
Abstract
Necroptosis is a form of programmed necrosis that is mediated by various cytokines and pattern recognition receptors (PRRs). Cells dying by necroptosis show necrotic phenotypes, including swelling and membrane rupture, and release damage-associated molecular patterns (DAMPs), inflammatory cytokines, and chemokines, thereby mediating extreme inflammatory responses. Studies on gene knockout or necroptosis-specific inhibitor treatment in animal models have provided extensive evidence regarding the important roles of necroptosis in inflammatory diseases. The necroptosis signaling pathway is primarily modulated by activation of receptor-interacting protein kinase 3 (RIPK3), which phosphorylates mixed-lineage kinase domain-like protein (MLKL), mediating MLKL oligomerization. In the necroptosis process, these proteins are fine-tuned by posttranslational regulation via phosphorylation, ubiquitination, glycosylation, and protein–protein interactions. Herein, we review recent findings on the molecular regulatory mechanisms of necroptosis.
Similar content being viewed by others

Introduction
Cell death is a terminal physiological event and is intricately related to the maintenance of homeostasis in multicellular organisms. Cell death can be classified into two modes: regulated cell death (RCD) and accidental cell death (ACD). The representative RCD is apoptosis, which plays roles in many physiological processes, such as lymphocyte selection, embryonic development, and epithelium renewal, is modulated by signaling pathways and is identified by cell shrinkage, plasma membrane blebbing, nuclear condensation, and fragmentation of cellular compartments followed by membrane disruption1. Apoptosis is initiated by specific intrinsic or extrinsic triggers, including DNA damage, reactive oxygen species, loss of mitochondrial membrane potential, death ligands, and various cytokines, thereby executing cell death in a caspase-dependent manner1. In contrast, nonphysiological stimuli, such as physical, mechanical, and chemical stresses, prompt necrosis, which is representative of an ACD1. Cells dying by necrosis, which is not regulated by signaling pathways, exhibit swelling, membrane rupture, and secretion of cellular contents1. Apoptosis has been regarded as the sole type of RCD since its discovery more than 50 years ago; however, the cell death paradigm has been extended in the past two decades. Although evidence for caspase-independent programmed cell death has been known for a long time, the extensive underlying molecular mechanisms of nonapoptotic cell death have only emerged in the past decade2. Since the discovery of necrostatin-1, an inhibitor of receptor-interacting protein kinase 1 (RIPK1) kinase activity, many genetic and chemical inhibition studies have led to the identification of diverse forms of programmed necrosis, including necroptosis, ferroptosis, parthanatos, mitochondrial permeability transition-dependent necrosis, pyroptosis, pyronecrosis, and NETosis3,4,5. In contrast to the passive immune response of cells dying by apoptosis, cells dying by programmed necrosis actively release damage-associated molecular patterns (DAMPs), cytokines, and chemokines, leading to an inflammatory response5.
Necroptosis is the best-characterized programmed necrosis. Necroptosis is initiated by various cytokines or pattern recognition receptors (PRRs) and modulated in RIPK1 and RIPK3-dependent manner6. RIPK1, RIPK3, and their substrate, mixed-lineage kinase domain-like protein (MLKL), function as key proteins in executing necroptosis, forming a necrosome complex (also referred to as complex II c)6. Emerging evidence on the relevance of necroptosis in inflammatory diseases has expanded the roles of necroptosis from those studied in vitro to those studied under pathophysiological conditions7. Diverse pathogens, including cytomegalovirus, RNA viruses, and bacteria, are capable of triggering necroptosis by activating Toll-like receptors (TLRs) or Z-DNA-binding protein 1 (ZBP1)8,9. In addition to pathogenic infection, necroptosis plays a significant role in tissue injuries such as cardiac, brain, or kidney ischemia-reperfusion injury4,7,9. Furthermore, human inflammatory diseases, including atherosclerosis, inflammatory bowel disease, neuroinflammation, and autoimmune diseases, have been closely connected with necroptosis7. Since necroptosis plays significant physiological roles, more comprehensive analyses on the interactome effects on necroptosis are required for a thorough understanding of this process.
In the past decade, emerging biochemical and genetic studies, including knockout animal experiments, have illustrated that the necroptosis signaling pathway is fine-tuned by various posttranslational regulations, such as phosphorylation, ubiquitination, glycosylation, and protein–protein interactions. Here, we review the molecular mechanisms of the necroptosis signaling pathway and summarize the recent findings regarding necroptosis regulatory mechanisms.
Molecular mechanisms of necroptosis
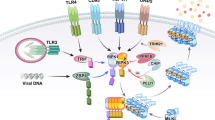
Most of the studies on the molecular mechanism of necroptosis involve the tumor necrosis factor (TNF) signaling pathway (Fig. 1). Generally, TNF induces an inflammatory response by activating proinflammatory genes through NF-κB signaling. Upon ligation of TNF to its receptor TNF receptor 1 (TNFR1), complex I, consisting of TNFR1-associated death domain protein (TRADD), TNF-receptor-associated factor 2 (TRAF2), RIPK1, cellular inhibitors of apoptosis (cIAP1 or cIAP2), and linear ubiquitin chain assembly complex (LUBAC), is recruited10. In complex I, cIAPs and LUBAC promote RIPK1 ubiquitination with K63-linked and linear ubiquitin chains that provide the platforms for recruiting downstream proteins such as TGF-activated kinase 1 (TAK1), TAK1-binding protein 2/3 (TAB2/3), and the IκB kinase (IKK) complex composed of IKKα, IKKβ, and NF-κB essential modulator (NEMO)10. The recruited downstream complex activates the NF-κB and mitogen-activated protein kinase (MAPK) pathways, leading to an increase in the expression of proinflammatory genes10. However, under certain conditions, such as destabilization of complex I or inhibition of the ubiquitination of RIPK1, TNF can trigger the formation of a cytosolic apoptotic complex (complex II a or II b), including Fas-associated protein with death domain (FADD) and caspase-8, and this complex executes apoptosis11.

TNF ligation induces complex I formation, which is composed of TRADD, TRAF2, cIAP1/2, RIPK1, TAK1, LUBAC, and the IKK complex, resulting in the activation of the NF-κB signaling pathway. When NF-κB target protein synthesis is inhibited by cycloheximide treatment, complex II a, consisting of TRADD, FADD, and caspase-8, is activated. Caspase-8, activated as part of complex II a, induces apoptosis through the cleavage of downstream molecules. The inhibition of RIPK1 ubiquitination or cytotoxicity-induced inhibition of phosphorylation, the early steps of TNF signaling, results in the induction of complex II b, which is composed of RIPK1, FADD, and caspase-8. Complex II b activation results in the induction of apoptosis through activated caspase-8. When caspase-8 is inhibited, RIPK1 and RIPK3 form necrosome complexes through homotypic interactions with RHIM, resulting in the activation of MLKL through a phosphorylation cascade. Phosphorylated MLKL undergoes oligomerization and migrates to the plasma membrane where it induces necroptosis by initiating membrane rupture or regulating ion flux. Death ligands, including FasL and TRAIL, initiate necroptosis by inducing necrosome complex formation. LPS, poly(I:C), double-stranded RNA, and viral RNA activate necroptosis by TRIF-mediated necrosome complex formation. Viral RNA or cellular endogenous RNA binding to ZBP1 results in RIPK1-independent necroptosis through the ZBP1–RIPK3 complex. Necroptosis factors are strictly regulated by ubiquitination, phosphorylation, glycosylation, and protein–protein interactions.
Disruption of the balance between caspase-8 and RIPK3 activities by caspase-8 inhibition or RIPK3 overexpression promotes conversion of complex II b to necrosome (Fig. 1)12,13,14. RIPK1 and RIPK3 are activated by autophosphorylation or cross-phosphorylation in the necrosome followed by the formation of a large amyloid-like structure15. The RIP homotypic interaction motif (RHIM)-dependent RIPK3 oligomer recruits MLKL to the necrosome, subsequently leading to the phosphorylation of MLKL on Thr357 and Ser358 located in the pseudokinase domain16. Phosphorylation of MLKL induces a conformational change in MLKL that exposes the 4-helical bundle (4HB) domain, which promotes MLKL oligomerization17,18,19. Oligomerized MLKL moves to the plasma membrane via the Golgi-microtubule-actin machinery, inducing membrane rupture by forming a pore cluster with tight junction proteins or regulating ion channel flux20,21,22,23,24,25,26.
In addition to TNF, other stimuli can initiate necroptosis (Fig. 1). Death receptors, including Fas (also referred to as CD95 or Apo-1), DR3 (also referred to as Apo-3), DR4 (also referred to as Apo-2 or TRAIL-R1), DR5 (also referred to as TRAIL-R2), and DR6, primarily recruit the membrane-associated death complex consisting of FADD and caspase-8, which is called the death-inducing signaling complex (DISC), upon ligation of the respective ligands27,28,29,30. In conditions where cIAP and caspase-8 are inhibited, death receptors promote the formation of necrosome, subsequently executing necroptosis in a RIPK3-dependent manner27,28,29,30. Furthermore, PRRs, such as TLR3 and TLR4, can trigger necroptosis by forming a necrosome through the Toll/IL-1 receptor (TIR) domain-containing adapter protein inducing interferon (IFN)-β (TRIF), which is another RHIM-containing protein31,32. In the presence of caspase inhibitors, polyinosine-polycytidylic acid (poly(I:C)) and lipopolysaccharide (LPS) stimulate TLR3 and TLR4 activation, respectively, and active TLRs promote the formation of TRIF-mediated necrosome composed of TRIF, RIPK3, and MLKL, thereby inducing necroptosis31,32. Another RHIM-containing protein, ZBP1 (also referred to as DAI), can also initiate necroptosis in response to viral infection33,34,35. Upon sensing viral RNA or cellular endogenous RNA, ZBP1 recruits RIPK3 through an RHIM–RHIM homotypic interaction and then prompts MLKL-dependent and RIPK1-independent necroptosis33,34,35. Recently, the possibility that ZBP1 can initiate necroptosis by the ligation of endogenous dsRNA derived from endogenous retroelements has been suggested36,37,38. RIPK1 deficiency increases ZBP1-mediated necroptosis and inflammation in mice, whereas crossing knock-in mice expressing Zα domain-deleted ZBP1 recovered the inflammatory phenotypes in RIPK1-deficient mice. ZBP1 constitutively binds to endogenous Z-nucleic acid through its Zα domain, suggesting that the Z-nucleic acid sensing of ZBP1 through the Zα domain might be a novel necroptosis initiation mechanism.
Posttranslational regulation of RIPK1 as a cell fate determinant
RIPK1 is a multidomain protein that contains a serine/threonine-protein kinase domain at the N-terminus, an RHIM domain at the intermediate region, and a death domain (DD) at the C-terminus11. Following TNF signaling activation, RIPK1 is recruited to complex I via homotypic interactions with other DD-containing proteins11. In complex I, RIPK1 is posttranslationally modified by various E3 ligases, deubiquitinases, and kinases, which function as cell fate determinants, regulating the prosurvival, inflammatory, and death signaling pathways (Fig. 2)10.

Overview of RIPK1, RIPK3, and MLKL posttranslational modifications, indicating the amino acid sites for ubiquitination, phosphorylation, and glycosylation with the enzymes have been reported to date.
cIAP1 and cIAP2 are RING finger E3 ligases and are recruited to complex I via interaction with TRAF26. cIAP1 and cIAP2 promote K63-linked ubiquitination on RIPK1 Lys377 and their own lysine sites, providing a platform for NF-κB activation (Fig. 2)39. The K63-linked ubiquitin chains of cIAPs stabilize complex I. However, when the function of cIAP1 and cIAP2 is inhibited by gene deletion or pharmacological treatment, such as second mitochondria-derived activator of caspase (SMAC) mimetics, the transition of complex I to complex II is promoted, subsequently initiating cell death signaling activation40. In addition to K63-linked polyubiquitination of RIPK1, it was recently suggested that cIAP1 is able to regulate the cytotoxic potential of RIPK1 by promoting K48-linked ubiquitination41. Cells expressing cIAP1 with a ubiquitin-associated (UBA) domain deletion mutation are more susceptible to TNF-mediated death with a low level of RIPK1 K48-linked ubiquitination and an increase in complex II level41. Knock-in mice expressing UBA domain-mutant cIAP1 are more sensitive to TNF-mediated systemic inflammatory response syndrome (SIRS), implying that UBA domain-mediated RIPK1 K48-linked ubiquitination is an important step in modulating the cytotoxic potential of the TNF-mediated response. LUBAC, consisting of heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1), HOIL-1-interacting protein (HOIP), and shank-associated RH domain-interacting protein (SHARPIN), is a linear ubiquitin E3 ligase complex that engages complex I by interacting with the K63-linked ubiquitin chain of cIAPs in response to TNF activation42,43,44,45,46. In complex I, LUBAC conjugates linear ubiquitin chains to RIPK1 and NEMO, which function as docking sites for the IKK complex and another kinase complex composed of NEMO, TANK, NAP1, TBK1, and IKKε, phosphorylating RIPK1 and then restraining the formation of complex II 42,43,44,45,46,47,48,49. In contrast to cIAPs and LUBAC, which provide downstream signaling platforms by increasing the ubiquitination of complex I components, MIB2 modulates only the cytotoxic ability of RIPK1 by promoting K11-, K48-, and K63-linked polyubiquitination50. Under TNF-activation conditions, MIB2 engages complex I by binding to the linker region of oligomerized RIPK1, which increases RIPK1 ubiquitination on Lys377 and Lys634, thus suppressing the cytotoxic ability of RIPK1 (Fig. 2)50. In addition to E3 ligases, counteracting enzymes, deubiquitinases, are regarded as key determinants of cell fate to die or survive. A20, CYLD, and OTU deubiquitinase with linear linkage specificity (OTULIN), which are ubiquitin hydrolases, engage complex I, removing the K63-linked and linear ubiquitination of RIPK1 and other complex I components (Fig. 2)51,52,53,54,55,56. Consistent with their function, deficiency of these enzymes in mice solidifies complex I and the proinflammatory and survival signaling mechanism fails to be terminated, resulting in severe inflammatory phenotypes and eventual death in the early postnatal stage.
In contrast to the cell death-inhibiting ubiquitination described above, ubiquitination of Lys115 of RIPK1 by Pellino 1 promotes necroptosis (Fig. 2)57. Pellino 1 conjugates K63-linked polyubiquitination in a RIPK1 kinase activity-dependent manner, which increases the interaction between RIPK1 and RIPK3, subsequently promoting necroptosis57. In addition to Pellino 1, c-Cbl can mediate pro-death K63-linked polyubiquitination on RIPK158. When cells are inhibited by a TAK1 inhibitor, complex I recruits leucine-rich repeat serine/threonine-protein kinase 2 (LRRK2), anaphase-promoting complex subunit 11 (APC11), and c-Cbl, increasing K63-linked polyubiquitination of RIPK1. The ubiquitination of RIPK1 induces the formation of a large and insoluble RIPK1 complex (iuRIPK1) and executes RIPK1-dependent apoptosis (RDA) or necroptosis58.
Although most RIPK1 ubiquitination is regulated in complex I, cytosolic RIPK1 is also regulated by ubiquitination59. Under normal conditions, cytosolic RIPK1 interacts with the carboxy terminus of HSC70-interacting protein (CHIP), which mediates RIPK1 ubiquitination on Lys571, Lys604, and Lys627 (Fig. 2). Ubiquitinated RIPK1 is destabilized in a ubiquitin-lysosome-dependent manner. CHIP deficiency extends the half-life of the RIPK1 protein and increases cell sensitivity to TNF-mediated death, thereby suggesting that CHIP controls steady-state levels of RIPK159.
Although diverse ubiquitin chains on RIPK1 determine its functional roles in TNF-mediated signaling, various kinases are also able to regulate the RIPK1 cytotoxic potential by modulating its kinase activity. IKKα/β, which are recruited to complex I by NEMO, phosphorylate RIPK1 on Ser25, inhibiting RIPK1 kinase-dependent cell death (Fig. 2)60,61. Because Ser25 of RIPK1 is located at a Gly-rich loop in the ATP-binding pocket, phosphorylation of RIPK1 at Ser25 seems to inactivate RIPK1 kinase activity by providing electrostatic repulsion at the ATP-binding pocket61. Deficiency of either IKKα or IKKβ was insufficient to inhibit RIPK1 phosphorylation or affect RIPK1 kinase-dependent cell death, thereby indicating that these two kinases show functional redundancy60. In addition to IKKα/β, TANK-binding kinase 1 (TBK1), also recruited to complex I, can modulate RIPK1 phosphorylation and inhibit its cytotoxic effect (Fig. 2)62,63. TBK1/IKKε are recruited to complex I through their interaction with NEMO, NAP1, and TANK, which phosphorylate RIPK1, subsequently suppressing RIPK1 kinase-dependent cell death62,63. In contrast to IKKα/β, TBK1/IKKε-mediated RIPK1 phosphorylation does not require TAK1 activation, implying that TBK1/IKKε phosphorylates and modulates RIPK1 in an NF-κB signaling-independent manner62,63. Recently, three groups demonstrated that phosphorylation of RIPK1 at Ser321 and Ser336 by MK2 controls RIPK1-dependent cell death (Fig. 2)64,65,66. Under TNF signaling activation, TAK1 phosphorylates p38 and then activates MK264,65,66. Active MK2 phosphorylates both cytosolic RIPK1 and complex I-recruited RIPK1, restraining the formation of complex II b by blocking its interaction with FADD64,65,66. Although TAK1 indirectly phosphorylates RIPK1 by activating IKKα/β and MK2, direct phosphorylation of RIPK1 by TAK1 has also been demonstrated67.
In contrast to the prosurvival effect of RIPK1 phosphorylation mediated by other kinases, the autophosphorylation of RIPK1 causes its enzymatic activation, thus activating its cytotoxic function. Mass spectrometry analysis revealed Ser14/15, Ser20, Ser161, and Ser166 of RIPK1 as autophosphorylation sites (Fig. 2)68. Among these, phosphorylation of Ser161 located in the activated T-loop is presumed to induce the conformational change of RIPK1 from the closed form to the open form, leading to enzymatic activation. Consistent with this notion, cells expressing the S161A mutant of RIPK1 showed a slight decrease in kinase activity and RIPK1-dependent necroptosis. Furthermore, reconstitution of the RIPK1 K45A/S161E or D138N/S161E mutant in RIPK1-knockout L929 cells was sufficient to induce RIPK1-dependent necroptosis and necrosome formation, suggesting that autophosphorylation of RIPK1 on Ser161 is an essential step for RIPK1-dependent cell death69.
Posttranslational regulation of RIPK3 in necroptosis modulation
The discovery of the function of RIPK3 in TNF-mediated necrotic cell death was a historic event in cell death studies. For a decade since its discovery, RIPK3 was regarded as merely another RIP-like kinase capable of regulating TNF-mediated apoptosis70. However, in 2009, three groups identified RIPK3 as an essential factor in TNF-mediated necroptosis12,13,14. To date, considerable evidence supporting the indispensable role of RIPK3 in necroptosis has emerged.
RIPK3 is an RHIM domain-containing kinase that forms a complex with other RHIM domain-containing proteins, such as RIPK1, ZBP1, and TRIF, under necroptosis signaling activation6. In a necrosome, RIPK3 is phosphorylated at multiple sites; among these, autophosphorylation of Ser227 of RIPK3 triggers the recruitment of MLKL to the necrosome by forming a hydrogen bond with Ser404 of MLKL, thereby phosphorylating MLKL and triggering necroptosis (Fig. 2)16,71. Recently, two groups have found that members of the casein kinase 1 (CK1) family, serine/threonine kinases, are recruited to the necrosome during necroptosis and phosphorylate RIPK3 on Ser227 (Fig. 2)72,73. Loss of CK1 blocked necroptosis with a decrease in RIPK3 phosphorylation at Ser227, suggesting that CK1 family proteins may play important roles in the promotion of necroptosis73. Along with kinases and their counterenzymes, phosphatases have been identified as regulators of necroptosis. Using mass spectrometry, Ppm1b was isolated as a binding partner of RIPK3 that dephosphorylates RIPK3 (Fig. 2)74. Consistent with this finding, Ppm1b-deficient mice showed increased lethality in TNF-induced SIRS with augmented RIPK3 phosphorylation, implying that RIPK3 dephosphorylation by Ppm1b functions as a negative regulator under physiological conditions.
Ubiquitination of RIPK3 is required to stabilize the necrosome complex. Lys5 of RIPK3 was identified as the ubiquitination site by mass spectrometry analysis (Fig. 2)75. Cells expressing the K5A mutant of RIPK3 showed neither RIPK3 ubiquitination nor necroptotic cell death. Although the E3 ligase of RIPK3 remains unknown, the ubiquitination of RIPK3 on Lys5 was negatively regulated by A20. Furthermore, the necroptotic phenotypes of A20-deficient T cells were restored by the combination knockout that included abrogation of RIPK3, indicating that the ubiquitination of RIPK3 in the necrosome might be an important checkpoint in executing necroptosis.
Cytosolic RIPK3 is also regulated by ubiquitination. CHIP, which is also known as a RIPK1 E3 ligase, interacts with RIPK3, subsequently ubiquitinating Lys55 and Lys363 of RIPK3 (Fig. 2)59. Ubiquitinated RIPK3 is broken down by lysosome-mediated degradation. CHIP-deficient newborn mice die in 4 weeks with disruption of the intestine, whereas CHIP-deficient mice with RIPK3 knocked out overcome postnatal lethality with normal intestinal physiology. In addition to CHIP, Pellino 1 can regulate RIPK3 steady-state levels by increasing ubiquitination76. Thr182 phosphorylation of RIPK3, which is mediated by necroptotic stimuli, is required for the interaction between Pellino 1 and RIPK3, which is followed by RIPK3 ubiquitination. In contrast to CHIP-mediated ubiquitination, Pellino 1-mediated RIPK3 ubiquitination leads to protein degradation in a proteasome-dependent manner. Parkin was also identified as a RIPK3 E3 ligase77. During the necroptosis process, AMP-activated protein kinase (AMPK) is activated by RIPKs, subsequently phosphorylating Ser9 of Parkin. Phosphorylated Parkin causes K33-linked polyubiquitination on Lys197, Lys302, and Lys364 of RIPK3, thereby inhibiting necrosome formation (Fig. 2). Loss of Parkin in mice led to an increased incidence of colitis-associated cancer with elevated inflammation induced by DSS, and treatment with GSK′872, a RIPK3 inhibitor, reversed the effect of Parkin deficiency.
Recently, the O-GlcNAcylation of RIPK3 on Thr467 was identified as a posttranslational modification (PTM) inhibiting the homotypic interaction of the RHIM domain (Fig. 2)78. Under LPS treatment conditions, the interaction between O-GlcNAc transferase (OGT) and RIPK3 increased in a time-dependent manner with an increase in the O-GlcNAcylation of RIPK3, whereas the interaction between the two proteins was diminished with a decrease in O-GlcNAcylation of RIPK3 when cells were treated with LPS and z-VAD-FMK. GlcNAc on Thr467 is presumed to block the homotypic interaction of the RHIM domain by inducing steric hindrance. Consistent with this supposition, the inflammatory response of macrophage-specific OGT-deficient mice in response to septic shock was recovered by RIPK3 codeletion.
In addition to PTMs, several proteins can inhibit necroptosis by interfering with the RIPK1–RIPK3 interaction. Aurora kinase A (AURKA) and GSK3β interact with RIPK1 and RIPK3, thereby negatively regulating necroptosis by interfering with necrosome formation79. The heat-shock protein 90 (HSP90)–CDC37 complex is also able to regulate RIPK3 by protein–protein interactions and is required for the interaction between RIPK1 and RIPK3 in response to necroptotic stimuli80. HSP90 inhibitor treatment alleviates TNFα-induced tissue damage in vivo. In addition to HSP90–CDC37, MYC, which is an oncogene, can negatively regulate necroptosis by blocking the interaction between RIPK1 and RIPK381. MYC directly interacts with RIPK3 under normal conditions and interferes with necrosome formation. Loss of MYC sensitizes cells to necroptosis with an increase in RIPK1 and RIPK3 interaction, and reconstitution of MYC in MYC-knockout cells restores cell sensitivity to necroptotic stimuli such that it is similar to that of normal cells. Interestingly, the expression of MYC mutants with deficient nuclear function in MYC-knockout cells can also reestablish the cytotoxic effect of MYC, implying that MYC negatively regulates necroptosis in a transcriptional function-independent manner. Consistent with this notion, xenograft analysis showed that MYC deficiency increased leukemia cells sensitivity to SMAC mimetic and pan-caspase inhibitor treatment, thus suggesting that the combination treatment of a MYC inhibitor with a SMAC mimetic and a pan-caspase inhibitor might be a good therapeutic strategy for leukemia patients.
Posttranslational regulatory mechanism of MLKL
In 2012, a mass spectrometry analysis revealed that MLKL and phosphoglycerate mutase family 5 (PGAM5) were downstream molecules of RIPK316,82. Although the necroptotic function of PGAM5 has been challenged by gene-deletion studies, MLKL is an unquestionable downstream executor of necroptosis. MLKL contains 4HB in its N-terminus and a pseudokinase domain at its C-terminus. Under RIPK3 activation, MLKL is recruited to the necrosome through its interaction with RIPK3 and is subsequently phosphorylated at the Thr357 and Ser358 in the pseudokinase domain (Fig. 2)16. This MLKL phosphorylation by RIPK3 leads to a conformational change in which the 4HB domain changes from the masked form to the exposed form, causing MLKL oligomerization17,19,20,21,22,23,24,71,83. Recently, TYRO3, AXL, and MER receptor kinases (TAMs) were identified as MLKL kinases84. TAMs phosphorylate Tyr376 of MLKL under necroptotic stress, which promotes MLKL oligomerization (Fig. 2). Notably, loss of TAMs attenuated necroptosis without changing the phosphorylation of RIPK1, RIPK3, or Ser358 in MLKL, implying that the phosphorylation of MLKL by TAMs might be the final modification step in the necroptosis process.
In addition to kinases, several proteins can regulate MLKL function through protein–protein interactions. TRAF2, an adapter protein in complex I, constitutively interacts with MLKL under normal conditions and suppresses the recruitment of MLKL to the necrosome in response to necroptotic stimuli85. While TRAF2-inducible knockout mice died within a week of induction because of an increase in necrosome formation, RIPK3 codeletion delayed the lethality of the TRAF2-inducible knockout mice. HSP90, a molecular chaperone, has also been identified as a regulator of MLKL86,87. The inhibition of HSP90 by specific inhibitors blocks necroptosis with a decrease in MLKL oligomerization and protein stability, suggesting that HSP90 may be required for the maintenance of MLKL oligomerization and protein stability. In addition HSP90, HSP70 has recently been found to be a positive regulator of MLKL88. The substrate-binding domain (SBD) of HSP70 is able to bind to the N-terminal domain (NTD) of MLKL, promoting MLKL polymerization and stabilization. The inhibition of HSP70 by treatment with NBC1, which can bind to the SBD of HSP70, prevents MLKL polymerization without inhibiting MLKL tetramerization, suggesting that HSP70 regulates necroptosis by modulating the MLKL polymerization step after MLKL tetramerization.
MLKL contains protein-binding regions such as the BH3-like motif and the coiled-coil domain (CCD) in its N-terminal region. Bcl-2, which is a BH3 motif-containing antiapoptotic protein, was identified as a negative regulator of MLKL89. Bcl-2 can bind to the BH3-like motif located at Lys165 to Lys177 of MLKL, interfering with RIPK3-mediated MLKL phosphorylation and MLKL oligomerization. Recently, the autophagic core protein Beclin 1 was identified as an inhibitory member of the necrosome90. Beclin 1 engages the necrosome during necroptosis by interacting with MLKL. The CCD is critical for the interaction of Beclin 1 with MLKL, inhibiting necroptosis by suppressing MLKL oligomerization. The necroptosis-inhibiting function of Beclin 1 proceeds in an autophagy-independent manner. MLKL phosphorylation by RIPK3 is required for the Beclin 1 and MLKL interaction, implying that Beclin 1 functions as the final barrier to the necroptosis process. In addition to in vitro analysis, tests on the inhibitory function of Beclin 1 in vivo were performed using a leukemia cell-based xenograft animal model. Beclin 1-defective leukemia cells were more sensitive to necroptotic stimuli in vitro and in vivo with increased MLKL oligomerization, suggesting that Beclin 1 might be a good target for the treatment of leukemia patients.
Concluding remarks
Currently, necroptosis is recognized as a representative RCD, as is apoptosis. In the past decade, the relevance of necroptosis to inflammatory diseases has been proven using animal models, including gene knockout mice and necroptosis-inhibiting treatment7. The inhibition of necroptosis by gene knockout or pharmacological treatment alleviated SIRS, ischemic-reperfusion injury, and other inflammatory responses7. Diverse necroptosis inhibitors have been discovered because of the increasing recognition of their importance under physiological conditions. Necrostatin-1 and its derivative were identified as the first necroptosis inhibitors that block RIPK1 kinase activity68,91. After the discovery of necrostatin-1, GlaxoSmithKline (GSK) found highly specific necroptosis inhibitors, namely, GSK′963 (a RIPK1 kinase inhibitor), GSK′843, and GSK′872 (RIPK3 kinase inhibitors)92,93. Moreover, necrosulfonamide has been reported as an MLKL inhibitor16. In addition to these newly generated chemicals, many approved medicines have been found to be necroptosis inhibitors94. Most of the approved medicines seem to target RIPK1 or RIPK3, indicating the importance of RIPKs in necroptosis. Currently, several RIPK1 inhibitors, GSK′2982772, DNL747, and DNL758, are in phase 1 or 2 clinical trials targeting inflammatory diseases such as psoriasis, ulcerative colitis, severe rheumatoid arthritis, Alzheimer’s disease, amyotrophic lateral sclerosis, and COVID-1995,96.
As diverse diseases continue to be connected with necroptosis, the significance of understanding necroptosis regulatory mechanisms is increasing (Table 1). Although various factors have been revealed as posttranslational regulatory molecules since the discovery of necroptosis, several posttranslational modifications, such as the ubiquitination of RIPK3 and MLKL in the necrosome, remain unclear. Furthermore, cross talk between other signaling molecules and necroptosis has recently been reported, suggesting that diverse cellular signaling pathways might be intricately entangled with the necroptosis signaling pathway. Thus, elucidation of these regulatory networks in necroptosis is required to obtain a comprehensive understanding of the pathophysiological roles of necroptosis in inflammatory diseases, which will ultimately result in the development of novel therapeutic strategies for the management of inflammatory diseases.
References
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Wallach, D., Kang, T. B. & Kovalenko, A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat. Rev. Immunol. 14, 51–59 (2014).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Choi, M. E., Price, D. R., Ryter, S. W. & Choi, A. M. K. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 4, e128834 (2019).
Kim, E. H., Wong, S. W. & Martinez, J. Programmed necrosis and disease:we interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 26, 25–40 (2019).
Seo, J. et al. The roles of ubiquitination in extrinsic cell death pathways and its implications for therapeutics. Biochem. Pharm. 162, 21–40 (2019).
Khoury, M. K., Gupta, K., Franco, S. R. & Liu, B. Necroptosis in the pathophysiology of disease. Am. J. Pathol. 190, 272–285 (2020).
Xia, X., Lei, L., Wang, S., Hu, J. & Zhang, G. Necroptosis and its role in infectious diseases. Apoptosis 25, 169–178 (2020).
Galluzzi, L., Kepp, O., Chan, F. K. & Kroemer, G. Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. 12, 103–130 (2017).
Annibaldi, A. & Meier, P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol. Med. 24, 49–65 (2018).
He, S. & Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 19, 912–922 (2018).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
Li, J. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Murphy, J. M. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013).
Davies, K. A. et al. The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ. 25, 1567–1580 (2018).
Petrie, E. J. et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 9, 2422 (2018).
Su, L. et al. A plug release mechanism for membrane permeation by MLKL. Structure 22, 1489–1500 (2014).
Hildebrand, J. M. et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl Acad. Sci. USA 111, 15072–15077 (2014).
Quarato, G. et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601 (2016).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014).
Chen, X. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121 (2014).
Xia, B. et al. MLKL forms cation channels. Cell Res. 26, 517–528 (2016).
Samson, A. L. et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 11, 3151 (2020).
Bittner, S., Knoll, G. & Ehrenschwender, M. Death receptor 3 mediates necroptotic cell death. Cell Mol. Life Sci. 74, 543–554 (2017).
Strilic, B. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 (2016).
Geserick, P. et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187, 1037–1054 (2009).
Feoktistova, M. et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
He, S., Liang, Y., Shao, F. & Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl Acad. Sci. USA 108, 20054–20059 (2011).
Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012).
Thapa, R. J. et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681 (2016).
Maelfait, J. et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017).
Lin, J. et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128 (2016).
Newton, K. et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133 (2016).
Jiao, H. et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395 (2020).
Bertrand, M. J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Varfolomeev, E. et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131, 669–681 (2007).
Annibaldi, A. et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 69, 566–580 e565 (2018).
Tokunaga, F. et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 11, 123–132 (2009).
Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
Ikeda, F. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 471, 637–641 (2011).
Tokunaga, F. et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636 (2011).
Peltzer, N. et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 9, 153–165 (2014).
Taraborrelli, L. et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 9, 3910 (2018).
Peltzer, N. et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–117 (2018).
Feltham, R. et al. Mind bomb regulates cell death during TNF signaling by suppressing RIPK1’s cytotoxic potential. Cell Rep. 23, 470–484 (2018).
Wertz, I. E. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699 (2004).
Enesa, K. et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J. Biol. Chem. 283, 7036–7045 (2008).
Draber, P. et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 13, 2258–2272 (2015).
Brummelkamp, T. R., Nijman, S. M., Dirac, A. M. & Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424, 797–801 (2003).
Kovalenko, A. et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424, 801–805 (2003).
Keusekotten, K. et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153, 1312–1326 (2013).
Wang, H. et al. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc. Natl Acad. Sci. USA 114, 11944–11949 (2017).
Amin, P. et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc. Natl Acad. Sci. USA 115, E5944–E5953 (2018).
Seo, J. et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 18, 291–302 (2016).
Dondelinger, Y. et al. NF-kappaB-independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729 (2019).
Xu, D. et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491 e1419 (2018).
Lafont, E. et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Jaco, I. et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710 e695 (2017).
Menon, M. B. et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 (2017).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 (2017).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Zhang, Y. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329 (2017).
Yu, P. W. et al. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr. Biol. 9, 539–542 (1999).
Xie, T. et al. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 5, 70–78 (2013).
Lee, S. Y. et al. Casein kinase-1gamma1 and 3 stimulate tumor necrosis factor-induced necroptosis through RIPK3. Cell Death Dis. 10, 923 (2019).
Hanna-Addams, S., Liu, S., Liu, H., Chen, S. & Wang, Z. CK1alpha, CK1delta, and CK1epsilon are necrosome components which phosphorylate serine 227 of human RIPK3 to activate necroptosis. Proc. Natl Acad. Sci. USA 117, 1962–1970 (2020).
Chen, W. et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat. Cell Biol. 17, 434–444 (2015).
Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627 (2015).
Choi, S. W. et al. PELI1 selectively targets kinase-active RIP3 for ubiquitylation-dependent proteasomal degradation. Mol. Cell 70, 920–935 e927 (2018).
Lee, S. B. et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat. Cell Biol. 21, 940–951 (2019).
Li, X. et al. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity 50, 576–590 e576 (2019).
Xie, Y. et al. Inhibition of aurora kinase A induces Necroptosis in Pancreatic Carcinoma. Gastroenterology 153, 1429–1443.e1425 (2017).
Li, D. et al. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc. Natl Acad. Sci. USA 112, 5017–5022 (2015).
Seong, D. et al. Identification of MYC as an antinecroptotic protein that stifles RIPK1-RIPK3 complex formation. Proc. Natl Acad. Sci. USA 117, 19982–19993 (2020).
Wang, Z., Jiang, H., Chen, S., Du, F. & Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012).
Tanzer, M. C. et al. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem. J. 471, 255–265 (2015).
Najafov, A. et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol. Cell 75, 457–468 e454 (2019).
Petersen, S. L. et al. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 22, 1846–1857 (2015).
Zhao, X. M. et al. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 7, e2089 (2016).
Jacobsen, A. V. et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 7, e2051 (2016).
Johnston, A. N. et al. Necroptosis-blocking compound NBC1 targets heat shock protein 70 to inhibit MLKL polymerization and necroptosis. Proc. Natl Acad. Sci. USA 117, 6521–6530 (2020).
Shi, C. S. & Kehrl, J. H. Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Disco. 5, 151 (2019).
Seo, J. et al. Beclin 1 functions as a negative modulator of MLKL oligomerisation by integrating into the necrosome complex. Cell Death Differ. 27, 3065–3081 (2020).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Berger, S. B. et al. Characterization of GSK’963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Disco. 1, 15009 (2015).
Mandal, P. et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014).
Molnar, T. et al. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis. 10, 860 (2019).
Harris, P. A. et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261 (2017).
Mifflin, L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Disco. 19, 553–571 (2020).
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea [NRF-2015R1A3A2066581 and 2020R1C1C1006833], Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program, and BK21 PLUS program. All figures were created with biorender.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seo, J., Nam, Y.W., Kim, S. et al. Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators. Exp Mol Med 53, 1007–1017 (2021). https://doi.org/10.1038/s12276-021-00634-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00634-7
This article is cited by
-
A matter of new life and cell death: programmed cell death in the mammalian ovary
Journal of Biomedical Science (2024)
-
HIST3H2A promotes the progression of prostate cancer through inhibiting cell necroptosis
BMC Cancer (2024)
-
LUBAC-mediated M1 Ub regulates necroptosis by segregating the cellular distribution of active MLKL
Cell Death & Disease (2024)
-
RIPK3 signaling and its role in regulated cell death and diseases
Cell Death Discovery (2024)
-
Entosis: the core mechanism and crosstalk with other cell death programs
Experimental & Molecular Medicine (2024)
