
Abstract
Brattforsite is an approved mineral (IMA2019-127), with ideal formula Mn19(AsO3)12Cl2. Associated minerals in the type specimen from the Brattfors mine, Nordmark (Värmland, Sweden) include jacobsite, alleghanyite, phlogopite, calcite and dolomite. Brattforsite, forming subhedral, mostly equant crystals up to 0.5 mm across, is orange to reddish-brown with a white streak, and translucent with a resinous to vitreous lustre. The fracture is uneven to subconchoidal, and no cleavage is observed. It is very weakly pleochroic in yellow, optically biaxial (–) with 2V = 44(5)° and has calculated mean refractive index of 1.981. Measured and calculated density values are 4.49(1) and 4.54(1) g·cm− 3, respectively. Chemical analyses yields (in wt%): MgO 0.62, CaO 1.26, MnO 48.66, FeO 0.13, As2O3 46.72, Cl 2.61, H2Ocalc 0.07, O ≡ Cl –0.59, sum 99.49, corresponding to the empirical formula (Mn17.67Ca0.58Mg0.40Fe0.05)∑18.70As12.17O35.90Cl1.90(OH)0.20, based on 38 (O + Cl + OH) atoms per formula unit. The five strongest Bragg peaks in the powder X-ray diffraction pattern are [d (Å), I (%), (hkl)]: 2.843,100, (\( \overline{4} \)44); 2.828, 99, (444); 1.731, 32, (880); 2.448, 28, (800); 1.739, 25, (088). Brattforsite is monoclinic and pseudotetragonal, space group I2/a, with unit-cell parameters a = 19.5806(7), b = 19.5763(7), c = 19.7595(7) Å, β = 90.393(3)°, V = 7573.9(5) Å3 and Z = 8. The crystal structure was solved and refined to an R1 index of 3.4 % for 7445 reflections [Fo > 4σ(Fo)]. Brattforsite has the same overall structural topology as magnussonite (i.e., the species can be considered as homeotypic), but with 12 independent tetrahedrally coordinated As sites and 21 Mn sites with varying (4–8) coordination. The Mn-centered polyhedra, bonded through edge- and face-sharing, give rise to a three-dimensional framework. The (AsO3)3− groups are bonded to this framework through corner- and edge-sharing. Spectroscopic measurements (optical absorption, Raman, FTIR) carried out support the interpretation of the compositional and structural data.
Similar content being viewed by others


Introduction
Arsenites form a subclass of rare, but significantly diverse, minerals with about 20 known species. They are most commonly characterized by the isolated (AsO3)3− anion in their crystal structures, and typically formed during the late-stage evolution of Mn or base-metal polymetallic ores (Majzlan et al. 2014). Here we describe a recently approved arsenite mineral from Brattforsgruvan, Filipstad municipality, Värmland, Sweden, named brattforsite (IMA2019-127). The mineral has been the subject to previous studies, and was long considered a variety of magnussonite, a presumed cubic arsenite mineral, first described by Gabrielson (1956) from the Långban deposit (also in Filipstad, Värmland), with the current IMA-formula Mn10As6O18(OH,Cl)2 (Pasero 2021). Moore (1970a) found that the mineral from Brattforsgruvan was pseudocubic, with a tetragonal unit cell, a = 19.58(2), c = 19.72(2) Å, space group I41/amd. Dunn and Ramik (1984) noted a high Cl content of 2.7-3.0 wt% for the same mineral, and obtained the provisional formula Mn10As6O18Cl(OH).
The mineral name is given for the type locality, the mine Brattforsgruvan. This name was previously used informally, on a label to the type specimen, and mentioned by Paul B. Moore in a letter from 1967 (F.E. Wickman file, Archives of the Royal Swedish Academy of Sciences, Stockholm). “Brattfors” is the name of a neighbouring parish, and of an old iron smelter. Brattforsite corresponds to UM1984-09 in the list of valid unnamed mineral species (Smith and Nickel 2007). The holotype specimen is deposited in the type mineral collection of the Department of Geosciences, Swedish Museum of Natural History, Box 50007, SE-10405 Stockholm, Sweden, under collection number GEO-NRM 19100303. The single crystal used for X-ray diffraction intensity data measurement (a part of the holotype) is kept in the mineralogical collection of the Natural History Museum of Pisa University, under catalogue number 19912.
Occurrence and paragenesis
Brattforsgruvan (59°49.99’ N, 14°7.41’ E, 230 m above shore-line) belongs to the Nordmark ore field (Nordmarks odalfält), which comprises about twenty old, small mines and prospects (Magnusson 1929). The principal ore mineral was magnetite, but a few of the old mines intersect enclaves with Mn oxides (dominantly hausmannite) and associated skarns, including Mn-bearing silicates and a number of exotic As-, Sb-, and B-oxyminerals (e.g., Sjögren 1884; Holtstam and Langhof 1995; Holtstam et al. 1998; Cooper et al. 2018). This particular mineralisation is related to the Långban-type of deposits in the Paleoproterozoic Bergslagen ore region (Moore 1970b). The ores and skarns of the Nordmark field are closely associated to a marble (dolomite-calcite) body adjacent to felsic metavolcanic rocks (Björck 1986). The Långban-type carbonate-hosted deposits primordially formed as Fe-Mn-Pb-As-Sb-rich precipitates from volcanogenic hydrothermal solutions in a shallow submarine environment at ca. 1.9 Ga (Boström et al. 1979; Holtstam and Mansfeld 2001). Subsequent regional metamorphism, deformation and intrusive events have affected the synvolcanic-synsedimentary protoliths until ca. 1.75 Ga (Stephens et al. 2009). Peak metamorphic conditions are estimated as T = 600–650°C and P = 0.3–0.4 GPa (Sandström and Holtstam 1999; Christy and Gatedal 2005).
The original occurrence of the present sample in the mine is unknown; it was purchased by the Swedish Museum of Natural History from Olof Backelin, a mining foreman and local mineral collector, in 1910. Backelin is noted as the source for two other type mineral specimens from this mine, katoptrite (Flink 1917) and manganhumite (Moore 1978). The mines of the Nordmark field closed in 1962 and are now inaccessible.
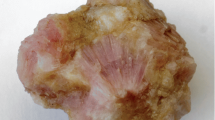
Arsenite minerals are typically confined to late-stage mineralization in fissures and druses in Långban-type deposits (Nysten et al. 1999); in the present case, it is probable that the new mineral has replaced a Mn-rich precursor mineral in the skarn mass, from alteration by a late- or post-metamorphic, As- and Cl-bearing fluid. Slightly reducing conditions in these rocks are indicated by the presence of manganosite and alabandite, observed in minor amounts in similar skarn matrices. Brattforsite occurs as crystals forming aggregates up to 8 mm across, in a granular matrix of (Fig. 1) jacobsite, calcite, dolomite, phlogopite and alleghanyite. Minor associated minerals identified by energy-dispersion microanalysis are rhodochrosite and johnbaumite. Subrounded grains of jacobsite commonly appear as inclusions in brattforsite. A Mn-As-(OH) mineral, tentatively identified as allactite, occurs filling fine fractures of brattforsite. Allactite is a late-stage vein mineral in the Mn mineralizations at Nordmark (Sjögren 1884).

Image of the brattforsite (arrow points to the largest aggregate) type specimen with jacobsite (black) and calcite (whitish), GEO-NRM #19100303. Inset image shows a detail on the specimen (red frame). Photos: Torbjörn Lorin
Physical and optical properties
Individual crystals of brattforsite are somewhat irregular in shape but essentially equant, and up to 0.5 mm in size. The macroscopic colour is orange to reddish brown, with a white streak. It has a vitreous to resinous lustre. Brattforsite is translucent and transparent in thin section. No fluorescence effects were detected in ultra-violet light. The hardness (Mohs) is estimated to 3–4. Neither an obvious cleavage nor parting is observed. Brattforsite is brittle and shows an uneven to subconchoidal fracture. Moore (1970a) reported 4.49 g·cm− 3 for the density; a calculated value of 4.54(1) g·cm− 3 is obtained for the ideal formula and unit-cell volume from single-crystal X-ray diffraction data. The mineral dissolves in 30 % hydrochloric acid at room temperature.
The refractive indices were not measured conventionally, because found higher than available reference liquids (> 1.8); the average calculated n is 1.981 from Gladstone-Dale constants (Mandarino 1981). The mineral is optically biaxial (–), with 2Vmeas = 44(5)°. The estimated birefringence is small, ~0.002. The dispersion is weak, with v > r. Brattforsite is very weakly pleochroic, in yellow hues. Polysynthetic twinning has been observed in a couple of grains. The angle of extinction between twin lamellae, up to 80 μm wide, is 27(1)°.
Mineral chemistry
The mineral was mounted in epoxy resin and polished prior to analysis. Electron probe microanalysis (EPMA) was performed with a Cameca Camebax SX-50 instrument, running at 20 kV, with the sample current 12 nA, spot size = 2 μm and a take-off angle of 40°. Corrections of the raw data were executed with Cameca’s PAP-routine, a modified ZAF procedure (Pouchou and Pichoir 1984). Used reference materials and the results from four spot analyses are given in Table 1. The concentration values for CuO and ZnO were below or close to the detection limit. Minor (OH)− in the crystal structure is indicated by infra-red spectra (see the following). The empirical formula for brattforsite, calculated on the basis of 38 (O + Cl + OH) atoms per formula unit (apfu), with As as a trivalent cation, is (Mn17.67Ca0.58Mg0.40Fe0.05)∑18.70As12.17O35.90Cl1.90(OH)0.20. The simplified formula may be written (Mn, Ca, Mg, Fe)19As12O36(Cl, OH)2 and the ideal formula is Mn19As12O36Cl2 or Mn19(AsO3)12Cl2, which corresponds to (in wt%) MnO 52.04, As2O3 45.84, Cl 2.74, O ≡ Cl -0.62, total 100.00.
Infra‐red, Raman and optical absorption spectroscopy
Infrared spectroscopy
Fourier-transform infrared (FTIR) spectra were measured on a double-side polished single crystal (thickness = 116 μm) using a Bruker Vertex spectrometer equipped with a Hyperion II microscope, a Globar source, a CaF2 beam-splitter, and an InSb detector. Data were acquired during 64 scans in the wavenumber range 2000–12,000 cm− 1 at a spectral resolution of 4 cm− 1. No absorption bands apart from very weak ones in the OH stretching region (Fig. 2) were observed. From comparison with the intensity of OH stretching bands in the infrared spectrum of a magnussonite single crystal (Fig. 2) from the holotype specimen GEO-NRM #g32215, the H2O-concentration in the brattforsite crystal is estimated to be ≤ 0.07 wt %, corresponding to ≤ 0.2 OH apfu. A relative low energy, with initial absorption at ca. 2700 cm− 1, and a broadness of the envelope of the bands marking OH stretching modes may be due to relatively strong hydrogen bonding in magnussonite and in brattforsite. Alternatively, these features are due to OH stretching bands superimposed on an absorption band caused by a low-energy d-d transition in a transition metal cation as e.g. Fe2+.

FTIR spectra of brattforsite (blue line) and holotype magnussonite (red line) in the OH-stretching region
Micro‐Raman spectroscopy
A Raman spectrum of brattforsite (Fig. 3) was collected from a randomly oriented, polished crystal fragment on a LabRAM HR 800 micro-spectrometer, using a 514 nm Ar-ion laser source at < 1 mW power, a Peltier-cooled (–70 °C) 1024 × 256 pixel CCD detector (Synapse), an Olympus M Plan N 100×/0.9 NA objective and laser spot of ca. 3 μm. A 600 grooves/cm grating was used, and the resolution is about 1 cm− 1. Spectral positions were corrected against the Raman band at 789 cm− 1 of a SiC-6H crystal measured on {0001}. Instrument control and data acquisition (range 50-4000 cm− 1, 10 s exposure time in 20 cycles) were made with the LabSpec 5 software. No laser-induced degradation of the sample was observed. A sample of magnussonite (type specimen) was measured for a comparison (Fig. 3).

Raman spectra of brattforsite and holotype magnussonite (GEO-NRM #g32215) recorded using a 514-nm laser
The spectra were found to be featureless in the range 4000–1000 cm− 1; prominent Raman peaks are identified for brattforsite at 786, 760 (shoulder), 708, 660, 510, 203 and 117 cm− 1 (Fig. 3). The most intense band at 786 cm− 1 is ascribed to symmetric stretching of the (AsO3)3− groups and the shoulder at 760 cm− 1 to antisymmetric stretching (cf. Frost and Bahfenne 2010). The corresponding bands in magnussonite are found at 811 and 794 cm− 1. The peaks between 700 and 500 cm− 1, including minor ones, for both minerals are resulting either from stretching vibrations of Mn2+-O(Cl,OH) bonds or from bending vibrations of (AsO3)3−. Specifically, the band at 510 cm− 1 is consistent with octahedrally coordinated Mn2+, whereas the one at 660 cm− 1 would be compatible with lower coordination numbers for this cation (Bernardini et al. 2021). Peaks in the low-frequency region from ca. 200 cm− 1 are probably related to lattice modes.
Optical absorption spectroscopy
Polarized optical absorption spectra (Fig. 4) were measured on two double-side polished oriented crystal sections (one XZ and one YZ section), both 70 μm thick, of brattforsite. The crystals were oriented by means of optical microscopy. The spectra were recorded in the range 10,000–32,000 cm− 1 with an AVASPEC-ULS2048 × 16 spectrometer attached via a 400-µm UV fibre cable to a Zeiss Axiotron UV-microscope. A 75 W Xenon arc lamp served as a light source and Zeiss Ultrafluar 10× lenses were used as objective and condenser. The size of the circular measure aperture was 64 μm in diameter. An UV-quality Glan-Thompson prism with a working range from 250 to 2700 nm (40,000 to 3704 cm− 1) was used as polarizer. The recorded spectra show a set of absorption bands caused by spin-forbidden electronic d-d transitions in Mn2+ superimposed on an UV absorption edge. The most prominent of these bands, at 23,650 cm− 1, marks the field independent 6A1(S) →4A14E(G) transition in Mn2+. The calculated molar absorption coefficient (ε) of the band is approximately 1 l·mole− 1·cm− 1, which is comparable to the ε-value recorded for the corresponding band in spectra of magnussonite (Hålenius and Lindqvist 1996). An additional, broad and strongly polarized band at ~ 13,700 cm− 1 is tentatively assigned to a spin-allowed d-d transition in Fe2+ in four-fold coordination or in a strongly axial distorted octahedral coordination. Based on the molar absorption coefficient (~ 165 l·mole− 1·cm− 1) of a corresponding absorption band at ~ 15,300 cm− 1 in spectra of magnussonite (Hålenius and Lindqvist 1996), the FeO concentration in brattforsite is calculated at 0.15 wt%, which compares well with a concentration of 0.13 wt% determined by EPMA (Table 1).

Polarized optical absorption spectra of brattforsite
X-ray crystallography
X-ray powder diffraction data were collected with a PANalytical X’Pert3 powder diffractometer equipped with an X’celerator silicon-strip detector and operated at 40 mA and 45 kV (CuKα-radiation, λ = 1.5406 Å). Bragg-peak positions (Table 2) were determined with the PANalytical HighScore Plus 4.6 software and corrected against an external Si standard (NBS 640b). The monoclinic (space group I2/a, #15) unit-cell parameters refined by a least-squares method from the powder data are: a = 19.592(2) Å, b = 19.586(2) Å, c = 19.770(2) Å, β = 90.413(7)º and V = 7586(1) Å3 for Z = 8.
Single-crystal X-ray intensity data were collected using a Bruker Smart Breeze diffractometer equipped with a Photon II CCD detector and graphite monochromatized MoKα radiation. The detector-to-crystal working distance was set at 50 mm. Data were collected using ω scan mode in 0.5° slices, with an exposure time of 20 s per frame. Correction for Lorentz and polarization factors, absorption, and background were applied using the package of software Apex3 (Bruker AXS Inc. 2016). Brattforsite is monoclinic, with unit-cell parameters a = 27.7223(9), b = 19.5763(7), c = 19.5806(7) Å, β = 134.5410(10)°, V = 7573.9(5) Å3. The statistical tests on the distribution of |E| values (|E2 – 1| = 1.006) indicated the occurrence of an inversion center. The examination of systematic absences suggested the space group symmetry C2/c. The crystal structure of brattforsite was solved in this space group through direct methods using Shelxs-97 and refined using Shelxl-2018 (Sheldrick 2015). Following the recommendation of Mighell (2003), the C-centered monoclinic cell was transformed in a conventional I-centered cell, through the matrix [0 0–1 | 0 1 0 | 1 0 1], obtaining the pseudotetragonal unit-cell parameters a = 19.5806(7), b = 19.5763(7), c = 19.7595(7) Å, β = 90.393(3)°, V = 7573.9(5) Å3, space group I2/a. These unit-cell parameters can be compared with those reported by Moore (1970a), a = 19.58(2), c = 19.72(2) Å, space group I41/amd. The following neutral scattering curves, taken from the International Tables for Crystallography (Wilson 1992), were used: Mn vs. Ca at Mn sites, As at As sites, O at O sites, and Cl vs. O at Cl sites. Several Mn sites were found fully occupied by Mn and their site occupancy factors (s.o.f.) were fixed to the full occupancy by Mn. The anisotropic structural model of brattforsite converged to R1 = 0.0343 for 7445 reflections with Fo > 4σ(Fo) and 640 refined parameters. A crystallographic information file (CIF) is available as electronic supplementary material. Details of data collection and refinement are given in Table 3. Fractional atom coordinates and displacement parameters are reported in Table 4, whereas Table 5 contains interatomic distances and bond-valence sums (BVS) for cation sites, and Table 6 the BVS sums for the anion sites. BVS were calculated using the bond parameters of Gagné and Hawthorne (2015) for As–O, Ca–O, and Mn–O bonds, and those of Brese and O’Keeffe (1991) for Mn–Cl and Ca–Cl bonds.
Crystal structure description
General features
The crystal structure of brattforsite (Fig. 5) shows the occurrence of thirty-three independent cation sites (twelve As sites and twenty-one Mn-dominant sites) and thirty-eight anion positions.

The crystal structure of brattforsite, as seen down a. Symbols: yellow polyhedra are Mn-centered sites. Circles: dark violet = As sites; green = Cl sites; red = O sites. As–O bonds are shown as red sticks. Unit-cell is shown by dashed lines. L0, L1, and L2 indicate the structural layers shown in Fig. 7
The crystal structure can be described as formed by polyhedral layers stacked along c. Within a unit-cell, eight layers occur; three symmetrically distinct layers, labelled L0, L1, and L2, alternate in the unit-cell according to the sequence shown in Fig. 5. Figure 6 shows each single layer as seen down c. The layer L0 is formed by rows of Mn-centered polyhedra running along [110] and [-110]. These rows show the sequence ···Mn(2)-Mn(6)-Mn(13)-Mn(4)-Mn(3)-Mn(8)-Mn(13)-Mn(9)-Mn(2)···, with Mn(13) occurring at the intersection between perpendicular rows. The intersection of the rows form a square cavity where a trimer composed of Mn(7)-Mn(20)-Mn(5) occurs, along with (AsO3) groups. Two As sites, i.e., As(1) and As(5), point in the + c direction, whereas As(4) and As(8) point in the –c direction. The layer L1 shows clusters formed by four independent Mn sites, i.e., Mn(10), Mn(11), Mn(15), and Mn(16). This cluster forms chains along a, obtained through corner-sharing between Mn(15) and Mn(16) belonging to consecutive tetramers, and through (AsO3) groups [As(2) and As(7)]. Along b, the connection between these chains is due to (AsO3) groups [As(9) and As(12)]. The layer L2 has two independent tetramers, formed by Mn(1), Mn(21), and two Mn(7) and by Mn(14), Mn(18), and two Mn(12), respectively. These clusters are bonded along a through As(10) and As(11), whereas along b the connection is due to the occurrence of As(3) and As(6).

The polyhedral layers occurring in the crystal structure of brattforsite: L0 (a), L1 (b), and L2 (c), as seen down c (a horizontal)
Notwithstanding such a description, the large number of connections between Mn-centered polyhedra, bonded through edge- and face-sharing, give rise to a three-dimensional framework. The (AsO3)3− groups are bonded to this three-dimensional framework through corner- and edge-sharing. They are located on the walls of the structural cavities, with their 4s2 lone-electron pairs pointing towards the center of the cavities.
Atom coordination
Arsenic displays the typical asymmetric trigonal pyramidal coordination, showing < As–O > distances ranging between 1.764 and 1.793 Å, in agreement with the < As–ϕ > value of 1.782 Å reported by Majzlan et al. (2014). The BVS at these sites range between 2.88 and 3.08 valence units (v.u.), in agreement with the occurrence of As3+.
Manganese, along with minor Ca, Mg, and Fe, shows coordination numbers ranging from four to eight. Ten sites show a variably distorted octahedral coordination; among them, MnO6, MnO5Cl and MnO4Cl2 polyhedra can be recognized. MnO6 octahedra have < Mn–O > distances ranging between 2.193 and 2.234 Å, with individual distances varying between 2.098 and 2.385 Å. Gagné and Hawthorne (2020) gave an average bond distance for six-fold coordinated Mn2+ of 2.20(8) Å, in agreement with the observed values. BVS at these sites are in the range 1.89–2.07 v.u., thus agreeing with the occurrence of divalent cations, mainly represented by Mn2+, with only a negligible substitution by Ca2+. Two sites, i.e., Mn(19) and Mn(20), are coordinated by five O2− anion and one Cl−. Whereas the former has a pure Mn occupancy, the latter is a mixed (Mn,Ca) position, agreeing with an average bond distance larger than that shown by Mn(19). Both sites have a BVS of 1.92 v.u. Finally, the Mn(14) site displays four short bonds with O2− (average < Mn–O > = 2.102 Å) and two longer Mn–Cl distances (at ca. 2.94 Å). A six-fold coordination is assumed for four other Mn sites. Two of them, i.e., Mn(2) and Mn(3), have a distorted trigonal prismatic coordination, with bond distances ranging between 2.044 and 2.386 Å, and < Mn–O > distances of 2.219 and 2.229 Å. Their BVS are 1.92 and 2.00 v.u. Other two six-fold coordinated sites, Mn(10) and Mn(12), have a very distorted trigonal prismatic coordination, with five shorter bonds (< Mn–O > distances of 2.149 and 2.190 Å, respectively), and a longer one, at 2.914 and 2.657 Å, respectively. Indeed, the coordination of Mn(10) may be described as a [5 + 1] coordination, forming a distorted square pyramid. The < Mn–O > distance observed at Mn(10) is fully consistent with the average distance reported by Gagné and Hawthorne (2020) for five-fold coordinated Mn2+, i.e., 2.14(5) Å. In addition, the range of observed bond distances for such a coordination, 2.024–2.267 Å (Gagné and Hawthorne 2020), agrees with the Mn–O distances observed for Mn(10). This is a bonding environment similar to that observed for Mn(7) and Mn(11); these sites can be described as square pyramids, with Mn–O distances ranging between 1.996 and 2.296 Å. For the Mn(11) site, the addition of a definitely longer bond at 3.098 Å increases the coordination number to six, allowing the description of this site as a very distorted trigonal prism; for the Mn(7), the sixth ligand is further away, at ~ 3.20 Å. The BVS at Mn(7), Mn(10), Mn(11), and Mn(12) are 2.02, 1.94, 1.92, and 1.87 v.u., respectively.
Mn(13) and Mn(15) show a seven-fold coordination. The former has six short bonds, ranging between 2.153 and 2.366 Å. A longer bond at 2.905 Å increases its coordination number to seven. Additionally, another O atom is located at 3.111 Å from Mn(13). Similarly, Mn(15) has six short bonds, varying between 2.187 and 2.437 Å, and a seventh one at 2.714 Å. Both these sites show a very minor Ca content, and their BVS are 1.81 and 1.82 v.u., respectively. These relatively low BVS values may indicate a higher Ca content than the refined one. Ideal BVS of 2.00 v.u. would be achieved assuming the site occupancies (Mn0.75Ca0.25) and (Mn0.92Ca0.08) at Mn(13) and Mn(15), respectively. Following Gagné and Hawthorne (2020), Mn(15) can be considered as a typical seven-fold coordinated site, because its bond distances are in the range 2.119–2.782 Å; in contrast, Mn(13), having the seventh ligand at 2.905 Å, may be more correctly considered as a six-fold coordinated site.
The largest Mn-centered polyhedron in the crystal structure of brattforsite is represented by the Mn(18) site. This position shows an eight-fold coordination, with < Mn–O > distance of 2.428 Å, comparable with the average distance of 2.420 Å reported by Moore and Araki (1979a) for the distorted cubic polyhedron surrounding the Mn(1) site of magnussonite. Mn(18) is a mixed (Mn,Ca) position, with BVS of 1.89 v.u.
Finally, there are two split cation positions, i.e., Mn(16) and Mn(21). The Mn(16) site is split into two sub-positions, Mn(16)a and Mg(16)b, having site occupancies Mn0.93 and Mg0.07, respectively. The former has a distorted square pyramidal coordination, with four O atoms forming the base and Cl occupying the pyramidal vertex. Assuming the full-occupancy of this position, the BVS is 1.91 v.u., agreeing with the occurrence of a divalent cation. Mg(16)b sub-site was refined using the scattering curve of Mg, owing to shorter bonds with ligands. It has a four-fold coordination with O atoms, at distances ranging between 1.94 and 2.27 Å. Two longer distances with Cl atoms (~ 3.28 and 3.42 Å) increase the coordination number to six, giving rise to a distorted octahedron. BVS at the Mg(16)b position is 1.84 v.u. Mn(21) is split into Mn(21)a and Mn(21)b. The coordination of Mn(21)a is similar to that observed for Mg(16)b, with four short bonds with O atoms, in the range 2.04–2.06 Å, and two long distances with Cl at 3.64 Å. Mn(21)b has a five-fold coordination, with four O atoms at distances in the range between 1.99 and 2.22 Å, and a Cl atom at 3.28 Å. BVS at these two sites are 1.91 and 1.84 v.u.
The oxygen atoms are usually four-fold coordinated, being bonded to three Mn sites and one As site. The O(4) and O(31) sites show three slightly different bonding configurations, owing to the split nature of the Mn(21) site. Three O sites have not a four-fold coordination: O(18) and O(23) are five-fold coordinated, forming bonds with four Mn sites (one Mn–O distance being larger than 2.90 Å) and one As site, whereas O(21) has a three-fold coordination with two Mn and one As sites. BVS at the O sites range between 1.86 and 2.06 v.u., confirming the occurrence of O2− at these sites. Chlorine and minor (OH) groups are hosted at two sites, Cl(1) and Cl(2). These two sites are bonded to two Mn atoms, achieving a relatively low BVS of 0.55 and 0.65 v.u., respectively. Such an underbonding is related to the long Mn–Cl distances. Moore and Araki (1979a) found similar long distance in magnussonite, stressing that the observed distance is 0.30 Å longer than that predicted on the basis of ionic radii, i.e., 2.51 Å.
The structural formula derived through the crystal structure refinement of brattforsite is (Mn18.24Ca0.69Mg0.07)Σ19.00(As3+O3)12 [Cl1.89(OH)0.11] (Z = 8). The total site scattering refined at the Mn sites is 470.6 electrons per formula unit (epfu), to be compared with a calculated site scattering of 466.8 epfu derived from the chemical analysis after the normalization of Σ(Mn, Ca, Mg, Fe) = 19 apfu. The actual partitioning of Mg and Ca is not determined. It is likely that they could be disordered over several mixed (Mn, Ca, Mg) sites, with Ca preferentially ordered at the larger Mn positions and Mg within the smaller Mn polyhedra. The minor amount of Fe was not located either, owing to the similar scattering factor to Mn; taking into account the results of Hålenius and Lindqvist (1996) for the related mineral magnussonite, as well as the result of optical absorption spectroscopic measurements performed on brattforsite, Fe may be hosted in the four-fold coordination or in a strongly axially distorted octahedral coordination.
Discussion
Brattforsite and its relationships with magnussonite
Brattforsite is related to the manganese arsenite mineral magnussonite. The definition of this latter mineral has yet not been fully clarified. Its crystal structure was solved by Moore and Araki (1979a) in the cubic space group Ia\( \overline{3} \)d, with unit-cell parameter a = 19.680(4) Å. The authors proposed the ideal formula Mn2 +18[As3+6Mn+O18]2Cl2 (Z = 8) or Mn20As12O32Cl2. This was questioned by Dunn and Ramik (1984), who highlighted that their analyses of magnussonite, as well as those given by Gabrielson (1956), showed (OH) > Cl. Similar results were obtained by Hålenius and Lindqvist (1996). Despite the apparent cubic character of magnussonite, some details suggest that further studies are needed to fully characterize this mineral. For example, crystals are optically anisotropic and biaxial (–) in sufficiently thick sections (~ 0.1 mm), with a very small 2V and birefringence. In addition, the stability of the Mn+ ion in Nature is questioned, and no features ascribed to this rare cationic species have observed in optical spectra of magnussonite (Hålenius and Lindqvist 1996). Finally, the role of minor elements (e.g., Cu) has to be clarified. Figure 7 compares the crystal structure of magnussonite with that of brattforsite. One feature common to both structures is an As-octahedron, with As–As distances of 3.64 Å in magnussonite (actually ranging between 3.56 and 3.71 Å) and 3.66 Å in brattforsite (ranging between 3.49 and 3.78 Å). This octahedron is occupied by Mn1+ (at 0, 0, 0) in magnussonite (according to Moore and Araki 1979a), with <Mn–As> distance of 2.65 Å. In brattforsite, this position is empty, and the shortest Mn–As distance was observed between As(12) and Mn(19), with a distance of 2.9482(10) Å. No residuals were found in the difference-Fourier maps at coordinates (0, 0, 0). Neglecting minor differences in the cation coordinations, this is the main difference between brattforsite and magnussonite, which can be considered as having a homeotypic relationship. The spectroscopic data strengthen the picture of a close structural relationship between these two minerals, with, e.g., only small shifts in Raman bands (ca. 25 cm− 1) mainly related to the variation in overall < As–O > bond distances, 1.778 Å for brattforsite and 1.760 Å for magnussonite.

Projection of the crystal structure of brattforsite (a) and magnussonite (b) down b (a horizontal). Circles represent As (violet), Cl (green), and O (red) sites. In magnussonite, the Mn(5) site is shown as yellow circles. As–O bonds are shown as red thick lines, whereas As–As contacts are shown by thick black lines. In the center of the Fig., the As6-octahedra occurring in brattforsite (above) and magnussonite (below) are shown. In this latter image, Mn–As contacts are shown as dotted grey lines
Consequently, based on structural details, the overall crystal symmetry, the chemical composition and the optical characteristics, brattforsite is distinct from magnussonite, and a bona fide Cl-analogue. At present there is no suggestion of the upper limit of Cl incorporation in magnussonite. Nysten (2003) identified a Cl-rich specimen (1.9 wt% Cl) from the Garpenberg Norra deposit, Dalecarlia, Sweden, with no indication of lower symmetry than cubic from single-crystal X-ray diffraction data.
Brattforsite in the domain of Mn arsenite minerals
Currently, ca. 70 mineral species have Mn and As as essential chemical constituents. However, the large majority of these minerals has As in its pentavalent state, forming (As5+ϕ4) oxyanions. Other phases have mixed (As5+ϕ4) and (As3+ϕ3) arrangements. Ten mineral species containing Mn and As3+ are currently known; brattforsite is the eleventh mineral having species-defining Mn and As3+. Table 7 lists these arsenite minerals.
Armangite is another example of fluorite-derivative structure (Moore and Araki 1979b), along with brattforsite and magnussonite. Its crystal structure is formed by a framework of Mn-centered polyhedra, with cavities lined by six (AsO3) groups, which point their lone-electron-pairs into the center of such cavities, similarly to what is observed in brattforsite. Along with nelenite, schallerite, and trigonite, armangite is characterized by the occurrence of [As3+O2(OH)] groups, together with (AsO3) groups. Nelenite and schallerite are dimorphs of the compound Mn2+16As3+3Si12O36(OH)17. Whereas the crystal structure of the latter was solved by Kato and Watanabe (1992), the crystal structure of nelenite is still unknown, although Dunn and Peacor (1984) proposed a relation with friedelite. Both are T-O phyllosilicates, with (AsO3) groups hosting at partially occupied positions, and residing within large twelve-membered rings.
Manganarsite is another mineral related to schallerite. Also in this case the crystal structure is only hypothetical, and Peacor et al. (1986) proposed the occurrence of rings or chains of (AsO3) groups. Trigonite displays an open heteropolyhedral network, with (010) heteropolyhedral sheets cross-linked by additional (Asϕ3) groups; the large cavities of this framework host Pb2+ cations (Pertlik 1978). Dunn et al. (1986) proposed that trigonite is structurally related to rouseite, whose crystal structure is currently unknown. It is worth noting that all these minerals have their type localities at Långban-type ore deposits (i.e., Långban and Nordmark, in Sweden), or at Franklin-Sterling Hill-type deposits (in USA). Another mineral found in the same kind of occurrence (Långban) is wiklundite, a mineral representing a transition towards the mixed As3+-As5+ species (Cooper et al. 2017). Indeed, As5+ occurs only as a minor substituent of Si4+ and consequently only As3+ can be considered as species-defining. Its complex layered crystal structure is characterized by the presence of isolated (AsO3) groups. Like brattforsite, wiklundite contains Mn and Cl as species-defining elements. An additional example is the recently described mineral cuyaite (Kampf et al. 2020), although with a different Mn valency (3+ instead of 2+). Its crystal structure can be described as an arsenite framework, with (AsO3) groups sharing corners. In contrast, brattforsite can be considered as a Mn-polyhedral framework. It is worth noting that Cl in cuyaite is severely underbonded (only 0.26 v.u.), and Kampf et al. (2020) attributed this discrepancy to the inability of the bond-valence theory to take the effect of the lone-electron-pair in account. In fact, Cl is hosted at the centre of channels accommodating the lone-electron-pairs of As sites. The underbonding of the Cl sites in brattforsite cannot be explained in this a way, since the lone-electron-pairs of the As3+ sites do not point towards Cl atoms. Cuyaite is one of the two manganese arsenites not found in Långban-type occurrences, with the type locality in the Camarones Valley, Chile. The other one is lepageite, discovered in the Szklary pegmatite, Poland (Pieczka et al. 2019). Its crystal structure is characterized by a finite [Sb4As4O19] cluster and by isolated (AsO3) groups.
Manganese arsenites are apparently very rare in Nature, with reduced mineral assemblages of Långban or Franklin-Sterling Hill types of deposits representing their preferred kind of occurrence. Brattforsite, showing a close relationship with magnussonite and some structural similarity with armangite, is thus an interesting new addition to this category of minerals. In terms of mineral classification, it belongs to the Strunz group 4.JB, i.e., Arsenites, antimonites, bismuthites, with additional anions, without H2O (Strunz and Nickel 2001).
Change history
22 September 2021
Funding note should be added: Open access funding provided by Swedish Museum of Natural History.
References
Bernardini S, Bellatreccia F, Della Ventura G, Sodo A (2021) A reliable method for determining the oxidation state of manganese at the microscale in Mn oxides via Raman spectroscopy. Geostand Geoanal Res 45:223–244
Björck L (1986) Beskrivning till Berggrundskartan Filipstad NV. Sverig Geolog Undersök Af 147:1–110
Boström K, Rydell H, Joensuu O (1979) Långban - An exhalative sedimentary deposit? Econ Geol 74:1002–1011
Brese NS, O’Keeffe M (1991) Bond-valence parameters for solids. Acta Crystallogr B 47:192–197
Bruker AXS, Inc. (2016) APEX 3. Bruker Advanced X-ray Solutions, Madison
Christy AG, Gatedal K (2005) Extremely Pb-rich rock-forming silicates including a beryllian scapolite and associated minerals in a skarn from Långban, Värmland, Sweden. Mineral Mag 69:995–1018
Cooper MA, Hawthorne FC, Langhof J, Hålenius U, Holtstam D (2017) Wiklundite, ideally Pb2[4](Mn2+,Zn)3(Fe3+,Mn2+)2(Mn2+,Mg)19(As3+O3)2[(Si,As5+)O4]6(OH)18Cl6, a new mineral from Långban, Värmland, Sweden: Description and crystal structure. Mineral Mag 81:841–855
Cooper MA, Raade G, Ball NA, Abdu YA, Hawthorne FC, Rowe R (2018) Folvikite, Sb5+Mn3+(Mg, Mn2+)10O8(BO3)4, a new oxyborate mineral from the Kitteln mine, Nordmark ore district, Värmland, Sweden: description and crystal structure. Mineral Mag 82:821–836
Dunn PJ, Peacor DR (1984) Nelenite, a manganese arsenosilicate of the friedelite group, polymorphous with schallerite, from Franklin, New Jersey. Mineral Mag 48:271–227
Dunn PJ, Ramik RA (1984) Magnussonite: new chemical data, an occurrence at Sterling Hill, New Jersey, and new data on a related phase from the Brattfors mine, Sweden. Am Mineral 69:800–802
Dunn PJ, Peacor DR, Sturman BD, Wicks FJ (1986) Rouseite, a new lead manganese arsenite from Långban, Sweden. Am Mineral 71:1034–1036
Flink G (1917) Einige Neuigkeiten in schwedischer Mineralogie. Katoptrit, ein neues Mineral vor Nordmarken. Geol Foren Stock For 40:426–452
Frost RL, Bahfenne S (2010) Raman spectroscopic study of the arsenite minerals leiteite ZnAs2O4, reinerite Zn3(AsO3)2 and cafarsite Ca5(Ti, Fe, Mn)7(AsO3)12· 4H2O. J Raman Spectrosc 41:325–328
Gabrielson O (1956) Magnussonite, a new arsenite mineral from the Långban mine in Sweden. Ark Min Geol 2:133–135
Gagné OC, Hawthorne FC (2015) Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen. Acta Crystallogr B 71:562–578
Gagné OC, Hawthorne FC (2020) Bond-length distributions for ions bonded to oxygen: results for the transition metals and quantification of the factors underlying bond-length variation in inorganic solids. IUCrJ 7:581–629
Hålenius U, Lindqvist B (1996) Chromophoric divalent iron in optically anisotropic magnussonite. Eur J Mineral 5:25–34
Holtstam D, Langhof J (1995) Metamorphic harkerite from Nordmarks odalfält. Värmland Sweden GFF 117:151–152
Holtstam D, Mansfeld J (2001) Origin of a carbonate-hosted Fe-Mn-(Ba-As-Pb-Sb-W) deposit of Långban-type in Central Sweden. Miner Deposita 36:641–657
Holtstam D, Nysten P, Gatedal K (1998) Parageneses and compositional variations of Sb oxyminerals from Långban-type deposits in Värmland, Sweden. Mineral Mag 62:395–407
Kampf AR, Mills SJ, Nash B, Dini M, Donoso AAM (2020) Cuyaite, Ca2Mn3+As3+14O24Cl, a new mineral with an arsenite framework from near Cuya, Camarones Valley, Chile. Mineral Mag 84:477–484
Kato T, Watanabe I (1992) The crystal structure of schallerite and friedelite. Yamaguchi University, College of Arts Bulletin 26:51–63
Magnusson NH (1929) Nordmarks malmtrakt. Sverig Geol Undersök Ca 13:1–98
Majzlan J, Drahota P, Filippi M (2014) Parageneses and crystal chemistry of arsenic minerals. Rev Mineral Geochem 79:17–184
Mandarino J (1981) The Gladstone-Dale relationship: part IV. The compatibility concept and its application. Can Mineral 19:441–450
Mighell AD (2003) Conventional cells: monoclinic I- and C-centered cells. Acta Crystallogr B59:300–302
Moore PB (1970a) Stenhuggarite, a new mineral from Långban and new data on magnussonite. Ark Min Geol 5:55–62
Moore PB (1970b) Mineralogy and chemistry of Långban-type deposits in Bergslagen, Sweden. Mineral Rec 1:154–172
Moore PB (1978) Manganhumite, a new species. Mineral Mag 42:133–136
Moore PB, Araki T (1979a) Magnussonite, manganese arsenite, a fluorite derivative structure. Am Mineral 64:390–401
Moore PB, Araki T (1979b) Armangite, Mn2 + 26[As3 + 6(OH)4O14][As3 + 6O18]2[CO3], a fluorite derivative structure. Am Mineral 64:748–757
Nysten P (2003) Yeatmanite and magnussonite from the Garpenberg Norra mine, Bergslagen ore province, Sweden. Can Mineral 41:201–206
Nysten P, Holtstam D, Jonsson E (1999) The Långban minerals. In: Holtstam D, Langhof J (eds) Långban: the mines, their minerals, geology and explorers. Naturhistoriska riksmuseet and Raster Förlag, Stockholm
Pasero M (2021) The new IMA list of minerals – A work in progress – Updated: March 2021. http://cnmnc.main.jp/IMA_Master_List_%282021-03%29.pdf
Peacor DR, Dunn PJ, Simmons WB, Wicks FJ (1986) Arsenites related to layer silicates: Manganarsite, the arsenite analogue of manganpyrosmalite, and unnamed analogues of friedelite and schallerite from Långban, Sweden. Am Mineral 71:1517–1521
Pertlik F (1978) The crystal structure of trigonite, Pb3Mn(AsO3)2(AsO2OH). Tscher Miner Petrog 25:95–105
Pieczka A, Cooper MA, Hawthorne FC (2019) Lepageite, Mn2+3(Fe3+7Fe2+4)O3[Sb3+5As3+8O34], a new arsenite-antimonite mineral from the Szklary pegmatite, Lower Silesia, Poland. Am Mineral 104:1043–1050
Pouchou JL, Pichoir F (1984) A new model for quantitative X-ray microanalysis. I. Application to the analysis of homogeneous samples. Rech Aerospatiale 3:13–36
Sandström F, Holtstam D (1999) Geology of the Långban deposit. In: Holtstam D, Langhof J (eds) Långban: the mines, their minerals, geology and explorers. Naturhistoriska riksmuseet and Raster Förlag, Stockholm
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr C71:3–8
Sjögren H (1884) Om manganarseniaternas från Nordmarken förekomstsätt och paragenesis. Geol Foren Stock For 7:407–416
Smith DGW, Nickel EH (2007) A system for codification for unnamed minerals: report of the subcommittee for unnamed minerals of the IMA Commission on New Minerals. Nomenclature Classification Can Mineral 45:983–1055
Stephens MB, Ripa M, Lundström I, Persson L, Bergman T, Ahl M, Wahlgren CH, Persson PO, Wickström L (2009) Synthesis of the bedrock geology in the Bergslagen region, Fennoscandian Shield, south-central Sweden. Sverig Geol Undersök Rapp Med Ba 58:1–259
Strunz H, Nickel E (2001) Strunz Mineralogical Tables. Chemical-Structural Mineral Classification system. E. Schweizerbart’sche Verlagsbuchhandlung, Stuttgart
Wilson AJC (ed) (1992) International Tables for Crystallography, Volume C: Mathematical, physical and chemical tables. Kluwer Academic, Dordrecht
Acknowledgements
Torbjörn Lorin helped with colour photography of the type specimen. The paper benefited from reviews made by Adam Pieczka and an anonymous referee, and suggestions from the journal editor Maarten A.T.M. Broekmans.
Funding
Open access funding provided by Swedish Museum of Natural History.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: M. A.T.M. Broekmans
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(CIF 2.65 mb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holtstam, D., Biagioni, C. & Hålenius, U. Brattforsite, Mn19(AsO3)12Cl2, a new arsenite mineral related to magnussonite, from Brattforsgruvan, Nordmark, Värmland, Sweden. Miner Petrol 115, 595–609 (2021). https://doi.org/10.1007/s00710-021-00749-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00710-021-00749-9