876–881. https://doi.org/10.1107/S0108767383001762
Yin C-L, Hu Z-B, Long Q-Q, Wang H-S, Li J, Song Y, Zhang Z-C, Zhang Y-Q, Pan Z-Q (2019) Single molecule magnet behaviors of Zn4Ln2 (Ln = DyIII, TbIII) complexes with multidentate organic ligands formed by absorption of CO2 in air through in situ reactions. Dalton Trans 48(2):512–522. https://doi.org/10.1039/C8DT03849J
Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J Chem Soc, Dalton Trans 7:1349–1356. https://doi.org/10.1039/DT9840001349
Addison CC, Logan N, Wallwork SC, Garner CD (1971) Structural aspects of co-ordinated nitrate groups. Q Rev Chem Soc 25(2):289–322. https://doi.org/10.1039/QR9712500289
Morozov IV, Serezhkin VN, Troyanov SI (2009) Modes of coordination and stereochemistry of nitrate groups in organic and organometallic nitrates. Russ Chem Bull 58(12):2407–2417. https://doi.org/10.1007/s11172-009-0336-4
Alvarez S, Alemany P, Casanova D, Cirera J, Llunell M, Avnir D (2005) Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord Chem Rev 249(17):1693–1708. https://doi.org/10.1016/j.ccr.2005.03.031
M. Llunell, D. Casanova, J. Cirera, J. Bofill, P. Alemany, S. Alvarez, M.Pinsky, D. Avnir, SHAPE v. 2.1. Program for the calculation of continuous shape measures (CShM) of polygonal and polyhedral molecular fragments; CShM = 2.420 (1) and 2.433 (2).
Spek AL (2009) Structure validation in chemical crystallography. Acta Crystallogr D Biol Crystallogr 65(Pt 2):148–155. https://doi.org/10.1107/S090744490804362X
Spek AL (2018) What makes a crystal structure report valid? Inorg Chim Acta 470:232–237. https://doi.org/10.1016/j.ica.2017.04.036
Schwalbe CH (2018) Should we remediate small molecule structures? If so, who should do it? Crystallogr Rev 24(4):217–235. https://doi.org/10.1080/0889311X.2018.1508209
Noor S, Suda S, Haraguchi T, Khatoon F, Akitsu T (2021) Chiral crystallization of a zinc(II) complex. Acta Cryst E 77(5):542–546. https://doi.org/10.1107/S2056989021003650
Morozov IV, Serezhkin VN, Troyanov SI (2008) Modes of coordination and stereochemistry of the NO3− anions in inorganic nitrates. Russ Chem Bull 57(3):439–450. https://doi.org/10.1007/s11172-008-0071-2
Sreejith SS, Mohan N, Kurup MRP (2018) Experimental and theoretical analysis of a rare nitrato bridged 3d–4f complex containing LaZn2 core synthesized from a Zn(II) metalloligand. J Mol Struct 1153:85–95. https://doi.org/10.1016/j.molstruc.2017.10.008
Deacon GB, Phillips RJ (1980) Relationships between the carbon-oxygen stretching frequencies of carboxylato complexes and the type of carboxylate coordination. Coord Chem Rev 33(3):227–250. https://doi.org/10.1016/S0010-8545(00)80455-5
Acknowledgements
The authors would like to thank Heike Schucht for technical assistance with the X-ray intensity data collections, Sylvia Palm for the EDX measurements, Dirk Kampen for recording the ESI mass spectra and Professor Christian W. Lehmann for providing access to the X-ray diffraction and electron microscopy facilities at the Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany. S.N. would like to thank the Head of the Department of Applied Sciences & Humanities, Faculty of Engineering & Technology, Jamia Millia Islamia, New Delhi, India, who facilitated this research.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by grants from the Council of Scientific and Industrial Research (CSIR), Government of India, New Delhi, India [No.:13(8967-A/2018-POOL)].
Author information
Authors and Affiliations
Contributions
Conceptualization: S.N. and S.K.; Data curation: R.G. and R.W.S.; Formal analysis: R.G. and R.W.S.; Funding acquisition: S.N.; Investigation: S.N. and R.G.; Methodology: R.G. and R.W.S.; Project administration: S.K. and R.W.S.; Resources: S.K., F.K. and R.G.; Supervision: S.K. and F.K.; Validation: R.G. and R.W.S.; Visualization: R.W.S.; Writing – original draft: R.W.S. and S.N.; Writing—review & editing: R.G. and R.W.S.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest/competing interests to declare.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All authors have seen the manuscript and agree to its publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noor, S., Goddard, R., Khatoon, F. et al. Structural Characterization of Heterodinuclear ZnII-LnIII Complexes (Ln = Pr, Nd) with a Ring-Contracted H2valdien-Derived Schiff Base Ligand. J Chem Crystallogr 52, 89–96 (2022). https://doi.org/10.1007/s10870-021-00891-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-021-00891-4