
Abstract
The systematics of Subantarctic and Antarctic near-shore marine benthic invertebrates requires major revision and highlights the necessity to incorporate additional sources of information in the specimen identification chart in the Southern Ocean (SO). In this study, we aim to improve our understanding of the biodiversity of Kidderia (Dall 1876) through molecular and morphological comparisons of Antarctic and Subantarctic taxa. The microbivalves of the genus Kidderia are small brooding organisms that inhabit intertidal and shallow subtidal rocky ecosystems. This genus represents an interesting model to test the vicariance and dispersal hypothesis in the biogeography of the SO. However, the description of Kidderia species relies on a few morphological characters and biogeographic records that raise questions about the true diversity in the group. Here we will define the specimens collected with genetic tools, delimiting their respective boundaries across provinces of the SO, validating the presence of two species of Kidderia. Through the revision of taxonomic issues and species delimitation, it was possible to report that the Antarctic species is Kidderia subquadrata and the species recorded in the Subantarctic islands Diego Ramirez, South Georgia and the Kerguelen Archipelago is Kidderia minuta. The divergence time estimation suggests the origin and diversification of Kidderia lineages are related to historical vicariant processes probably associated with the separation of the continental landmasses close to the late Eocene.
Similar content being viewed by others
Introduction
In the light of global change, understanding the evolutionary and ecological processes underlying the origin, distribution, and maintenance of Southern Ocean (SO) biodiversity has pivotal importance in mitigating the consequences on biodiversity in this area (Aronson and Blake 2001; Linse et al. 2006; Rogers 2007; Griffiths et al. 2009; Chown et al. 2015; Hawkins et al. 2018). The species of the genus Kidderia (Dall 1876) is one of the few members of the family Cyamiidae in the SO. These small bivalves have an average shell length of 4.5 mm (Simpson 1977) with a maximum size of 7 mm, and belong to a widely distributed group of microbivalves that inhabit across the SO. The current biodiversity value of this group is required to increase knowledge of the diversification and biogeography patterns in the SO, as this is still scarce in various taxa (Geiger and Thacker 2008; Ghiglione et al. 2017). It is especially important as future projections indicate a reduction in the richness of marine species in the SO due to climate change (Griffiths et al. 2017).
Kidderia have elongated, flat shells with an internal ligament located behind the cardinal tooth. They are gregarious with direct development and inhabit intertidal and shallow subtidal rocky ecosystems (Shabica 1974; Simpson 1977). Consequently, a low potential for dispersal and a spatial distribution limited to the ice-free rocky coast of Antarctica is expected. The taxonomy of the species in this group has been developed exclusively using morphological characters and biogeographic records in Subantarctic and Antarctic localities in the SO.The type species of the genus is Kidderia minuta (Dall 1876), originally described in the Kerguelen Islands. Kidderia bicolor (Martens 1885) has been reported in South Georgia, the South Orkney, Heard and Kerguelen Islands as well as in the Weddell Sea and South Shetland Islands (Martens 1885; Hedley 1916; Simpson 1977; Clarke 1996; Zelaya 2005; Barnes et al. 2006; Engl et al. 2016). The species Kidderia subquadrata (Pelseneer 1903) and Kidderia pusilla (Gould 1850) have been reported in Subantarctic ecosystems including Patagonia, Cape Horn, the Falkland/Malvinas and South Georgia Islands (Zelaya 2005, 2015; Barnes et al. 2006). Shabica (1974) reported the presence of K. subquadrata at Doumer Island, Antarctic Peninsula and the most recent record in the literature reports the presence of K. subquadrata in the Antarctic region, on King George Island, South Shetland Islands (Aghmich et al. 2016). These species correspond to the principal species has been described inhabiting in SO (Online Resource 1). Given this pattern of biogeographical reports, new questions arise: Are the same Kidderia species distributed in Subantarctic and Antarctic areas? What is the effectiveness of the physical barrier, such as the geographic distance that separates the Antarctic continent from the others to gene flow in this genus? What has been the role of the Antarctic circumpolar current (ACC) in promoting speciation and divergence in this genus, an isolation barrier or dispersal vector?
Two main hypotheses have been proposed to explain the current biogeography and species diversification in the SO (Patarnello et al. 1996; Linse et al. 2007; Pearse et al. 2009; Dalziel et al. 2013; Taylor et al. 2018). The vicariance hypothesis is supported by the high degree of genetic differentiation and old divergence-times in some marine taxa such as mollusks, echinoderms, crustaceans and algae (Patarnello et al. 1996; Held 2000; Linse et al. 2007; Medlin and Zingone 2007; Hunter and Halanych 2008; Jackson et al. 2015) distributed across the SO, initiated by the continental separation of the Antarctic from the others in the Jurassic period (Jokat et al. 2003; König and Jokat 2006; Eagles and Vaughan 2009), followed by the opening of Drake passage 42 Ma (Crame 1999; Clarke and Crame 2010; Hayanich and Mahon 2018), and the subsequent deepening of the Tasman Sea (34–33 Ma) (Stickley et al. 2004). These changes modified the oceanographic circulation of the SO (Stickley et al. 2004), the establishment of the ACC and the Antarctic Polar Front (32 Ma) around the Oligocene/Eocene boundary (33.9 Ma) (Clarke and Crame 1989, 1992; Orsi et al. 1995; Clarke and Johnston 2003).
The dispersal hypothesis is related to the function that the ACC fulfills by moving marine organisms around the Antarctic; it is considered the primary vector of dispersal, especially in taxa with high dispersal ability (Clarke et al. 2005; Fraser et al. 2009; Hüne et al. 2015; Hayanich and Mahon 2018). The dispersal hypothesis is also supported by molecular studies that reveal the importance of dispersal and recent climatic/oceanographic processes associated with more recent divergences that help to understand the connectivity in the distribution and diversification of species present across the SO (Díaz et al. 2011; González-Wevar et al. 2012, 2019; Near et al. 2012; Poulin et al. 2014; Dueñas et al. 2016). In this study, using molecular analyses we aim to reveal the current biogeographic distribution of species Kidderia to test the hypothesis that best fits the diversity and distribution of this group across in Subantarctic and Antarctic localities in the SO.
Materials and methods
Sample collection and initial specimen identification
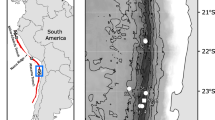
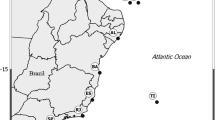
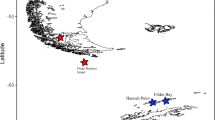
Specimens of Kidderia were collected from rocky intertidal ecosystems in different localities of the SO during summer 2016–2019 (Table 1, Fig. 1). In some of these sampled sites, Kidderia species were absent, however, we included records for other microbivalves species found as a confirmation of their distribution, this is the case for Lasaea sp. and Lissarca sp. (Table 1). The initial taxonomic identification was made based on a complete review of the current literature (Güller and Zelaya 2015; Zelaya 2015), as well as using classic taxonomic works (Martens 1885; Martens and Pfeffer 1886; Pelseneer 1903; Odhner 1923), systematic studies on specific taxa (Soot-Ryen 1951; Ponder 1971; Osorio and Arnaud, 1984), and academic databases available on the Internet (Morris and Rosenberg 2005; WORMS editorial board 2018). Finally, for the correct identification of the species we followed the last revision of the group (Zelaya et al. 2020), examining morphological aspects such as: shell outline, sculpture, the position of the umbones and the number and morphology of the hinge teeth.

a General map shows Subantarctic localities b Map of Antarctic Peninsula localities sampled in this study
To assist the morphological identification, we also performed scanning electron microscopy (SEM) analysis to identify key characters in the shell. The SEM analysis was performed using a Hitachi SU3500 scanning microscope at the Universidad Catolica del Norte–Chile.
DNA extraction, amplification, sequencing and alignment
Collected samples were stored in 70% alcohol until DNA extraction. Total genomic DNA was isolated using the commercial kit Quick—DNA Plus according to the manufacturer’s protocol (Zymo research). Following this, different fragments of nuclear 28S rRNA (ribosomal large subunit), ITS2 (internal transcribed spacer 2) and mitochondrial (Cox1) markers were amplified using polymerase chain reactions (PCR). In the case of 28S rRNA forward primer described by Littlewood et al. (2000) and reverse primer described by Williams et al. (2003) were used. The primers for ITS2 are described by Cheng et al. (2006). Given the absence of genetic information for the genus Kidderia, the strategy to generate molecular markers was to perform MiSeq Illumina sequencing using the genomic DNA and an in-silico enrichment to obtain fragments of the mitochondrial genome. Then, a species-specific primer was designed to amplify a partial fragment of Cox1 (approximate 1000 pb): kg8F (TTG GGC TGG GTT AAT AGG TACA-3′) as a forward primer and kg7R (5′-GAA AAC CAG CAA ACA TAG CA-3′) as a reverse primer.
All PCR were performed in a final volume of 25 μl containing 1X PCR buffer, 3.5 mM MgCl2, 0.2 mM of each dNTP, 0.25 μM of each primer, 0.2X BSA, 1 μl DNA (10–50 ng), 0.6 units GoTaq DNA polymerase (Promega) and H2O to reach the final volume. For 28S rRNA, the thermocycling program included an initial denaturation step (95 °C for 3 min); 40 cycles of amplification (94 °C for 45 s), annealing at 52 °C for 45 s and extension (72 °C for 2 min), and a final extension step (72 °C for 10 min). For ITS2, the thermocycling program was according to Cheng et al. (2006). The Cox1 gene was amplified using the following thermocycling profile: an initial denaturation step (95 °C for 5 min); 35 cycles of amplification (94 °C for 30 s, 60 °C for 1 min and 72 °C for 2 min); and a final extension step (72 °C for 8 min). The annealing temperature was modified to 50º C (South Georgia and Kerguelen individuals) and 49 °C (Diego Ramirez individuals) for the PCR performed with Subantarctic individuals of Kidderia. The PCR products were purified using the E.Z.N.A. ® Cycle Pure PCR Purification Kit (Omega Bio-tek) and sequenced in both directions in the ABI PRISM® 310 Genetic Analyzer of the core-facility AUSTRAL-omics (www.austral-omics.cl).
All sequences were assembled, examined, and aligned in Geneious R10.2.4 software (Kearse et al. 2012) and manually edited to resolve unclear base calls. Sequences of each gene were aligned individually using the MAFFT v7.388 algorithm implemented in Geneious with G‐INS‐I strategy (Katoh and Standley 2013). Then, the Translate function offered by the Geneious program for coding genes was applied. Finally, the sequences were concatenated using the software Geneious.
Species delimitation and phylogenetic analysis
The following approaches were used to delimit the species of this study. First, the General Mixed Yule Coalescence (GMYC) single and multiple threshold model was used, applying the "Species Limits by Threshold Statistics" approach (SPLITS v1.0-19) in the R program (www.r-project.org) (Pons et al. 2006). A second method was used to delimit the species-the Bayesian implementation of the Tree Poisson Process (PTP) model, using a distance-based tree to delimit species implemented in the web server for PTP (www.species.h-its.org/ptp/) (Zhang et al. 2013). The supports of this analysis are presented as posterior probability (PP). For GYMC and PTP, an ultrametric tree was generated in BEAST2. The analysis was run for 30 × 106 generations with parameters sampled every 1000 generations and a burn-in value of 10%. A tree file was generated with TreeAnnotator and visualized in FigTree v.1.4.2 (Rambaut 2014). The log file (produced in BEAST) was analyzed in Tracer V1.6 (Rambaut et al. 2016) to assess for convergence. Finally, the automatic barcode gap discovery method (ABGD) was used. This method delivers species boundaries based on patterns of pairwise genetic distances, providing estimates of a maximum limit for intraspecific genetic divergence and using this limit to group sequences belonging to the same species (with less divergence) from sequences belonging to different species (with greater divergence) (Puillandre et al. 2012).
We used maximum likelihood (ML), and Bayesian inference (BI) approaches to explore their phylogenetic relationship. The best evolutionary model was chosen through the JModeltest2 program (Darriba et al. 2012) in XSEDE, available on the CIPRES online platform (www.phylo.org/). The selection of the best model used corrected Akaike criterion (AIC) for ML and Bayesian criterion (BIC) for BI analysis. The selected models for the 28S rRNA gene were GTR+I for ML analysis and TIM3+I for BI analysis. For the ITS2 gene, the selected models were TPM3uf+G for the ML analysis, and TN93 for the BI analysis; and finally, for the Cox1 gene, the TIM1+G models were selected for the ML analysis and HKY+G for the BI analysis. The ML and BI analyses were performed by gene and with concatenated data (ITS2; 28S rRNA; Cox1), using the IQtree web server (http://iqtree.cibiv.univie.ac.at/) (Trifinopoulos et al. 2016) with 1000 bootstrap for ML analysis. BI analysis was implemented in the BEAST2 software package (Bouckaert et al. 2014) with four chains (three heated chains and one cold chain) running for 30 million generations and sampling every 10,000 generations. We discarded the first 10% of trees as “burn‐in” and the remaining trees were used to generate a majority‐rule consensus tree (Felsenstein 1985). Gaimardia and Lissarca specimens found in this study were sequenced and used as outgroups based on their relationships with Kidderia (sister genus and a more distant outgroup, respectively) (Odhner 1923; Soot-Ryen 1951; Ponder 1971).
Divergence time estimations
Strict clock analysis was used for 28S rRNA/ITS2 concatenated sequences with an uncorrelated lognormal (UCLN) model of molecular evolutionary rate heterogeneity and the GTR+G substitution model implemented in BEAST2. In the Gaimardia clade, an age prior with normal distribution was applied (mean 42.9; S 0.085) to the time of the oldest Antarctic Gaimardia fossil from the La Meseta Formation during the Eocene (Zinsmeister 1984; Stilwell 1992; Beu 2009). We also included another prior in the Kidderia clade from Antarctic specimens: Costokidderia (today synonymized with Kidderia sp.) (mean 3.95; S 0.085), a Pliocene fossil from King George Island (South Shetland Islands) (Gazdzicki and Pugaczewska 1984). We also specified the molecular clock prior in BEAST2 for 28S rRNA and ITS2 of 0.1% and 1% per Ma, respectively, (Anne 2006). A Yule model speciation prior was used for branching rates in the phylogeny and 50 × 106 generations were run; trees were sampled every 1000 generations. Given the identity of our Gaimardia sequence and the sequence of G. trapesina from Genbank (accession number KX713385), we also included the last one in the analysis.
We repeated the same analysis with mtDNA (Cox1) using relaxed clock analysis with a UCLN model of molecular evolutionary rate heterogeneity, the HKY+G substitution model and the same age priors described above. We performed several initial runs considering a range of Cox1 rates that have been proposed for several other marine bivalves from 0.10 to 2.0% per Ma as lower and upper thresholds (Marko and Moran 2002, 2009; Wilke et al. 2009). We used a Bayes factor analysis to define and select a conservative molecular clock rate of 1% per Ma (Wilke et al. 2009; Li and Drummond 2011). The convergence of model parameters was reviewed in the log file in TRACER v1.6.
Results
In ten of the 15 sample sites, Kidderia species were found (Table 1; Fig. 1). The Subantarctic individuals (Diego Ramírez, South Georgia and Kerguelen Islands) were identified morphologically as K. minuta as well as those from the Antarctic islands King George, Penguin, Greenwich, Livingston, Deception and Doumer identified as K. subquadrata (Table 1). Individuals of K. subquadrata have a small, inequilateral, oblong, rather compressed laterally, white and reddish-brown shell; the sculpture consists of fine dense concentric striae and includes two cardinal teeth in the left valve (Fig. 2a–c). The representative character of the species K. minuta is the presence of rounded, inflated, nearly terminal umbones; the surface of the shell has minute rounded incremental concentric ridges and the cardinal teeth are very minute (Fig. 2d–h).

Images of taxonomic identification. a Copy of the original drawing from the Ponder’s description was made (1971) of the left valve (internal view) c Electron microscopy image of external view of left valve (zoom) b Electron microscopy image of internal view of left valve of K. subquadrata d and e Copy of the original drawing from the Ponder’s description of the left valve (internal view) g Copy of external view of right valve h Electron microscopy image of external view of right valve. f Electron microscopy image of internal view of left valve of K. minuta i Phylogenetic concatenated tree of Antarctic (A) and Subantarctic (SA) lineages of Kidderia. Numbers along branches indicate the support values from the different analyses in the order BI/ ML. Bars indicate the summary of the results obtained in the species delimitation analysis
In total, 22 individuals from Subantarctic (Diego Ramirez, South Georgia and Kerguelen islands) and Antarctic localities (Penguin, King George, Greenwich, Livingston, Deception and Doumer) were sequenced successfully. The alignment of the three concatenated genes was a total of 2639 bp. The 28 s rRNA gene is composed of 1305 bp, with 52 variable sites and 56 parsimonious sites; 473 bp of the alignment correspond to the ITS2 gene, with 88 variable sites and 86 parsimonious sites and finally the Cox1 gene is composed of 861 bp, where 168 sites were variable and 147 parsimoniously informative.
Species delimitation and phylogenetic analysis
We used the concatenated database for species delimitation analysis and constructed unrooted and ultrametric trees (Fig. 2). The summary bars of species delimitation results are shown in Fig. 2i. The GMYC analysis identified three putative species, two in the Antarctic clade (A) and one in the Subantarctic clade (SA). Two groups were delimited with the single-threshold method (confidence limits = 2–4; LR test value = 0.0009, p < 0.01) while the multiple thresholds delimited three groups (confidence limits = 3–4; LR test value = 0.0001, p < 0.01). The likelihood of the GMYC model for the single and multiple threshold models (LGMYC = 154.40) was compared to the likelihood of the null model (L0 = 147.57). The result of the Bayesian implementation of the Process Tree Poisson model (bPTP) with concatenated rooted trees detected 2 putative species with good bayesian support, Species 1 (PP = 0.91) composed of the Subantarctic clade (SA) (Subantarctic Islands Kerguelen, South Georgia and Diego Ramírez), and Species 2 (PP = 0.99) composed of an Antarctic clade (A) (Antarctic Islands: Signy, King George, Penguin, Greenwich, Livingston, Deception and Doumer). The ABGD analysis showed a tri-modal pairwise genetic distance (K2P) distribution with a clear and wide barcode gap located between 1 and 14% genetic distance. This method detected two stable putative species with estimated prior maximum divergences of intraspecific diversity as large as 10% (Fig. 2i) (one-tailed 95% confidence interval).
Phylogenetic analysis based on multiple markers and methods recognized the presence of two main clades within Kidderia with high values of bootstrap support and posterior probability (Fig. 2i). The first includes all the Subantarctic specimens analyzed (Diego Ramirez, South Georgia, Kerguelen), while the second includes the Antarctic specimens (Fig. 2i). The genetic divergence between the Subantarctic and Antarctic lineages ranged from 4.0 to 4.3% (28S rRNA), 18.3 to 19.8% (ITS2) and 16.1 to 18.0% (Cox1). Phylogenetic reconstructions based on each separate gene are shown in Online resource 2.
Divergence time estimations
The time estimation analysis for the divergence among Subantarctic and Antarctic Kidderia lineages with 28S rRNA/ITS2 concatenated dataset revealed a time around 36.43 Ma (44.87–28.35 Ma; 95% Height posterior density, HPD) that indicated an ancient divergence during the late Eocene (Fig. 3). A similar result was obtained when the analysis was performed using mtDNA (Cox1), 34.08 Ma in the Eocene/Oligocene boundary (14.94–56.53 Ma; 95% HDP; Online resource 3).

Bayesian maximum clade credibility tree based on the concatenated nuclear marker 28S rRNA/ITS2 showing the divergence time estimates within Subantarctic and Antarctic Kidderia lineages. Posterior probabilities are shown above the clades. Gray shaded regions depict 95% HPD for relative divergence times. Stars represents point calibrations and arrow the node ages estimation
Discussion
The present study revealed morphological and genetic evidence that differentiates two Kidderia lineages, with a time of divergence associated with historical events that occurred millions of years ago in the SO. Many of the microbivalve species that inhabit the rocky intertidal and shallow subtidal areas were described between 1850 and 1980, since then there has not been an exhaustive revision in the genus Kidderia (Philippi 1845; Gould 1850; Dall 1876; Martens 1885; Pfeffer 1886; Pelseneer 1903; Odhner 1923; Soot-Ryen 1951; Dell 1964; Ponder 1971). The records of the classical literature are not clear enough to specify the type locality for all described species of Kidderia. In addition, some relatively recent studies based on species checklists may be erroneous, especially if there is no evidence that they were effectively sampled in a specific location. The changes of names and taxonomic assignments generate errors in the identification of species that were described a long time ago and whose presence is currently being re-evaluated. Zelaya (2005) reports that there are 12 species of Cyamiidae inhabiting the Subantarctic ecoregions, of which 5 species belong to the genus Kidderia (Online resource 1).
In the revision of Ponder (1971) of the superfamily Cyamiacea, he describes that the species K. minuta, K. pusilla, K. bisulcata (EA Smith 1877), K. campbellica (Odhner 1924) and K. bicolor belong to the type genus of Kidderia which does not present teeth, or only one in each valve, of which K. minuta is the type species of the genus. However, in the bivalve catalog of Bernard (1983), he reported that K. minuta and K. bicolor are junior synonyms for K. pusilla. Therefore, throughout the history of this genus and by the material reviewed in this study, it has been observed that there are certain morphological similarities between the species of the type genus of Kidderia (Ponder 1971). However, as mentioned by Dall (1876) and observed in this study, the color of the individuals of K. minuta is different (more whitish in the umbones and more reddish in the upper posterior surface), and the shape of the cardinal tooth is also different.
Regarding the present taxonomic status of the Kidderia species, it is important to note that in the Ponder review (1971), he indicated that the species Cyamium subquadratun should be included within the genus Kidderia. The accepted name of K. subquadrata in WORMS is Mysella subquadratum (Engl 2012). This genus belongs to the family Lasaeidae (MolluscaBase 2018), although the status is considered as “taxon inquerendum” (MolluscaBase 2018). However, Dell (1964) and Soot-Ryan (1951) considered the species K. subquadrata to be a member of the family Cyamiidae. However, in the latest review by Zelaya et al. (2020), these authors concluded that based on morphology, anatomy and DNA, the species of the genus Kidderia and Gaimardia belong to the family Gaimardiidae. However, these authors did not include DNA sequences of Kidderia species. Finally, our results would indicate that the Antarctic lineage would correspond to the species K. subquadrata. In future research of the group, phylogenetic relationship with other taxa such as Heteromactra and Cyamium should be addressed.
Species delimitation
To generate new information and to advance in our understanding of microbivalve evolution, here we introduced the use of a DNA based-identification approach to clarify the taxonomic issues previously mentioned. The multilocus trees provide a valuable source of information for inferring the pattern and processes of diversification (Fujisawa and Barraclough 2013). While PTP analysis is considered faster and more robust (Zhang et al. 2013), ABGD is a simple method to split a sequence alignment dataset based on the distribution of pairwise differences, as such these approaches to species delimitation were concordant in identifying two species of Kidderia. With morphological identification, the Subantarctic lineage is assigned as K. minuta while the Antarctic lineage correspond to K. subquadrata (Fig. 2i). However, the GYMC analysis showed three putative species, specifically in the Antarctic clade. The GYMC analysis may suggest a population genetic structure or a more complex lineage classification (Pons et al. 2006; Reid and Carstens 2012; Talavera et al. 2013; Tang et al. 2014). Furthermore, when a single specimen that differs from the others by one nucleotide substitution is found, GMYC tends to diverge in the presence of a low intraspecific divergence (Pentinsaari et al. 2017; Luo et al. 2018). The occurrence of multiple lineages in the Antarctic could be a consequence of a speciation process explained by repeated glacial–interglacial events that have left characteristic signatures of limited gene flow and the occurrence of multiple independent refuges, fragmented populations, and isolation, especially in brooders (Pearse et al. 2009; Chown et al. 2015; Riesgo et al. 2015; Hayanich and Mahon 2018; Lau et al. 2020).
A serious limitation to the utility of DNA barcoding as a practical resource for regulation and molecular diagnostics is human error and uncertainty in creating and curating reference libraries (Collins and Cruikshank 2013). The microbivalves from Antarctic are not the exception. In fact, to have more confidence in our results we decided to generate sequences from the sister group Gaimardia to deal with unambiguous species-level identification. Moreover, in a recent work about review the taxonomy of the Cyamioidea confirm the placement of Gaimardia and Kidderia in Gaimardiidae (Zelaya et al. 2020). We are confidence that the extensive sampling design, the use of three molecular markers, and the integrative morphology-based and molecular approach are the main strengths of our study. The results presented in our work will contribute to clarifying the systematics, taxonomy and biogeography of the genus along with its diversity across the SO.
Divergence time estimation
The divergence time estimation between Subantarctic and Antarctic Kidderia lineages is related to past geographic and oceanographic events. Relevant tectonic events initiated in Jurassic times, where the South American and Antarctic continents started moving since at least 150 Ma (Jokat et al. 2003; König and Jokat 2006; Eagles and Vaughan 2009). The period between Paleocene and Eocene has been correlated with vicariance hypothesis caused by continental drift (Pearse et al. 2009). Later, a significant cooling event occurred at 37 Ma, well known as the Antarctic cooling, a climatic process started at the Paleocene-Eocene boundary (55 Ma) (Crame 1999; Thatje 2012; Hayanich and Mahon 2018). In the Eocene, the breakup of continents was complete with the opening of the Drake Passage (42 Ma) (Crame 1999; Eagles et al. 2006; Scher and Martin 2006; Clarke and Crame 2010; Hayanich and Mahon 2018), which led to the deepening of the sTasman Sea (34–33 Ma) (Stickley et al. 2004) and the establishment of the Antarctic Circumpolar Current and the Antarctic Polar Front around Oligocene/Eocene boundary (34 Ma) (Clarke and Crame 1989, 1992; Orsi et al. 1995; Pudsey and Howe 1998; Rintoul et al. 2001; Clarke and Johnston 2003; Stickley et al. 2004; Pfuhl and McCave 2005).
The role of the ACC has been very important in understanding the richness of species in the SO. Its influence on the existence and formation of several species with a varied time of divergence is widely accepted (Pearse et al. 2009). The ACC has been flowing through and around the Antarctic for more than 30 million years with occasional dispersal events, greatly impacting incubator species and establishing new isolated populations that could drive a speciation process before secondary contact of isolated populations, however, it is also known that the ACC currently maintains the link between spatially isolated populations in different taxa inhabiting the SO (Allcock and Strugnell 2012). While the ACC acted as a barrier between Subantarctic and Antarctic populations in species such as Parborlasia corrugatus (Thornhill et al. 2008) and species of the genus Ophionotus) (Hunter and Halanych 2010; Janosik et al. 2011) other species remain genetically connected across the SO, such as Astronoma agassizi (Hunter and Halanych 2008) and Siphonaria species (González-Wevar et al. 2018). In other phylogeographic studies in non-buoyant macroalgae species, evidence of recent colonization explained by exceptional events of rafting and current reproduction and recruitment occurring on a very small scale (Guillemin et al. 2020) has been reported. In addition, recent studies have reported a relaxation of the ACC which has been observed through kelp rafting (Durvillaea antarctica and Macrocystis pyrifera), suggesting that the ACC could act as a dispersion agent for marine organisms, specifically, for species that at present are non-native in the Antarctic (Avila et al. 2020; Macaya et al. 2020).
Many successful colonizations appear to have preceded Pliocene intensification of the ACC (Chown et al. 2015). In bivalves, there are some cases reported in the literature. For example, Linse et al. (2007) studied different populations of the brooding bivalve Lissarca notorcadensis and found a divergence between Subantarctic and Antarctic groups. This study presented one of the first pieces of evidence of the action of physical barriers that limit the gene flow between these provinces, particularly in these types of marine organisms. In another study, Jackson et al. (2015) estimated the origins of Philobryidae species around 66 Ma for the. A recent report about the divergence time estimation in the bivalve species Aequiyoldia eightsii by González-Wevar et al. (2019) suggests that the populations from Kerguelen Island separated from the South American and Antarctic lineages during the middle Miocene (16.5 Ma), while the separation between Antarctic and South American A. eightsii populations occurred at the end of the Miocene (8.5 Ma). In other examples, Page and Linse (2002) concluded a more recent speciation in Limatula species (divergence time of 8.03–5.79 Ma) and questioned the vicariance hypothesis. These examples show the relevance to incorporate and evaluate more species with contrasting development modes. In recent years, molecular tools have revealed that each species from the SO appears to have a particular life history. In this way, obtaining a general biogeographic pattern of marine organisms that inhabit the SO is more complex than previously thought (Eastman and McCune 2000; Clarke et al. 2007; Hunter et al. 2008; Leese et al. 2010; Macaya and Zuccarello 2010; Hoffman et al. 2012; Hayanich and Mahon 2018). Our evidence revealed that the divergence of Kidderia species is related to the complete separation of the Antarctic from the other continents and the establishment of the ACC, in the timeframe that supports the vicariance hypothesis, around late Eocene.
Conclusion
Here, we used classical and molecular approaches to identify two Kidderia species. This study allowed us to contribute new genetic records to advance the understanding of microbivalve evolution. With the verification of their presence in these environments, it was possible to investigate their evolutionary history, and therefore, provide useful information to clarify the evolutionary patterns of diversity on a broad scale (Avise 2009). We found the species K. subquadrata in the Antarctic Peninsula region and K. minuta in Subantarctic islands. Greater sampling effort is necessary to verify the presence of other species described in Subantarctic environments such as K. bicolor, K. pusilla and K. bisulcata and other diversity of the genus described in New Zealand.
References
Aghmich A, Taboada S, Toll L, Ballesteros M (2016) First assessment of the rocky intertidal communities of Fildes Bay, King George Island (South Shetland Islands, Antarctica). Polar Biol 39:189–198. https://doi.org/10.1007/s00300-015-1814-9
Allcock AL, Strugnell JM (2012) Southern Ocean diversity: new paradigms from molecular ecology. Trends Ecol Evol 27:520–528. https://doi.org/10.1016/j.tree.2012.05.009
Anne C (2006) Choosing the right molecular genetic markers for studying biodiversity: from molecular evolution to practical aspects. Genetica 127:101–120. https://doi.org/10.1007/s10709-005-2485-1
Aronson RB, Blake DB (2001) Global climate change and the origin of modern benthic communities in Antarctica. Am Zool 41:27–39. https://doi.org/10.1093/icb/41.1.27
Avila C, Angulo-Preckler C, Martín-Martín RP, Figuerola B, Griffiths HJ, Waller CL (2020) Invasive marine species discovered on non–native kelp rafts in the warmest Antarctic island. Sci Rep 10:1–9. https://doi.org/10.1038/s41598-020-58561-y
Avise JC (2009) Phylogeography: retrospect and prospect. J Biogeogr 36:3–15. https://doi.org/10.1111/j.1365-2699.2008.02032.x
Barnes DK, Linse K, Waller C, Morely S, Enderlein P, Fraser KP, Brown M (2006) Shallow benthic fauna communities of South Georgia Island. Polar Biol 29:223–228. https://doi.org/10.1007/s00300-005-0042-0
Bernard ER (1983) Catalogue of the living Bivalvia of the Eastern Pacific Ocean: bering strait to cape horn. Sci Can Spec Publ Fish Aquatic 61:102
Beu AG (2009) Before the ice: biogeography of Antarctic Paleogene molluscan faunas. Palaeogeogr Palaeoclimatol Palaeoecol 284:191–226. https://doi.org/10.1016/j.palaeo.2009.09.025
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537. https://doi.org/10.1371/journal.pcbi.1003537
Cheng HL, De-Quan XIA, Ting-Ting WU, Xue-Ping MENG, Hong-Ju JI, Zhi-Guo DONG (2006) Study on sequences of ribosomal DNA internal transcribed spacers of clams belonging to the Veneridae family (Mollusca: Bivalvia). Acta Genet Sin 33:702–710. https://doi.org/10.1016/S0379-4172(06)60102-9
Chown SL, Clarke A, Fraser CI, Cary SC, Moon KL, McGeoch MA (2015) The changing form of Antarctic biodiversity. Nature 522:431–438. https://doi.org/10.1038/nature14505
Clarke A (1996) The distribution of Antarctic marine benthic communities. Antarct Res Ser 70:219–230
Clarke A, Crame JA (1989) The origin of the Southern Ocean marine fauna. Geol Soc Spec Publ 47:253–268. https://doi.org/10.1144/GSL.SP.1989.047.01.19
Clarke A, Crame JA (1992) The Southern Ocean benthic fauna and climate change: a historical perspective. Philos T Roy Soc B 338:299–309. https://doi.org/10.1098/rstb.1992.0150
Clarke A, Crame JA (2010) Evolutionary dynamics at high latitudes: speciation and extinction in polar marine faunas. Philos T Roy Soc B 365:3655–3666. https://doi.org/10.1098/rstb.2010.0270
Clarke A, Johnston NM (2003) Antarctic marine benthic diversity. Oceanogr Mar Biol 41:47–114
Clarke A, Barnes DK, Hodgson DA (2005) How isolated is Antarctic? Trends Ecol Evol 20:1–3. https://doi.org/10.1016/j.tree.2004.10.004
Clarke A, Griffiths HJ, Linse K, Barnes DK, Crame JA (2007) How well do we know the Antarctic marine fauna? A preliminary study of macroecological and biogeographical patterns in Southern Ocean gastropod and bivalve molluscs. Divers Distrib 13:620–632. https://doi.org/10.1111/j.1472-4642.2007.00380.x
Collins RA, Cruickshank RH (2013) The seven deadly sins of DNA barcoding. Mol Ecol Resour 13:969–975. https://doi.org/10.1111/1755-0998.12046
Crame JA (1999) An evolutionary perspective on marine faunal connections between southernmost South America and Antarctica. Sci Mar 63:1–14
Dall W (1876) Mollusca from South Georgia. Sci Bull Brooklyn Inst 2:67–70
Dalziel IWD, Lawver LA, Pearce JA, Barker PF, Hastie AR, Barfod DN, Davis MB (2013) A potential barrier to deep Antarctic circumpolar flow until the late miocene? Geology 41:947–950. https://doi.org/10.1130/G34352.1
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. https://doi.org/10.1038/nmeth.2109
Dell RK (1964) Antarctic and sub-Antarctic Mollusca: amphineura, scaphopoda and bivalvia. Discov Rep 33:99–250
Díaz A, Féral JP, David B, Saucède T, Poulin E (2011) Evolutionary pathways among shallow and deep-sea echinoids of the genus Sterechinus in the Southern Ocean. Deep Sea Res 58:205–211. https://doi.org/10.1016/j.dsr2.2010.10.012
Dueñas LF, Tracey DM, Crawford AJ, Wilke T, Alderslade P, Sánchez JA (2016) The Antarctic circumpolar current as a diversification trigger for deep-sea octocorals. BMC Evol Biol 16:1–17. https://doi.org/10.1186/s12862-015-0574-z
Eagles G, Vaughan APM (2009) Gondwana breakup and plate kinematics: business as usual. Geophys Res Lett 36:L10302. https://doi.org/10.1029/2009GL037552
Eagles G, Livermore R, Morris P (2006) Small basins in the Scotia Sea: the Eocene Drake passage gateway. Earth Planet Sci Lett 242:343–353. https://doi.org/10.1016/j.epsl.2005.11.060
Eastman JT, McCune AR (2000) Fishes on the Antarctic continental shelf: evolution of a marine species flock? J Fish Biol 57:84–102. https://doi.org/10.1111/j.1095-8649.2000.tb02246.x
Engl W (2012) Shells of Antarctica. ConchBooks, Hackenheim, p 402
Engl W, Aldea C, Kohlberg G, Schories D, Schroedl M, Schories D, Kohlberg G (eds) (2016) Marine wildlife, King George Island, Antarctica. Dirk Schories Publications, p 156
Fraser CI, Nikula R, Spencer HG, Waters JM (2009) Kelp genes reveal effects of subantarctic sea ice during the last glacial maximum. Proc Natl Acad Sci 106:3249–3253. https://doi.org/10.1073/pnas.0810635106
Fujisawa T, Barraclough TG (2013) Delimiting species using single-locus data and the Generalized mixed yule coalescent approach: a revised method and evaluation on simulated data sets. Syst Biol 62:707–724. https://doi.org/10.1093/sysbio/syt033
Gazdzicki A, Pugaczewska H (1984) Biota of the “pecten conglomerate” (polonez cove formation, pliocene) of King George Island (South Shetland Islands, Antarctica). Stud Geol Polon 79:59–120
Geiger DL, Thacker CE (2008) Micromolluscs in molecular systematics: experiences and best practices. Zoosymposia 1:39–45
Ghiglione C, Alvaro MC, Piazza P, Bowden D, Griffiths HJ, Carota C, Schiaparelli S (2017) Mollusc species richness and abundance from shelf to abyssal depths in the Ross Sea (Antarctica): the importance of fine-mesh-towed gears and implications for future sampling. Polar Biol 40:1989–2000. https://doi.org/10.1007/s00300-017-2117-0
González-Wevar CA, Díaz A, Gerard K, Cañete JI, Poulin E (2012) Divergence time estimations and contrasting patterns of genetic diversity between Antarctic and southern South America benthic invertebrates. Rev Chil Hist Nat 85:445–456
González-Wevar CA, Segovia NI, Rosenfeld S, Ojeda J, Hüne M, Naretto J, Poulin E (2018) Unexpected absence of island endemics: long-distance dispersal in higher latitude sub-Antarctic Siphonaria (Gastropoda: Euthyneura) species. J Biogeogr 45:874–884. https://doi.org/10.1111/jbi.13174
González-Wevar CA, Gérard K, Rosenfeld S, Saucède T, Naretto J, Díaz A, Poulin E (2019) Cryptic speciation in Southern Ocean Aequiyoldia eightsii (Jay, 1839): mio-pliocene trans-drake passage separation and diversification. Progr Oceanogr 174:44–54. https://doi.org/10.1016/j.pocean.2018.09.004
Gould AA (1850) Descriptions of new species of shells from the United States exploring expedition. Proc Boston Soc Nat Hist 7:277
Griffiths HJ, Barnes DK, Linse K (2009) Towards a generalized biogeography of the Southern Ocean benthos. J Biogeogr 36:162–177. https://doi.org/10.1111/j.1365-2699.2008.01979.x
Griffiths HJ, Meijers AJ, Bracegirdle TJ (2017) More losers than winners in a century of future Southern Ocean seafloor warming. Nat Clim Change 7:749–754. https://doi.org/10.1038/nclimate3377
Guillemin ML, González-Wevar C, Cárdenas L, Dubrasquet H, Garrido I, Montecinos A, Robles KF (2020) Comparative phylogeography of Antarctic seaweeds: genetic consequences of historical climatic variations. In: Gomez I, Zelaya DG (eds) Antarctic Seaweeds. Springer, Cham, pp 103–127
Güller M, Zelaya DG (2015) Bivalves from the Chilean fjords region: knowns and unknowns. Am Malacol Bull 33:233–245. https://doi.org/10.4003/006.033.0218
Halanych KM, Mahon AR (2018) Challenging dogma concerning biogeographic patterns of Antarctica and the Southern Ocean. Annu Rev Ecol Evol Syst 49:355–378. https://doi.org/10.1146/annurev-ecolsys-121415-032139
Hawkins SJ, Evans AJ, Dale AC, Firth LB, Smith IP (2018) Implications of long-term climate change for biogeography and ecological processes in the Southern Ocean. Oceanogr Mar Biol 56:1–72
Hedley C (1916) Mollusca. Australian Antarctic expedition. Series C zoology and botany IV. Rogers, Adelaide, pp 1–80
Held C (2000) Phylogeny and biogeography of serolid isopods (Crustacea, Isopoda, Serolidae) and the use of ribosomal expansion segments in molecular systematics. Mol Phylogenet Evol 15:165–178. https://doi.org/10.1006/mpev.1999.0739
Hoffman JI, Clarke A, Clark MS, Fretwell P, Peck LS (2012) Unexpected fine-scale population structure in a broadcast-spawning Antarctic marine mollusc. PLoS ONE 7:e32415. https://doi.org/10.1371/journal.pone.0032415
Hüne M, González-Wevar C, Poulin E, Mansilla A, Fernández DA, Barrera-Oro E (2015) Low level of genetic divergence between Harpagifer fish species (Perciformes: Notothenioidei) suggests a quaternary colonization of patagonia from the Antarctic Peninsula. Polar Biol 38:607–617. https://doi.org/10.1007/s00300-014-1623-6
Hunter RL, Halanych KM (2008) Evaluating connectivity in the brooding brittle star Astrotoma agassizii across the Drake passage in the Southern Ocean. J Hered 99:137–148. https://doi.org/10.1093/jhered/esm119
Hunter RL, Halanych KM (2010) Phylogeography of the Antarctic planktotrophic brittle star Ophionotus victoriae reveals genetic structure inconsistent with early life history. Mar Biol 157:1693–1704. https://doi.org/10.1007/s00227-010-1443-3
Jackson JA, Linse K, Whittle R, Griffiths HJ (2015) The evolutionary origins of the Southern Ocean philobryid bivalves: hidden biodiversity, ancient persistence. PLoS ONE 10:e0121198. https://doi.org/10.1371/journal.pone.0121198
Janosik AM, Mahon AR, Halanych KM (2011) Evolutionary history of Southern Ocean Odontaster sea star species (Odontasteridae; Asteroidea). Polar Biol 34:575–586. https://doi.org/10.1007/s00300-010-0916-7
Jokat W, Boebel T, König M, Meyer U (2003) Timing and geometry of early gondwana breakup. J Geophys Res. https://doi.org/10.1029/2002JB001802
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Thierer T (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
König M, Jokat W (2006) The mesozoic breakup of the weddell sea. J Geophys Res. https://doi.org/10.1029/2005JB004035
Lau SC, Wilson NG, Silva CN, Strugnell JM (2020) Detecting glacial refugia in the Southern Ocean. Ecography 43:1639–1656. https://doi.org/10.1111/ecog.04951
Leese F, Agrawal S, Held C (2010) Long-distance island hopping without dispersal stages: transportation across major zoogeographic barriers in a Southern Ocean isopod. Naturwissenschaften 97:583–594. https://doi.org/10.1007/s00114-010-0674-y
Li WLS, Drummond AJ (2011) Model averaging and Bayes factor calculation of relaxed molecular clocks in Bayesian phylogenetics. Mol Biol Evol 29:751–761. https://doi.org/10.1093/molbev/msr232
Linse K, Griffiths HJ, Barnes DK, Clarke A (2006) Biodiversity and biogeography of Antarctic and sub-Antarctic mollusca. Deep Sea Res 53:985–1008. https://doi.org/10.1016/j.dsr2.2006.05.003
Linse K, Cope T, Lörz AN, Sands C (2007) Is the Scotia Sea a centre of Antarctic marine diversification? Some evidence of cryptic speciation in the circum-Antarctic bivalve Lissarca notorcadensis (Arcoidea: Philobryidae). Polar Biol 30:1059–1068. https://doi.org/10.1007/s00300-007-0265-3
Littlewood DTJ, Curini-Galletti M, Herniou EA (2000) The interrelationships of Proseriata (Platyhelminthes: Seriata) tested with molecules and morphology. Mol Phylogenet Evol 16:449–466. https://doi.org/10.1006/mpev.2000.0802
Luo A, Ling C, Ho SY, Zhu CD (2018) Comparison of methods for molecular species delimitation across a range of speciation scenarios. Syst Biol 67:830–846. https://doi.org/10.1093/sysbio/syy011
Macaya EC, Zuccarello GC (2010) Genetic structure of the giant kelp Macrocystis pyrifera along the southeastern Pacific. Mar Ecol Prog Ser 420:103–112. https://doi.org/10.3354/meps08893
Macaya EC, Tala F, Hinojosa IA, Rothäusler E (2020) Detached seaweeds as important dispersal agents across the Southern Ocean. Antarctic Seaweeds. Springer, Cham, pp 59–81
Marko PB, Moran AL (2002) Correlated evolutionary divergence of egg size and a mitochondrial protein across the Isthmus of Panama. Evolution 56:1303–1309. https://doi.org/10.1111/j.0014-3820.2002.tb01442.x
Marko PB, Moran AL (2009) Out of sight, out of mind: high cryptic diversity obscures the identities and histories of geminate species in the marine bivalve subgenus Acar. J Biogeogr 36:1861–1880. https://doi.org/10.1111/j.1365-2699.2009.02114.x
Martens E (1885) Vorläutige Mittheilungen über die Mollusken-fauna von Süd Georgien S.B. Ges.naturf. Fr. Berlin, pp 89–94.
Martens E, Pfeffer G (1886) Die mollusken von süd-georgien nach der ausbeute der deutschen station 1882–83. Jahrbuch Der Hamburgischen Wissenschaftlichen Anstalten 3:65–135
Medlin L, Zingone A (2007) A taxonomic review of the genus Phaeocystis. Biogeochemistry 83:3–18. https://doi.org/10.1007/978-1-4020-6214-8_2
MolluscaBase (2018). Mysella subquadrata (Pelseneer, 1903). World Register of Marine Species. http://www.marinespecies.org/aphia.php?p=taxdetails&id=714457. Accessed 24 Aug 2018
Morris PJ, Rosenberg G (2005) Search interface and documentation for Malacolog, an online database of Western Atlantic marine Mollusks. http://www.malacolog.org [WWW database (version 4.1.1).
Near TJ, Dornburg A, Kuhn KL, Eastman JT, Pennington JN, Patarnello T, Jones CD (2012) Ancient climate change, antifreeze, and the evolutionary diversification of Antarctic fishes. Proc Natl Acad Sci 109:3434–3439. https://doi.org/10.1073/pnas.1115169109
Odhner N (1923) Paper from Dr. Th Mortensen`s Pacific Expedition 1914–16. Stephensen, K. Revideret Fortegnelse over Danmarks Arter af Amphipoda (1. Del). (Hyperiidea; Gammaridea: Lysianassidae). Videnskabelige Meddelelser fra Dansk Naturhistorisk Forening i Kjöbenhavn, 76, 5–20.
Orsi AH, Whitworth T, Nowlin WD (1995) On the meridional extent and fronts of the Antarctic circumpolar current. Deep Sea Res 42:641–673. https://doi.org/10.1016/0967-0637(95)00021-W
Osorio C, Patrick M (1984) Gaimardia bahamondei, spec. nov., from Central Chile (Mollusca: Bivalvia: Cyamiidae: Gaimardiinae). Veliger 26:311–315
Page TJ, Linse K (2002) More evidence of speciation and dispersal across the Antarctic polar front through molecular systematics of Southern Ocean Limatula (Bivalvia: Limidae). Polar Biol 25:818–826. https://doi.org/10.1007/s00300-002-0414-7
Patarnello T, Bargelloni L, Varotto V, Battaglia B (1996) Krill evolution and the Antarctic ocean currents: evidence of vicariant speciation as inferred by molecular data. Mar Biol 126:603–608. https://doi.org/10.1007/BF00351327
Pearse JS, Mooi R, Lockhart SJ, Brandt A (2009) Brooding and species diversity in the Southern Ocean: selection for brooders or speciation within brooding clades? In: Pearse JS, Mooi R, Lockhart SJ, Brandt A (eds) Smithsonian at the poles: contributions to international polar year science. Smithsonian Institution Scholarly Press, Washington, DC. https://doi.org/10.5479/si.097884601X.13
Pelseneer P (1903) Mollusques (Amphineures, Gastropodes et Lamellibranches) Expédition Antartique Belge: Résultats Voyage du S. Y. Belgica en 1897–1898–1899 7. pls. 1–9, pp 85.
Pentinsaari M, Vos R, Mutanen M (2017) Algorithmic single-locus species delimitation: effects of sampling effort, variation and nonmonophyly in four methods and 1870 species of beetles. Mol Ecol Resour 17:393–404. https://doi.org/10.1111/1755-0998.12557
Pfeffer G (1886) Mollusken, Krebse und Echinodermen von Cumberland-Sund nach der Ausbeute der deutschen Nordexpedition 1882 und 1883 Jahrbuch der Hamburgischen Wissenschaftlichen Anstalten 3 23–50, 1 pl.
Pfuhl HA, McCave IN (2005) Evidence for late oligocene establishment of the Antarctic circumpolar current. Earth Planet Sci Lett 235:715–728. https://doi.org/10.1016/j.epsl.2005.04.025
Philippi A (1845) Diagnosen einiger neuen Conchylien Archiv für Naturgeschichte 11 50–71.
Ponder WF (1971) Some New Zealand and subantarctic bivalves of the Cyamiacea and Leptonacea with descriptions of new taxa. Rec Dom Mus 1971:119–141
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Vogler AP (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609. https://doi.org/10.1080/10635150600852011
Poulin E, González-Wevar C, Díaz A, Gérard K, Hüne M (2014) Divergence between Antarctic and South American marine invertebrates: what molecular biology tells us about Scotia Arc geodynamics and the intensification of the Antarctic circumpolar current. Glob Planet Change 123:392–399. https://doi.org/10.1016/j.gloplacha.2014.07.017
Pudsey CJ, Howe JA (1998) Quaternary history of the Antarctic circumpolar current: evidence from the Scotia Sea. Mar Geol 148:83–112. https://doi.org/10.1016/S0025-3227(98)00014-0
Puillandre N, Modica MV, Zhang Y, Sirovich L, Boisselier MC, Cruaud C, Samadi S (2012) Large-scale species delimitation method for hyperdiverse groups. Mol Ecol 21:2671–2691. https://doi.org/10.1111/j.1365-294X.2012.05559.x
Rambaut, A. (2014). FigTree v. 1.4. 2: Tree drawing tool [computer program].
Rambaut A, Suchard MA, Xie D, Drummond AJ (2016). Tracer v1. 6 [computer program].
Reid NM, Carstens BC (2012) Phylogenetic estimation error can decrease the accuracy of species delimitation: a Bayesian implementation of the general mixed Yule-coalescent model. BMC Evol Biol 12:196. https://doi.org/10.1186/1471-2148-12-196
Riesgo A, Taboada S, Avila C (2015) Evolutionary patterns in Antarctic marine invertebrates: an update on molecular studies. Mar Genomics 23:1–13. https://doi.org/10.1016/j.margen.2015.07.005
Rintoul S, Hughes C, Olbers D (2001) The Antarctic circumpolar current system. In: Siedler G, Church J, Gould J (eds) Ocean circulation and climate. Academic Press, New York, pp 271–302. https://doi.org/10.1016/S0074-6142(01)80124-8
Rogers AD (2007) Evolution and biodiversity of Antarctic organisms: a molecular perspective. Philos Trans R Soc B 362:2191–2214. https://doi.org/10.1098/rstb.2006.1948
Scher HD, Martin EE (2006) Timing and climatic consequences of the opening of drake passage. Science 312:428–430
Shabica SV (1974) Reproductive biology of the brooding Antarctic lamellibranch Kidderia Subquadrata Pelseneer (1903). MSc Thesis. School of Oceanography, Oregon.
Simpson RD (1977) The reproduction of some littoral molluscs from Macquarie Island (Sub-Antarctic). Mar Biol 44:125–142. https://doi.org/10.1007/BF00386953
Soot-Ryen T (1951) Antarctic pelecypoda. Sci Results nor Antarct Exped 1927–29(32):1–46
Stickley CE, Brinkhuis H, Schellenberg SA, Sluijs A, Röhl U, Fuller M, Williams GL (2004) Timing and nature of the deepening of the Tasmanian gateway. Paleoceanography. https://doi.org/10.1029/2004PA001022
Stilwell JD, Zinsmeister WJ (1992) Molluscan systematics and biostratigraphy: lower tertiary la meseta formation, Seymour Island, Antarctic Peninsula, vol 55. American Geophysical Union, Washington, DC
Talavera G, Dincă V, Vila R (2013) Factors affecting species delimitations with the GMYC model: insights from a butterfly survey. Methods Ecol Evol 4:1101–1110. https://doi.org/10.1111/2041-210X.12107
Tang CQ, Humphreys AM, Fontaneto D, Barraclough TG (2014) Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol Evol 5:1086–1094. https://doi.org/10.1111/2041-210X.12246
Taylor JD, Glover EA, Harper EM, Crame JA, Ikebe C, Williams ST (2018) Left in the cold? Evolutionary origin of Laternula elliptica, a keystone bivalve species of Antarctic benthos. Biol J Linn Soc 123:360–376. https://doi.org/10.1093/biolinnean/blx144
Thatje S (2012) Effects of capability for dispersal on the evolution of diversity in Antarctic benthos. Integr Comp Biol 52:470–482. https://doi.org/10.1093/icb/ics105
Thornhill DJ, Mahon AR, Norenburg JL, Halanych KM (2008) Open-ocean barriers to dispersal: a test case with the Antarctic polar front and the ribbon worm Parborlasia corrugatus (Nemertea: Lineidae). Mol Ecol 17:5104–5117. https://doi.org/10.1111/j.1365-294X.2008.03970.x
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast-online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44:W232–W235. https://doi.org/10.1093/nar/gkw256
Wilke T, Schultheiß R, Albrecht C (2009) As time goes by: a simple fool’s guide to molecular clock approaches in invertebrates. Am Malacol Bull 27:25–45. https://doi.org/10.4003/006.027.0203
Williams ST, Reid DG, Littlewood DTJ (2003) A molecular phylogeny of the Littorininae (Gastropoda: Littorinidae): unequal evolutionary rates, morphological parallelism, and biogeography of the Southern Ocean. Mol Phylogenet Evol 28:60–86. https://doi.org/10.1016/S1055-7903(03)00038-1
Zelaya DG (2005) The bivalves the Scotia Arc islands species richness and faunistic affinities. Sci Mar 69:113–212. https://doi.org/10.3989/scimar.2005.69s2113
Zelaya DG (2015) Marine bivalves from the Argentine coast and continental shelf: species diversity and assessment of the historical knowledge. Am Malacol Bull 33:245–262. https://doi.org/10.4003/006.033.0204
Zelaya DG, Güller M, Ituarte C (2020) Filling a blank in bivalve taxonomy: an integrative analysis of Cyamioidea (Mollusca: Bivalvia). Zool J Linn Soc 190:558–591. https://doi.org/10.1093/zoolinnean/zlz144
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2928. https://doi.org/10.1093/bioinformatics/btt499
Zinsmeister WJ (1984) Late eocene bivalves (mollusca) from the la meseta formation, collected during the 1974–1975 joint Argentine American expedition to Seymour Island, Antarctic Peninsula. J Paleontol 58:1497–1527
Acknowledgements
This work was supported and funded by the National Commission of Scientific and Technological Investigation of Chile through the Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias (FONDAP) program Research Center: Dynamics of High Latitude Marine Ecosystems (Grant No. 15150003) and by the Instituto Antartico Chileno through the INACh RT 02-15 and INACh MG 06-17 grants. LC acknowledge to Fondecyt 1170591. SR acknowledge to PIA CONICYT ACT172065 and PIA CONICYT APOYO CCTE AFB170008 from the Institute of Ecology and Biodiversity (IEB). We also acknowledge the support of Dr. Elie Poulin and his group in sampling and the project IPEV program PROTEKER (No. 1044) of which they are a part and we would like to thank the Subantarctic Conservation Program of the Universidad de Magallanes for all their logistical support in the expedition to Diego Ramirez. Finally, we would like to thank Dr. Claudio Gonzalez-Wevar for his first review and comments on the manuscript. We are thankful to Tomas Saucede and two anonymous reviewers for their meticulous revision that helped to improve this manuscript. We thank the Editor in Chief, Dieter Piepenburg for their work searching reviewers during these COVID-19 times, as well as their final comments to complete the editorial process. Finally, we thank the English language revision service offered by https://traduccionescientificas.cl/.
Author information
Authors and Affiliations
Contributions
LC and DL designed the project and LC, DL and SR collected the samples. DL performed the experiments and the data analysis. SR identified the species and revised the manuscript. DL and LC wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest was reported by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Levicoy, D., Rosenfeld, S. & Cárdenas, L. Divergence time and species delimitation of microbivalves in the Southern Ocean: the case of Kidderia species. Polar Biol 44, 1365–1377 (2021). https://doi.org/10.1007/s00300-021-02885-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-021-02885-6