
Abstract
Poorly soluble in the most part of organic solvents dimeric complexes \({\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (I) and \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (II) (L is 4,4'-(1,4-phenylenebis(azanylylidene))bis(3,6-di-tert-butyl-2-hydroxycyclohexa-2,5-dien-1-one dianion)) are synthesized by the reactions of magnesium and nickel acetates with the ditopic redox-active ligand of the hydroxy-para-iminoquinone type in a DMF solution. The molecular and crystal structures of the synthesized compounds are determined by X-ray diffraction analysis (CIF files CCDC nos. 2045665 (I) and 2045666 (II·3DMF)). The thermal stability is studied by thermogravimetry. The redox-active character of the organic bridging ligand in the dimeric complexes \({\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF and \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF is confirmed by the data of solid-phase electrochemistry.
Similar content being viewed by others

INTRODUCTION
The trend related to the construction of compounds based on di-, and tri-, and polytopic ligands is one of the most intensively developed and very promising fields in the modern coordination chemistry. The use of these ligand systems in the synthesis of new metal-organic derivatives makes it possible to produce polynuclear (both homo- and heterometallic) complexes [1–5], metal-organic frameworks (MOFs) [6–9], and metal-organic cages (MOCs) [10–12]. The presence of redox-active organic fragments in similar structures provides wide prospects for the practical use of the synthesized compounds, since they are characterized by such phenomena as valence tautomerism and spin crossover [9, 13–15].
Doubly deprotonated 2,5-dihydroxy-para-quinone and its analogs with various substituents in the 3,6-positions of the quinoid ring are often used as redox-active bridging ligands in the synthesis of the polynuclear derivatives and MOFs [7–9, 16]. The anilate ligands of this type were used to synthesize the binuclear complexes of various metals [1, 2, 4, 17] and linear [18–21], cross-linked [22–26], and framework [27–30] metal-organic coordination polymers with unique catalytic, sorption, magnetic, and luminescence properties. The heterometallic [3, 31–35] and mixed-ligand MOFs [36–38] were synthesized to extend the range of probable properties of the materials.
The extension of the range of the applied organic ligands substantially enhances the possibility of varying the structural and functional properties of both monomeric and polymeric coordination compounds, and the search for new redox-active ligands for the target design of metal-organic derivatives is an important task of the modern coordination chemistry.
The closest analog of anilate ligands containing the extended π-system, 4,4'-(1,4-phenylenebis(azanylylidene))bis(3,6-di-tert-butyl)-2-hydroxycyclohexa-2,5-dien-1-one (L2H2), is among the promising ditopic ligands for the synthesis of new binuclear derivatives and MOFs [5, 39]. This ligand is bis(p-iminobenzoquinone) in which the iminoquinone fragments are linked via the nitrogen atoms by the p-phenylene bridge. When bound in the complex with the metal, the ligand can potentially exist in seven different redox states (Scheme 1).

Scheme 1.
The dimeric derivatives of the divalent metals (Mg and Ni) based on L2H2 were synthesized and studied. The structures of the synthesized compounds were determined by various physicochemical methods, and the molecular and crystal structures were confirmed by X-ray diffraction analysis (XRD).
EXPERIMENTAL
Commercial N,N'-dimethylformamide (DMF), p‑phenylenediamine, Mg(CH3COO)2·H2O, and Ni(CH3COO)2·4H2O were used. Ligand L2H2 was synthesized according to a known procedure [39]. IR spectra in a range of 400–4000 cm–1 were recorded on an FSM-1201 FT-IR spectrometer (suspensions in Nujol, KBr pellets). Electronic absorption spectra (EAS) in a range of 200–1000 nm were recorded on a Varian Cary 50 spectrometer (suspensions in Nujol). Elemental analyses were carried out on an Elementar Vario El cube instrument. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TG) studies were carried out on a Shimadzu DTG-60H synchronous thermal analyzer.
Synthesis of dimeric complexes \({\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (I) and \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (II). Dark finely crystalline products (compounds I and II) poorly soluble in the most part of organic solvents were synthesized by the direct reactions of metal (Mg and Ni) acetates (0.04 mmol) with L2H2 (0.04 mmol) in DMF (5 mL) on heating to 120°С for 20 min followed by slow cooling to room temperature. The synthesized derivatives were collected on a Schott filter and dried in air.
Complex I was isolated as dark blue fine crystals in a yield of 70%.
For C80H112N8O12Mg2 | |||
|---|---|---|---|
Anal. calcd., % | C, 67.36 | H, 7.91 | N, 7.86 |
Found, % | C, 66.92 | H, 8.07 | N, 7.74 |
IR (Nujol), ν, cm–1: 1691 s, 1674 s, 1661 s, 1623 s, 1599 s, 1546 s, 1421 s, 1403 s, 1358 s, 1342 s, 1315 s, 1263 m, 1222 m, 1107 s, 1066 s, 1010 m, 949 m, 927 m, 910 s, 864 s, 699 m, 686 s, 671 m, 652 m, 630 m, 620 s, 603 s, 577 s, 537 s.
EAS (Nujol), λ, nm: 300, 335, 615.
Complex II was isolated as dark blue fine crystals in a yield of 78%.
The crystal unit cell of the nickel derivative contains uncoordinated DMF (three solvent molecules per dimer Ni2\({\text{L}}_{2}^{2}\)·4DMF). However, according to the elemental analysis data, the isolated finely crystalline product well dried in air contains no solvate molecules.
For C86H126N10O14Ni2 | |||
|---|---|---|---|
Anal. calcd., % | C, 64.26 | H, 7.55 | N, 7.49 |
Found, % | C, 63.91 | H, 7.75 | N, 7.41 |
IR (Nujol), ν, cm–1: 1676 s, 1655 s, 1587 s, 1541 m, 1484 m, 1387 s, 1358 s, 1347 s, 1317 s, 1267 s, 1221 s, 1105 s, 1069 s, 951 m, 925 m, 914 m, 894 m, 852 s, 733 m, 684 s, 649 s, 618 m, 549 s.
EAS (Nujol), λ, nm: 300, 330, 620, 710.
XRD data for complexes I and II·3DMF were obtained on the X-ray beamline of the Belok station at the Kurchatov Synchrotron Radiation Source of the Kurchatov Institute Russian Research Center (Moscow, Russia) in the φ scan mode using a Rayonix SX165 CCD detector at 100 K [40]. The primary indexing, refinement of unit cell parameters, reflection integration, and an intensity absorption correction were applied using the XDS program package [41]. The structures were solved by direct methods and refined by full-matrix least squares for F 2 in the anisotropic approximation for non-hydrogen atoms, except for the disordered DMF molecule in compound II, which was refined in the isotropic approximation using an idealized molecular geometry [42]. Hydrogen atoms were placed in the calculated positions and refined by the riding model with Uiso(H) = 1.5Ueq(C) for the hydrogen atoms of the methyl groups and 1.2Ueq(C) for other hydrogen atoms. For the structures of complexes I and II·3DMF, the coordinates of atoms, thermal parameters, and populations of the positions were refined without additional restraints of the model, except for the carbon atoms of the methyl groups in the tert-butyl group disordered over two positions (population ratio 0.558 : 0.442). The carbon atoms of the methyl groups were refined with restraints imposed on the C–C bond lengths for the structure of compound I. In the case of II·3DMF, restraints were also imposed on the bond lengths and thermal vibration parameters of the atoms of the carbonyl group in the completely disordered DMF molecule (population ratio 0.521 : 0.479). The calculations were performed using the SHELXTL program package [43] in the OLEX2 medium designed for structural data visualization and processing [44].
The crystallographic data for compounds I and II·3DMF were deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 2045665 and 2045666, respectively; deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk/data_request/cif).
Quantum-chemical calculations were performed using the Gaussian09 program package [45] by the density functional theory (DFT) using the B3LYP functional [46] in the 6-31(d) standard basis set for all atoms.
Electrochemical measurements were carried out on a BASiEpsilonE2P electrochemical analyzer (USA) with the Epsilon-EC-USB-V200 software. A standard three-electrode system with a carbon paste electrode (CPE) for powdered samples as the working electrode was used. An Ag/AgCl electrode (0.01 M) served as the reference electrode, and a platinum wire was used as the counter electrode. A supporting electrolyte for the determination of the volt–ampere characteristics was 0.1 M Et4NBF4. Acetonitrile was distilled over P2O5 and KMnO4 and then over molecular sieves. After purification, the solvent was stored in a dry argon atmosphere. The supporting salt Et4NBF4 was recrystallized from ethanol and dried at 100°C in vacuo for 2 days. The modified CPE was used as the working electrode for the study of the powdered samples and was prepared as follows. A graphite powder and phosphonium salt ((tri-tert-butyl)dodecylphosphonium tetrafluoroborate) in a ratio of 90 : 10 (wt/wt) were mixed and triturated in a mortar until a homogeneous mixture was formed [47–50]. The modified electrode was prepared similarly, except for the fact that about 5% graphite powder were replaced by the studied complex (I or II). A portion of the prepared paste was densely packed into the cavity (3 mm in diameter) of a Teflon holder.
RESULTS AND DISCUSSION
The reactions of divalent metal (Mg and Ni) acetates with L2H2 in a DMF solution afforded the dimeric complexes \({{{\text{M}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (М = Mg, Ni) (Scheme 2). The reaction occurs on heating to 120°С for 20 min. The heating of the reaction mixture for a longer time results in the degradation of the organic ligand due to the hydrolysis of the C=N imine bonds. The finely crystalline products obtained in the reaction are formed during the slow cooling of the reaction mixture to room temperature. The formed precipitates of the target product were dark blue (nearly black) and became analytically pure after drying in air. The synthesized compounds are resistant to air oxygen and moisture.
The compositions and structures of compounds I and II were confirmed by IR spectroscopy and elemental analysis data. The IR spectra of the complexes exhibit a strong absorption band at ~1670 cm–1 corresponding to vibrations of the carbonyl bond of DMF and a set of bands in a range of 1580–1660 cm–1 characterizing stretching vibrations in the system of the C=O and C=N bonds conjugated with C=C of the p‑iminoquinone ligands [51, 52].

Scheme 2.
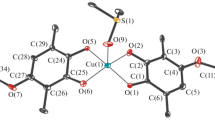
The molecular structures of \({\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (I) and \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF (II) are shown in Fig. 1. The crystallographic data and experimental XRD parameters for complexes I and II are given in Table 1, and selected bond lengths are listed in Table 2. In order to simplify the description of the structural characteristics of complexes I and II, we calculated the average bond lengths in the molecular fragments of the same type. The averaged bond lengths (Å) in complexes I and II are given in Table 3, and the numeration of atoms for the averaged values is presented in Scheme 1. It should be mentioned that the unit cell of the nickel complex (II) contains three solvate DMF molecules per dimer \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{{\text{2}}}^{{\text{2}}}\)·4DMF.

Molecular structures of dimeric derivatives (a) I and (b) II·3DMF.
Compound I crystallizes in the orthorhombic space group Pca21, and the \({\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF molecule is nonsymmetric and exists in the common position. Dimeric derivative II·3DMF crystallizes in the triclinic space group \(P\bar {1}\). The independent part of the crystal cell contains three DMF solvate molecules and two crystallographically independent molecules of complex \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF, each of which lies on the symmetry center. The geometric parameters of both \({\text{N}}{{{\text{i}}}_{{\text{2}}}}{\text{L}}_{2}^{2}\)·4DMF molecules are close to each other and, hence, the parameters of only one molecule are given in Table 2.
The general structure of the dimeric complexes remains unchanged for both metals. Two ditopic p‑iminoquinone ligands bind metal cations by two parentheses to form chelate five-membered metallocycles MOCCO (M = Mg, Ni) and to form thus a metal-organic cage. The metal atom significantly deviates from the OCCO planes by a distance 0.285(6)–0.699(5) Å in the magnesium compound and by 0.556(5)–0.566(5) Å in the nickel compound. The dihedral angles between the planes of the p-iminoquinone fragments coordinated to one metal atom are 51.39(6)°–52.28(6)° and 49.07(6)°–49.12(6)° for complexes I and II·3DMF, respectively. The dihedral angle between the planes of the p-phenylene bridges is only 1.0(2)° in complex I, whereas these planes are completely parallel in derivative II·3DMF. Note that the p-phenylene fragments themselves are significantly remote from each other within the same molecule: the distances between the centers of the rings are 9.143(2) Å in complex I and 8.832(4)–9.119(4) Å in complex II·3DMF. The metal atoms are remote by more than 14 Å in both complexes to form a cavity inside the MOC with a diameter of ~3.5 Å, which is principally accessible for small molecules (Fig. 2).

Cavity in the metal-organic cage of complex II·3DMF.
The crystal packing in the molecules of complexes I and II·3DMF is determined by many intermolecular contacts between the oxygen atoms of the p-quinoid ligands or DMF molecules and the hydrogen atoms of the tert-butyl substituents or N-methyl groups of DMF. In addition, the С–Н…π-arene contact between the N‑methyl group of DMF and aromatic p‑phenylene ring of another molecule (С–π-arene centroid distance 3.410(6) Å) is observed in compound I. Probably, many intermolecular contacts is a reason for the low solubility of the synthesized compounds in the most part of organic solvents.
The coordination environment of each of the magnesium and nickel cations in complexes I and II·3DMF, respectively, represents a distorted octahedron with four oxygen atoms of two p-iminoquinone ligands, and two oxygen atoms of the coordinated DMF molecules are arranged at the vertices. It should be mentioned that the M–O bond lengths in the chelate cycles expectedly differ between each other. The M–O1 bond lengths (~2.01 Å for both compounds, see Table 3) is less than the sum of covalent radii of the corresponding elements (2.18 and 2.11 Å for complexes I and II·3DMF, respectively [53], which is well consistent with covalent binding. The M–O2 coordination bonds of the metal with the carbonyl oxygen atom of the organic p-iminoquinone ligand (2.15 Å for I and 2.08 Å for II·3DMF) are longer by 0.14 and 0.07 Å, on the average, than covalent M–O1 bonds in the magnesium and nickel derivatives, respectively. The observed difference is substantially lower than that in the tin(IV) compounds (0.4 Å) based on the monotopic analog of ligand L2H2 [54], indicating a significant charge delocalization in the chelate cycles.
The p-iminoquinone structure of the initial ligand L2H2 is retained in dimeric derivatives I and II·3DMF [5]. The С1–С2 and С4–С5 bonds (Table 3) with the length ranging from 1.34 to 1.39 Å, respectively, can be characterized as double bonds, whereas the С1–С6, С2–С3, С3–С4, and С5–С6 bond lengths range from 1.45 to 1.50 Å. The coordination of the organic ligand to the metal atom results in only insignificant changes in the bond length distribution in the initial bis-para-iminobenzoquinone related to the negative charge delocalization in the p-iminoquinone fragment of the organic ligand. The С1–О1 bond (1.30 Å for both complexes) shortens compared to the C–OH bond in the initial ligand (1.352(2) Å), and the С2–О2 bond (1.25 Å for both complexes) elongates compared to L2H2 (C=O 1.230(2) Å). The C–C bond of the p‑phenylene bridge in complexes I and II·3DMF are equalized (1.38–1.40 Å) (Table 3), which is consistent with its aromatic nature.
The thermal stability of dimeric derivatives I and II was estimated by the TG and DSC methods (Fig. 3). According to the TG and DSC data, the decoordination of four DMF molecules from the magnesium atoms occurred for complex I in a temperature range of 170–250°С, and the mass loss was 20%. The further heating results in the destruction and complete decomposition of complex I. According to the TG data, compound II starts to decompose below 100°С and demonstrates a gradual mass loss (~4%) on heating to 130°С because of the removal of the occluded solvent from the sample. The decoordination of four DMF molecules from the nickel atoms occurs in a temperature range of 130–200°С, and the observed mass loss is ~18%. The further heating results in the decomposition of the nickel derivative.

Differential thermal analysis (DTA) curves of (solid line) TG and (dashed line) DSC for complexes I and II.
The low solubility of compounds I and II did not allow us to detect the EAS in solution. The absorption spectra in the UV and visible ranges of the studied dimeric magnesium and nickel complexes recorded in mineral oil are presented in Fig. 4 and show intense bands at 300 and 330–335 nm, as well as broad absorption bands at λ > 620 nm. The results obtained are in good agreement with the results of the quantum-chemical calculations performed for compound I in the experimental geometry at the B3LYP/6-31(d) level. The frontier orbitals of complex I (Fig. 5) consist of the orbitals of the ligand only. The LUMO and LUMO+1 orbitals are predominantly localized on the acceptor p-quinoid fragments, whereas the HOMO and HOMO–1 orbitals are delocalized over the whole system of conjugated double bonds of the ditopic ligands through the p-phenylene bridge. The intra- and interligand HOMO–LUMO electron transfer determines the long-wavelength absorption band in the EAS of compound I, and the differences in the frontier orbital energies (1.9–2 eV) are well consistent with the broad absorption band observed in the experimental spectrum. It is important that complex formation results in a significant bathochromic shift of the charge-transfer band for the bis-para-iminoquinone ligand L2H2 [5], which is 140 nm, and this shift is very sensitive to the complexing agent nature. In the presence of the weak Lewis acid (alkylmercury cation) the shift reached 40 nm only [5]. Magnesium(II) and nickel(II) cations favor the generation of a stronger negative charge on the ligand and thus bring together its frontier orbitals.

Electronic absorption spectra of complexes I and II.

Frontier orbitals in complex I.
The redox properties of compounds I and II in the solid state were studied by cyclic voltammetry (CV) using the modified CPE. The corresponding cyclic voltammograms are shown in Fig. 6. The potentials of the redox processes for complexes I and II are presented in Table 4.

CV curves of (a) reduction and (b) oxidation and their semidifferentiated forms for complexes I and II; CPE, CH3CN, 10–1 M Bu4NBF4, potential vs. Ag/AgCl.
The potentials of the first reduction and oxidation peaks for both studied complexes I and II are close (Table 4), and the electron transfer processes are irreversible under the CV conditions. The close potentials of the first steps of electron attachment and detachment for the derivatives of redox-active Ni(II) and redox-inactive Mg(II) confirm the ligand-centered character of the observed processes (Scheme 3). The peaks at higher reduction potentials of nickel complex II, which are not observed for magnesium complex I, should be attributed to the electron transfers to the nickel ions.
Thus, the metal-organic cages based on divalent magnesium and nickel cations and the redox-active ditopic ligand of the p-iminoquinone type were synthesized. The XRD study of the molecular and crystal structures of the compounds showed cavities inside the formed cages. The redox-active behavior of the ligand framework of the synthesized complexes was demonstrated by solid-state electrochemistry.

Scheme 3.
REFERENCES
Min, K.S., DiPasquale, A., Rheingold, A.L., et al., Inorg. Chem. Comm., 2007, vol. 46, p. 1048.
Min, K.S., DiPasquale, A.G., Rheingold, A.L., et al., J. Am. Chem. Soc., 2009, vol. 131, p. 6229.
Kanegawa, S., Shiota, Y., Kang, S., et al., J. Am. Chem. Soc., 2016, vol. 138, p. 14170.
Simonson, A.N., Kareis, C.M., Ovanesyan, N.S., et al., Polyhedron, 2018, vol. 139, p. 215.
Druzhkov, N.O., Meshcheryakova, I.N., Cherkasov, A.V., and Piskunov, A.V., Russ. Chem. Bull., 2020, vol. 69, p. 49.
Espallargas, G.M. and Coronado, E., Chem. Soc. Rev., 2018, vol. 47, p. 533.
D’Alessandro, D.M., Chem. Commun., 2016, vol. 52, p. 8957.
Kitagawa, S. and Kawata, S., Coord. Chem. Rev., 2002, vol. 224, p. 11.
Mercuri, M.L., Congiu, F., Concas, G., et al., Magnetochemistry, 2017, vol. 3, p. 1.
Castilla, A.M., Ramsay, W.J., and Nitschke, J.R., Acc. Chem. Res., 2014, vol. 47, p. 2063.
Schmidt, A., Casini, A., and Kuhn, F.E., Coord. Chem. Rev., 2014, vol. 275, p. 19.
Bloch, W.M. and Clever, G.H., Chem. Commun., 2017, vol. 53, p. 8506.
Darago, L.E., Aubrey, M.L., Yu, C.J., et al., J. Am. Chem. Soc., 2015, vol. 137, p. 15703.
Jeon, I.-R., Negru, B., Duyne, R.P.V., et al., J. Am. Chem. Soc., 2015, vol. 137, no. 50, p. 15699.
Chen, J., Sekine, Y., Komatsumaru, Y., et al., Angew. Chem., Int. Ed. Engl., 2018, vol. 57, p. 12043.
Benmansour, S. and Gomez-Garcia, C.J., Gen. Chem., 2020, vol. 6, p. 190033.
Benmansour, S., Lopez-Martinez, G., Canet-Ferrer, J., et al., Magnetochemistry, 2016, vol. 2, p. 1.
Chang, C.-H., Li, A.-C., Popovs, I., et al., J. Mater. Chem. A, 2019, vol. 7, p. 23770.
DeGayner, J.A., Wang, K., and Harris, T.D., J. Am. Chem. Soc., 2018, vol. 140, p. 6550.
Abrahams, B.F., Dharma, A.D., Dyett, B., et al., Dalton Trans., 2016, vol. 45, p. 1339.
Morikawa, S., Yamada, T., and Kitagawa, H., Chem. Lett., 2009, vol. 38, p. 654.
Kharitonov, A.D., Trofimova, O.Y., Meshcheryakova, I.N., et al., CrystEngComm, 2020, vol. 22, p. 4675.
Liu, L., DeGayner, J.A., Sun, L., et al., Chem. Sci., 2019, vol. 10, p. 4652.
Kingsbury, C.J., Abrahams, B.F., Auckett, J.E., et al., Chem.-Eur. J., 2019, vol. 25, p. 5222.
Gómez-Claramunt, P., Benmansour, S., Hernandez-Paredes, A., et al., Magnetochemistry, 2018, vol. 4, p. 6.
Kingsbury, C.J., Abrahams, B.F., D’Alessandro, D.M., et al., Cryst. Growth Des., 2017, vol. 17, p. 1465.
Abrahams, B.F., Hudson, T.A., McCormick, L.J., et al., Cryst. Growth Des., 2011, vol. 11, p. 2717.
Bondaruk, K. and Hua, C., Cryst. Growth Des., 2019, vol. 19, p. 3338.
Murase, R., Abrahams, B.F., D’Alessandro, D.M., et al., Inorg. Chem., 2017, vol. 56, no. 15, p. 9025.
Benmansour, S., Vallés-García, C., Gómez-Claramunt, P., et al., Inorg. Chem., 2015, vol. 54, p. 410.
Martínez-Hernández, C., Gómez-Claramunt, P., Benmansour, S., et al., Dalton Trans., 2019, vol. 48, p. 13212.
Martínez-Hernández, C., Benmansour, S., and Gómez-García, C.J., Polyhedron, 2019, vol. 170, p. 122.
Hernández-Paredes, A., Cerezo-Navarrete, C., Gómez-García, C.J., et al., Polyhedron, 2019, vol. 170, p. 476.
Atzori, M., Benmansour, S., Espallargas, G.M., et al., Inorg. Chem., 2013, vol. 52, p. 10031.
Abhervé, A.,Clemente-León, M., Coronado, E., et al., Inorg. Chem., 2014, vol. 53, p. 12014.
Sahadevan, S.A., Monni, N., Oggianu, M., et al., ACS Appl. Nano Mater., 2020, vol. 3, p. 94.
Atzori, M., Pop, F., Auban-Senzier, P., et al, Inorg. Chem., 2015, vol. 54, p. 3643.
Kabir, M.K., Kawahara, M., Kumagai, H., et al., Polyhedron, 2001, vol. 20, p. 1417.
Abakumov, G.A., Cherkasov, V.K., Kocherova, T.N., et al., Russ. Chem. Bull. (Int. Ed.), 2006, vol. 55, p. 1195.
Svetogorov, R.D., Dorovatovskii, P.V., and Lazarenko, V.A., Cryst. Res. Technol., 2020, vol. 55, p. 1900184.
Kabsch, W., Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, vol. 66, p. 125.
Guzei, I.A., J. Appl. Crystallogr., 2014, vol. 47, p. 806.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al., Wallingford: Gaussian, Inc., 2013.
Becke, A.D., J. Chern. Phys., 1993, vol. 93, p. 1372.
Khrizanforov, M.N., Arkhipova, D.M., Shekurov, R.P., et al., J. Solid State Electrochem., 2015, vol. 19, p. 2883.
Kataeva, O., Khrizanforov, M., Budnikova, Y., et al., Cryst. Growth Des., 2016, vol. 16, p. 331.
Khrizanforov, M.N., Fedorenko, S.V., Strekalova, S.O., et al., Dalton Trans., 2016, vol. 45, p. 11976.
Khrizanforov, M.N., Shekurov, R.P., Ermolaev, V.V., et al., Phosphorus Sulfur Silicon Relat. Elem., 2016, vol. 191, p. 1611.
Piskunov, A.V., Meshcheryakova, I.N., Fukin, G.K., et al., Russ. J. Coord. Chem., 2014, vol. 40, no. 4, p. 205. https://doi.org/10.1134/S1070328414040083
Khamaletdinova, N.M., Meshcheryakova, I.N., Piskunov, A.V., et al., J. Struct. Chem., 2015, vol. 56, p. 233.
Batsanov, S.S., Russ. J. Inorg. Chem., 1991, vol. 36, p. 1694.
Piskunov, A.V., Meshcheryakova, I.N., Fukin, G.K., et al., Russ. J. Coord. Chem., 2017, vol. 43, no. 12, p. 816. https://doi.org/10.1134/S1070328417120077
ACKNOWLEDGMENTS
The studies were carried out using the equipment of the Analytical Center at the G.A. Razuvaev Institute of Organometallic Chemistry (Russian Academy of Sciences). The DSC and TG studies were carried out using the equipment of the Center for Collective Use “New Materials and Energy Saving Technologies” (Lobachevsky State University of Nizhny Novgorod). Electronic absorption spectra recording was supported by the Ministry of Science and Higher Education of the Russian Federation using the scientific equipment of the Center of Molecular Structure Investigation at the Nesmeyanov Institute of Organoelement Compounds (Russian Academy of Sciences). The electrochemical properties were studied by M.N. Khrizanforov and Yu.G. Budnikova in terms of state assignment of the Kazan Research Center of the Russian Academy of Sciences.
Funding
This work supported by the Russian Foundation for Basic Research, project no. 18-29-04041-mk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Yablonskaya
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meshcheryakova, I.N., Trofimova, O.Y., Druzhkov, N.O. et al. Magnesium and Nickel Complexes with Bis(p-iminoquinone) Redox-Active Ligand. Russ J Coord Chem 47, 307–318 (2021). https://doi.org/10.1134/S1070328421050043
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328421050043