
Abstract
Viloxazine has a long history of clinical use in Europe as an antidepressant, and has recently been repurposed into an extended-release form for the treatment of attention-deficit/hyperactivity disorder in the USA. An immediate-release formulation was approved for the treatment of depression in the UK in 1974, and was subsequently marketed there and in several European countries for 30 years with no major safety concerns. In contrast to first-generation antidepressants (e.g., tricyclic antidepressants, monoamine oxidase inhibitors), viloxazine was associated with a relatively low risk for cardiotoxicity. Gastrointestinal symptoms were the most commonly reported side effects. The therapeutic effects of viloxazine are thought to be primarily the result of its action as a norepinephrine reuptake inhibitor, although in vitro and preclinical in vivo animal data suggest that viloxazine may also impact the serotoninergic system. This review summarizes the evolving knowledge of viloxazine based on information from previously published preclinical and clinical investigations, and acquired unpublished historical study reports from both open-label and blinded controlled clinical trials. We review the chemical properties, mechanism of action, safety, and tolerability across these studies, and discuss the contemporary rationale for the development of this agent as an extended-release oral formulation for the treatment of attention-deficit/hyperactivity disorder.
Similar content being viewed by others
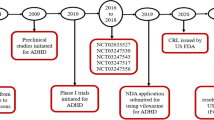


An immediate-release formulation of viloxazine was originally approved in the UK for the treatment of depression in adults in 1974, and was subsequently marketed in the UK and several European countries. |
Viloxazine immediate-release demonstrated an acceptable safety and tolerability profile in controlled clinical trials of adults with depression. There were no major safety concerns throughout the 30 years viloxazine was commercially available. The immediate-release formulation of viloxazine was removed from European markets during the early 2000s because of business reasons unrelated to safety or efficacy. |
The clinical efficacy of viloxazine is thought to be due to its action as a selective norepinephrine reuptake inhibitor, though in vivo and preclinical data have demonstrated direct or indirect effects on brain serotonin and dopamine systems. |
An extended-release formulation of viloxazine has recently been approved in the USA for the treatment of attention-deficit/hyperactivity disorder in children and adolescents. |
1 Introduction
Viloxazine extended-release (ER) is a novel nonstimulant that was approved in April 2021 by the US Food and Drug Administration under the trade name QelbreeTM for the treatment of attention-deficit/hyperactivity disorder (ADHD) in children and adolescents. The original immediate-release (IR) viloxazine formulation was discovered incidentally during an attempt to synthesize molecules chemically related to propranolol with central nervous system (CNS)-modulating properties [1] (see Fig. 1 for the chemical structure). Subsequently, an IR formulation was developed and approved for the treatment of adults with depression in the UK in 1974, and marketed there and in several European countries for over 30 years. While commercially available, viloxazine was used in both adult and elderly patients as a two-times or three-times daily treatment [2], and was distributed under the trade names Viloxazin [3], Emovit [4], Viloxazina [5], Viloxazinum [5], Vivarint [6], Vicilan [2], and Vivalan [7].

The chemical structure of viloxazine
In 1984, viloxazine was assigned an orphan drug designation in the USA under the brand name Catatrol for the treatment of cataplexy and narcolepsy, but, for unknown reasons, was never approved for either indication [8]. The IR formulation of viloxazine was discontinued in the early 2000s for business reasons unrelated to safety and efficacy [5, 9], and at the time of this publication has not been previously approved and marketed in the USA for any indication. Here, we review what is known about the chemical properties and clinical activity of viloxazine from its previous use in adult patients with depression, including individuals with comorbid conditions such as anxiety, alcoholism, and epilepsy, as well as recent clinical trials of the new ER formulation, viloxazine ER.
To review the basic and clinical science of viloxazine, preclinical and clinical studies and reviews were searched on PubMed (search terms: “Viloxazine,” “Viloxazin,” “Emovit,” “Viloxazina,” “Viloxazinum,” and “Vivalan”). In addition, meeting proceedings from the International Vivalan [Viloxazine] Symposium in England in 1975 [10], and study reports from 1973 to 1976 were obtained as part of the acquisition of viloxazine by Supernus Pharmaceuticals, Inc. From these study reports, we identified 23 open-label trials (total, N = 605) and 14 single-blind or double-blind (total, N = 821; viloxazine, n = 447) controlled trials investigating the utility and safety of viloxazine for depression and/or anxiety, all of which were conducted prior to 1976. Safety and tolerability data from the unpublished trials described in these study reports may be informative to the application of viloxazine ER in the treatment of ADHD, and are summarized in Tables 1 and 2, respectively.
2 Pharmacology and Mechanism of Action
The primary CNS targets of viloxazine appear to be in the monoaminergic systems, although viloxazine differs from many other known antidepressants in that its mechanism of action is not linked to inhibition of serotonin or dopamine reuptake [11, 12]. Viloxazine has been shown to increase brain norepinephrine concentrations [13] via norepinephrine reuptake blockade [11, 14], with no direct effect on in vitro norepinephrine release [14]. A contemporary study confirmed that viloxazine demonstrates moderate inhibitory activity at the norepinephrine transporter (half-maximal inhibitory concentration in the 0.3-µM range), significantly increases extracellular norepinephrine levels across several brain regions (assessed in the prefrontal cortex, nucleus accumbens, and amygdala), and possesses weak antagonistic effects at the adrenergic β2 and adrenergic α1B norepinephrine receptors [15].
Data from the 1970s showed that viloxazine does not significantly inhibit the uptake of serotonin into human blood platelets [1] or CNS tissue [11, 12]. Viloxazine increased neuronal sensitivity to serotonin [16, 17], and potentiated both the 5-hydroxytryptophan (precursor to serotonin)-induced behavioral syndrome in mice and the 5-hydroxytryptophan-induced hind-limb extensor reflex in rats, thought to reflect increased activation of serotonin receptors [12]. Notably, viloxazine has been shown to facilitate basal serotonin release from rat striatal brain slices [14]. A recent preclinical study showed that viloxazine increases serotonin levels in the prefrontal cortex, nucleus accumbens, and amygdala in vivo [15], and is an agonist at serotonergic 5-HT2C receptors, an antagonist at 5-HT2B receptors, and a weak antagonist at 5-HT7 receptors [15]. Importantly, this action on serotonin receptors may play a role in serotonin release: antagonism of 5-HT2B receptors located on GABAergic interneurons has been shown to disinhibit tonic inhibitory control of serotonin neurons innervating the medial prefrontal cortex, indirectly stimulating serotonin release in this region [18].
Initial reports suggested that viloxazine could produce modest and transient dopamine increases in the CNS, though to a lesser degree than norepinephrine or serotonin [13]. Other in vitro studies found no effect on dopamine release in striatal tissue [14], and little affinity for, or potency at, either dopamine receptors or the transporter [11, 14, 15, 19]. Recently, viloxazine was shown in a preclinical rodent model to increase dopamine levels in vivo appreciably in the prefrontal cortex, moderately in the amygdala, and minimally in the nucleus accumbens [15], which likely reflects a combination of direct (via the norepinephrine transporter, which also manages reuptake of dopamine in the prefrontal cortex) and indirect effects. The minimal impact of viloxazine on dopamine in the nucleus accumbens, a region known for its key role in substance use disorders [20, 21], may be associated with a low abuse liability [22]. Notably, dysregulated dopamine signaling in the prefrontal cortex and amygdala has been implicated in the pathophysiology of ADHD [23,24,25,26,27,28], and is a primary target of many effective ADHD medications [27, 29, 30].
In contrast to many first-generation antidepressants (i.e., tricyclic antidepressants [TCAs] and monoamine oxidase inhibitors), viloxazine demonstrates negligible activity in the cholinergic system [12, 13], and lacks any appreciable activity towards histamine [13, 31]. It reduces the responsiveness of rat cortical neurons to acetylcholine [32, 33], shows negligible binding to the choline transporter, and essentially no impact on acetylcholine levels in the prefrontal cortex, nucleus accumbens, or amygdala [15, 34]. Early in vitro studies suggested viloxazine was not a significant inhibitor of monoamine oxidase [1, 12], but it was later characterized as a very weak, competitive, and reversible inhibitor of monoamine oxidase A and B [35]. A recent study found viloxazine to demonstrate weak competitive inhibition (< 25%) at histamine receptors H1 and H2 and muscarinic receptors M1–4, and notably low affinity towards monoamine oxidase A [15].
3 Dosing
We were able to identify 37 clinical investigations assessing viloxazine dosing in adult patients with depressive disorders (including studies in subjects with comorbid anxiety disorders as well as epilepsy) [see Tables 1 and 2 for those that remain unpublished]. Across these trials, viloxazine was investigated at a dose range of 50–600 mg/day, administered two to four times daily. The typical target dose for depression was 300–400 mg/day in adults or 100–200 mg/day in elderly patients, generally administered twice or three times daily [4]. Contemporary studies in the treatment of ADHD have administered viloxazine ER at daily doses of 100–400 mg in children (aged 6–11 years), and 200–600 mg in adolescents (aged 12–17 years) and 200–600 mg in adults (aged 18–65 years) [36,37,38,39,40].
4 Pharmacokinetics
The pharmacokinetic profile of IR viloxazine has been well characterized in adult patients with depression. Orally administered viloxazine exhibited a short plasma half-life of 2.5 hours after being well absorbed through the small intestine [41]. Maximum blood concentrations were proportional to the oral dosage, achieved 1–4 h after a single dose [41, 42], with an average oral bioavailability of 85% [42]. The clearance rate after intravenous dosing was found to be 124 ± 11 mL/hour/kg, and the volume of distribution was 0.73 ± 0.28 L/kg [42]. Viloxazine is extensively metabolized, with only 12–15% of the drug being excreted unchanged, and, along with its metabolites, is rapidly eliminated via the kidneys [43, 44]. In both animal and human studies, viloxazine is thought to be responsible for all observed effects, as its metabolites are thought to be essentially inactive [43, 44]. Across species (including rodents, rabbits, canines, and primates) and in humans, viloxazine has a rapid clearance rate, with the majority (90%) of orally administered drug excreted through the urine within 24 hours, and only 2% excreted through fecal matter [43]. A clinical study of two healthy male volunteers given 40 or 100 mg of 14C-viloxazine indicated that the half-life for excretion in the urine was approximately 4–5 hours [43].
5 Efficacy and Safety of Viloxazine in CNS Conditions: Depression, Anxiety, Alcoholism, and Epilepsy
5.1 Clinical Efficacy
In numerous double-blind and open-label studies, viloxazine was effective in treating various types of depressive disorders (some of which are not included in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition [45]), including endogenous, neurotic, psychotic, and reactive depression, chronic and severe depression, and depression with comorbid conditions (reviewed in [2] and [46]). Across several studies in cohorts with depression, viloxazine reduced not only depressive symptoms but concomitant symptoms of anxiety ([7, 47, 48], also reviewed in [2]) and alcohol abuse in patients with comorbid alcohol dependence [49]. In a multicenter trial of 115 patients with moderate-to-severe depression, viloxazine was also effective in patients whose depression had not responded to prior antidepressant treatment [7]. Compared with other available antidepressants at the time (e.g., imipramine, amitriptyline), viloxazine was considered to be as effective in reducing symptoms of depression, but with better tolerability [2, 46, 47].
Viloxazine has only twice been directly compared with later antidepressants (and to our knowledge, never with contemporary antidepressants). A double-blind study comparing citalopram and viloxazine in severely depressed patients administered each drug via a slow-drop infusion for 2 weeks followed by oral administration for the following 4 weeks [50]. Both drugs improved scores on depression assessments (e.g., Montgomery–Åsberg Depression Rating Scale) and the Clinical Global Impressions–Improvement scale at both time points (i.e., 14 and 42 days), although this improvement was significantly greater for citalopram [50]. A single-blind trial comparing 4 weeks of nomifensin vs viloxazine (150 mg/day) administration in elderly depressed patients again showed significant improvements on the Hamilton Rating Scale for depression with both treatments [51].
5.2 Safety Profile
Because viloxazine was first developed in the 1970s and has been off the market since the 2000s (depending on the country), available toxicity data were obtained using the methodological standards and comparator treatments available at the time (i.e., TCAs). Viloxazine was generally considered to have a comparatively lower cardiovascular risk than the TCAs and was associated with normal electrocardiogram recordings (e.g., intra-cardiac conduction), even in cases of overdose [52, 53]. In historic studies, viloxazine also did not increase the risk of adrenaline-induced cardiac arrhythmias [2, 52]. In a contemporary study using supratherapeutic doses of viloxazine ER (1800 mg) in healthy adults, there was no effect on cardiac repolarization or other electrocardiogram parameters [54]. Minimal anticholinergic effects were found in human double-blind randomized studies of viloxazine 100 mg vs imipramine 50 mg [55, 56], though both drugs induced transient tachycardia 4 h post-dose [55].
Viloxazine was shown to have a relatively high median lethal dose (LD50) in animal studies, specifically, 500–1000 mg/kg in the mouse and 2000 mg/kg in the rat when administered orally, and 60 mg/kg in mice and 60–77 mg/kg in rats when administered intravenously [1, 5, 52]. There are relatively few documented suicide attempts by humans taking viloxazine [2, 52]. In a 1976 report of 12 cases of drug overdose, doses ranged from 1 to 6.5 g [52]. Gastric lavage and/or forced diuresis was performed and all 12 patients fully recovered [52]. However, a separate account reported one overdose death following ingestion of an unknown amount of medication where the patient received no medical treatment [2]. Given these data in the context of the long history of viloxazine of safe use as an antidepressant, additional studies to identify populations that may be particularly vulnerable to viloxazine toxicity may be informative in the future.
In preclinical rodent and nonhuman primate models, viloxazine administered at normal therapeutic concentrations was associated with anticonvulsant activity [1, 13, 57]. Clinical studies using viloxazine IR with or without concomitant anticonvulsant therapy described the drug to be non-epileptogenic or, at least, less epileptogenic than contemporaneous antidepressants; as a result, it was also studied as a possible treatment for depressed patients with epilepsy [42, 58, 59]. Importantly, viloxazine was found to induce elevations of plasma carbamazepine and carbamazepine-10,11-epoxide concentrations [59], while a later study showed that several epileptic drugs (administered in patients with epilepsy) did not alter the pharmacokinetics of viloxazine relative to controls [42]. Viloxazine was ultimately not contraindicated for depressed patients with epilepsy [46, 58].
In the aforementioned trial comparing nomifensin vs viloxazine in elderly patients, sinus tachycardia was reported in two patients (one in each group) [51]. Similarly, there were no major differences in safety outcomes when compared with citalopram [50]. Throughout the literature, there has been no mention of dose tapering, nor has it been evaluated in contemporary studies.
5.3 Tolerability
Viloxazine IR was generally well tolerated in clinical studies, with the most frequent side effects including gastrointestinal disturbances such as nausea and vomiting, as well as headache, dry mouth, and fatigue [2, 46,47,48, 50]. In a multicenter study of 115 adults with moderate-to-severe depression administered 150–300 mg of viloxazine, nausea and fatigue occurred in 13.2% and 9.4% of patients, respectively, with a relatively low incidence of headache (4.7%), vomiting (3.8%), and dry mouth (1.9%) [7]. In two separate studies of patients with mild-to-moderate depression, 150–200 mg/day of viloxazine was found to be efficacious in reducing symptoms of depression and anxiety, with mild side effects that were primarily gastrointestinal in nature [47, 48]. In an analysis of 771 patients across 29 studies, the dropout rate from gastrointestinal symptoms such as nausea and vomiting was 6.9% [60]. In some cases, viloxazine at twice-daily 100-mg doses disrupted sleep patterns (i.e., broken and reduced sleep, decreased rapid eye movement sleep) [61] and at 100 mg/day depressed the critical flicker frequency (a test of general CNS stimulation or depression), though this effect was not seen at lower doses [56]. Double-blind, placebo-controlled crossover studies of low doses (20–80 mg) in healthy volunteers indicated no significant differences between viloxazine and placebo on assessments of reaction time, coordination, or cognitive task performance [47, 62]. Of note, 150–300 mg viloxazine was found to have fewer anticholinergic effects and sedation compared with the TCAs, but nausea and vomiting were common complaints and thought to be dose related [2].
Although the mechanism of action responsible for the reported gastrointestinal side effects (e.g., nausea, vomiting) is unknown, these adverse events have been most pronounced at the onset of treatment and are generally transient in nature, frequently resolving as treatment continues [60], and have been shown to be minimized by gradual introduction of the drug [2]. Ongoing research has considered whether an ER formulation of viloxazine may help mitigate some of the aforementioned gastrointestinal side effects. Recent phase III [36,37,38,39] studies investigating the ER formulation at 100–600 mg/day in pediatric patients (i.e., children and adolescents, aged 6–17 years) diagnosed with ADHD found the incidence of nausea and vomiting for viloxazine to be 9.8%, vs 4.1% for placebo.
6 Future of Viloxazine: A Potential New Application in ADHD
The therapeutic effects of viloxazine for ADHD are thought to be the result of its action as a norepinephrine reuptake inhibitor. However, demonstrated in vitro and preclinical in vivo activity on norepinephrine and serotonin, as well as direct and downfield indirect effects on dopamine, may be relevant to current applications in ADHD, given the role these neurotransmitters have in regulating attentional and behavioral control [25, 63,64,65,66,67]. Norepinephrine has long been implicated in ADHD pathophysiology [25, 63, 66], and serotonin has also been suggested to play a role in the hyperactivity/impulsivity deficits associated with ADHD [64, 65]. In addition, the effects of viloxazine on dopamine, particularly in the prefrontal cortex, could also contribute to its therapeutic effects [23,24,25,26, 28]. Further research is required to address these questions.
Viloxazine was found to be effective for ADHD symptoms and generally well tolerated, as demonstrated in several recent randomized, double-blind, placebo-controlled, phase II and III clinical trials [36,37,38,39, 68]. In a recent phase II trial in children aged 6–12 years with ADHD (n = 222), treatment-emergent AEs (TEAEs) were generally mild or moderate, with only three severe TEAEs reported (tearfulness, irritability, and decreased appetite) [68]. In four subsequent phase III trials in children aged 6–11 years [36, 37] and adolescents aged 12–17 years [38, 39], viloxazine ER was well-tolerated over 6–8 weeks of treatment (pooled N = 1117). The most frequently reported AEs were somnolence (14.5%), headache (10.8%), and decreased appetite (8.1%), with low discontinuation rates because of TEAEs (4% averaged across all trials), less than 1% of subjects reporting a serious TEAE, and no deaths [36,37,38,39].
In three of these four phase III trials, statistically significant improvements in ADHD-Rating Scale, Fifth Edition total scores (the primary endpoint) were reported as soon as 1 week following the onset of once-daily treatment with viloxazine ER 100 mg, 200 mg, and 400 mg, relative to placebo [36,37,38]. In the fourth phase III trial (adolescents, N = 292), the primary efficacy endpoint (improvement on ADHD-Rating Scale, Fifth Edition total scores) did not reach statistical significance for once-daily viloxazine ER 600 mg, despite being statistically significant for patients treated with the 400-mg dose [39]. Why the 400-mg dose was more effective than the higher 600-mg dose relative to placebo in reducing ADHD symptoms is unclear.
The introduction of another nonstimulant option has potential utility in augmenting the therapeutic armamentarium. First-line treatment for ADHD usually includes stimulant pharmacotherapy, as stimulants are associated with near-immediate onset of effect and greater efficacy in improving ADHD outcomes than current nonstimulants [69,70,71]. However, there are also relative or labeled contraindications to stimulant use in the presence of several comorbid conditions (e.g., insomnia, anorexia), as stimulants are thought to possibly exacerbate these conditions, at least in some individuals [72,73,74,75,76]. Further, most stimulant formulations have a relatively short (e.g., 3–12 h) duration of action [76] (although new formulations such as Mydayis® and Adhansia XR® have labeled efficacy up to 16 h [77, 78]), thus consistent treatment throughout the day can be difficult to achieve. In certain situations, stimulants can fail to provide satisfactory symptom control [79], and have a known potential for abuse [22]. Several Food and Drug Administration-approved nonstimulant ADHD treatment options are currently available, including atomoxetine, guanfacine ER, and clonidine ER, and may be prescribed either alone or in combination with stimulant therapy [80,81,82,83]. However, nonstimulants, which are generally less effective and have a slower onset of effect than stimulants [84], are also not without limitations. For example, they have been associated with minor changes in blood pressure or cardiovascular risk [85,86,87], precipitation of secondary mania [88, 89], and suicidal ideation [90], although these events are rare and generally restricted to vulnerable populations with pre-existing conditions.
Viloxazine ER could help address these limitations and also provide an alternative non-stimulant option. To date, there have been no direct efficacy comparisons between viloxazine ER and other ADHD treatments (e.g., atomoxetine, stimulants), nor has their efficacy in combination been tested. Other studies of the efficacy and safety of viloxazine ER in adults are ongoing, as are long-term efficacy and safety studies in both pediatric and adult populations.
7 Conclusions, Future Perspectives, and Implications for Daily Practice
The pharmacokinetic, pharmacodynamic, and safety profiles of viloxazine have been investigated in numerous studies over several decades, predominantly in adults with depression, alone, or in the presence of comorbid conditions (e.g., anxiety disorders, alcoholism, epilepsy). During its period of commercial use in Europe beginning in the 1970s, the safety profile of viloxazine was well established [2, 46]. Although it has demonstrated in vitro and in vivo preclinical activity on both norepinephrine and serotonin, with additional direct and downstream indirect effects on dopamine [12, 91], the mechanism of action responsible for its clinical efficacy is thought to be the result of its properties as a selective norepinephrine reuptake inhibitor. Phase III clinical trials investigating viloxazine ER, the ER formulation of viloxazine, have recently been completed, and demonstrate efficacy in reducing ADHD symptomatology in children and adolescents [36,37,38,39]. These studies demonstrate an acceptable incidence of adverse events, including cardiac-related adverse events, and pooled discontinuation rates because of TEAEs of 4.0% in viloxazine ER-treated groups and 1.2% among placebo groups [36,37,38,39, 68]. Given its mechanism of action and excellent safety and efficacy profile, viloxazine may be a useful alternative to stimulant or other nonstimulant therapy for the treatment of ADHD.
References
Mallion KB, Todd AH, Turner RW, Bainbridge JG, Greenwood DT, Madinaveitia J, et al. 2-(2-Ethoxyphenoxymethyl)tetrahydro-1,4-oxazine hydrochloride, a potential psychotropic agent. Nature. 1972;238:157–8.
Pinder RM, Brogden RN, Speight TM, Avery GS. Viloxazine: a review of its pharmacological properties and therapeutic efficacy in depressive illness. Drugs. 1977;13:401–21.
Renfordt E, Busch H, Fähndrich E, Müller-Oerlinghausen B. Studies on a novel antidepressant (viloxazin) by means of time-series analysis of TV data. Arzneimittelforschung. 1976;26(6):1114–6.
Marsh W. Viloxazine. In: Enna SJ, Bylund DB, editors. xPharm: the comprehensive pharmacology reference. Amsterdam: Elsevier Inc; 2007.
National Center for Biotechnology Information, PubChem database. Viloxazine, CID = 5666. https://pubchem.ncbi.nlm.nih.gov/compound/Viloxazine. Accessed 17 Jan 2019.
Mulrow CD. Treatment of depression: newer pharmacotherapies. Rockville: Agency for Health Care Policy and Research; 1999.
Kabes J, Dostal T, Nahunek K, Svestka J, Molcan J, Caplová T, et al. Multicentre study with viloxazine (Vivalan®) in depressed patients. Int Pharmacopsychiatr. 1980;15:228–39.
US Food and Drug Administration. Orphan drug designations and approvals. Catatrol. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=2384. Accessed 30 Apr 2020.
Williams D. Antidepressants. In: Lemke TL, Williams DA, Roche VF, Zito SW, editors. Foye’s principles of medicinal chemistry. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2013.
Murphy JE. Vivalan: drug profile. J Int Med Res. 1975;3:122–5.
Blackburn TP, Foster GA, Greenwood DT, Howe R. Effects of viloxazine, its optical isomers and its major metabolites on biogenic amine uptake mechanisms in vitro and in vivo. Eur J Pharmacol. 1978;52:367–74.
Lippman W, Pugsley TA. Effects of viloxazine, an antidepressant agent, on biogenic amine uptake mechanisms and related activities. Can J Physiol Pharmacol. 1976;54:494–509.
Greenwood DT. Animal pharmacology of viloxazine (Vivalan). J Int Med Res. 1975;3:18–28.
Martin IL, Baker GB, Mitchell PR. The effect of viloxazine hydrochloride on the transport of noradrenaline, dopamine, 5-hydroxytryptamine and gamma-amino-butyric acid in rat brain tissue. Neuropharmacology. 1978;17(6):421–3.
Yu C, Garcia-Olivares J, Candler S, Schwabe S, Maletic V. New insights into the mechanism of action of viloxazine: serotonin and norepinephrine modulating properties. J Exp Pharmacol. 2020;12:285–300.
Jones RSG, Roberts MHT. Potentiation of responses to monoamines by antidepressants after destruction of monoamine afferents. Br J Pharmacol. 1979;65(3):501–10.
Jones RSG, Roberts MHT. Potentiation of monoamine responses of denervated cells by a noradrenaline uptake inhibitor (viloxazine). Br J Pharmacol. 1978;62(3):403P-P404.
Cathala A, Devroye C, Drutel G, Revest JM, Artigas F, Spampinato U. Serotonin2B receptors in the rat dorsal raphe nucleus exert a GABA-mediated tonic inhibitory control on serotonin neurons. Exp Neurol. 2019;311:57–66. https://doi.org/10.1016/j.expneurol.2018.09.015.
Hall H, Sällemark M, Wedel I. Acute effects of atypical antidepressants on various receptors in the rat brain. Acta Pharmacol Toxicol (Copenh). 1984;54(5):379–84.
Kalivas PW, Volkow N. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13.
Volkow N, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction. Arch Neurol. 2007;64(11):1575–9.
Clemow DB, Walker DJ. The potential for misuse and abuse of medications in ADHD: a review. Postgrad Med. 2014;126(5):64–81. https://doi.org/10.3810/pgm.2014.09.2801.
Arnsten A. Fundamentals of attention-deficit/hyperactivity disorder: circuits and pathways. J Clin Psychiatry. 2006;67:7–12.
Arnsten AF. The emerging neurobiology of attention deficit hyperactivity disorder: the key role of the prefrontal association cortex. J Pediatr. 2009;154(5):I-S43. https://doi.org/10.1016/j.jpeds.2009.01.018.
Curatolo P, D’Agati E, Moavero R. The neurobiological basis of ADHD. Ital J Pediatr. 2010;36(1):79. https://doi.org/10.1186/1824-7288-36-79.
Levy F. The dopamine theory of attention deficit hyperactivity disorder (ADHD). Aust N Z J Psychiatry. 1991;25(2):277–83. https://doi.org/10.3109/00048679109077746.
Levy F. Pharmacological and therapeutic directions in ADHD: specificity in the PFC. Behav Brain Funct. 2008;4:12. https://doi.org/10.1186/1744-9081-4-12.
Swanson JM, Kinsbourne M, Nigg J, Lanphear B, Stefanatos GA, Volkow N, et al. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev. 2007;17(1):39–59. https://doi.org/10.1007/s11065-007-9019-9.
Faraone SV. The pharmacology of amphetamine and methylphenidate: relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci Biobehav Rev. 2018;87:255–70. https://doi.org/10.1016/j.neubiorev.2018.02.001.
Hutson PH, Pennick M, Secker R. Preclinical pharmacokinetics, pharmacology and toxicology of lisdexamfetamine: a novel d-amphetamine pro-drug. Neuropharmacology. 2014;87:41–50. https://doi.org/10.1016/j.neuropharm.2014.02.014.
Hall H, Ogren SO. Effects of antidepressant drugs on histamine-H1 receptors in the brain. Life Sci. 1984;34(6):597–605.
Jones RSG, Roberts MHT. Modification of cortical neuron responses to acetylcholine by viloxazine. J Pharm Pharmacol. 1979;31:87–90.
Jones RSG. Long-term administration of atropine, imipramine, and viloxazine alters responsiveness of rat cortical neurones to acetylcholine. Can J Physiol Pharmacol. 1980;58(5):531–5.
Golds PR, Przyslo FR, Strange PG. The binding of some antidepressant drugs to brain muscarinic acetylcholine receptors. Br J Pharmacol. 1980;68:541–9.
Martinez C, Dominiak P, Kees F, Grobecker H. Inhibition of monoamine oxidase by viloxazine in rats. Drug Res. 1986;36(1):800–3.
Nasser A, Liranso T, Adewole T, Fry N, Hull JT, Chowdhry F, et al. A phase 3, randomized, placebo-controlled trial to assess the efficacy and safety of once-daily SPN-812 (viloxazine extended release) in the treatment of ADHD in school-age children. Clin Ther. 2020;42(8):1452–66.
Nasser A, Liranso T, Adewole T, Fry N, Hull JT, Busse GD, et al. Once-daily SPN-812 200 and 400 mg in the treatment of ADHD in school-aged children: a phase III randomized, controlled trial. Clin Ther. 2021. https://doi.org/10.1016/j.clinthera.2021.01.027.
Nasser A, Liranso T, Adewole T, Fry N, Hull JT, Busse GD, et al. A phase 3, placebo-controlled trial of once-daily viloxazine extended-release in adolescents with attention-deficit/hyperactivity disorder. J Clin Psychopharmacol. 2021.
Nasser A, Liranso T, Adewole T, Fry N, Hull JT, Chowdhry F, et al. A phase 3 placebo-controlled trial of once-daily 400-mg and 600-mg SPN-812 (viloxazine extended-release) in adolescents with ADHD. Psychopharm Bul. 2021;51(2):43–64.
Supernus Pharmaceuticals Inc. Supernus announces positive results from phase III study for SPN-812 in adults with ADHD. 2020. https://ir.supernus.com/news-releases/news-release-details/supernus-announces-positive-results-phase-iii-study-spn-812-0. Accessed 10 May 2021.
Bayliss PFC, Case DE. Blood level studies with viloxazine hydrochloride in man. Br J Clin Pharmacol. 1975;2:209–14.
Pisani F, Fazio A, Spina E, Artesi C, Pisani B, Russo M, et al. Pharmacokinetics of the antidepressant drug viloxazine in normal subjects and in epileptic patients receiving chronic anticonvulsant treatment. Psychopharmacology. 1986;90:295–8.
Case DE, Reeves PR. The disposition and metabolism of I.C.I. 58,834 (viloxazine) in humans. Xenobiotica. 1975;5(2):113–29.
Case DE, Illston H, Reeves PR, Shuker B, Simons P. The disposition and metabolism of I.C.I. 58,834 (viloxazine) in animals. Xenobiotica. 1975;5(2):83–111. https://doi.org/10.3109/00498257509056096.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, D.C.: American Psychiatric Publishing; 2013.
Ban TA, McEvoy JP, Wilson WH. Viloxazine: a review of the literature. Int Pharmacopsychiatr. 1980;15:118–23.
Bayliss PFC, Dewsbury AR, Donald JF, Harcup JW, Mayer M, Million R, et al. A double-blind controlled trial of “Vivalan” (viloxazine hydrochloride) and imipramine hydrochloride in the treatment of depression in general practice. J Int Med Res. 1974;2:260–4.
Bayliss PFC, Harcup JW, Mayer M, Million R, Murphy JE, Plant B, et al. An open study of two dose levels of ‘Vivalan’ (viloxazine hydrochloride ICI 58 834) in depression in general practice. J Int Med Res. 1974;2:253–9.
Altamura AC, Mauri MC, Girardi T, Panetta B. Alcoholism and depression: a placebo controlled study with viloxazine. Int J Clin Pharmacol Res. 1990;10(5):293–8.
Bouchard JM, Strub N, Nil R. Citalopram and viloxazine in the treatment of depression by means of slow drop infusion: a double-blind comparative trial. J Affect Disord. 1997;46:51–8.
Moizeszowicz J, Subirá S. Controlled trial of nomifensin (HOE 984) and viloxazine in the treatment of depression in the elderly. J Clin Pharmacol. 1977;17(1):81–3.
Brosnan RD, Busby AM, Holland RP. Cases of overdosage with viloxazine hydrochloride (Vivalan). J Int Med Res. 1976;4:83–5.
Corona G, Frattini P, Cucchi M, Santagostino G, Schinelli S, Zerbi F, et al. Viloxazine in depressed women: clinical response and cardiovascular effects. Int J Clin Pharmacol Ther Toxicol. 1987;25(6):322–7.
Nasser A, Faison SL, Liranso T, Adewole T, Busse GD, Fava M, et al. Evaluation of the effect of SPN-812 (viloxazine extended release) on QTc interval in healthy adults. J Clin Psychiatry. 2020;81(6):e1-6.
Bayliss PFC, Duncan SM. The clinical pharmacology of viloxazine hydrochloride: a new anti-depressant of novel chemical structure. Br J Clin Pharmacol. 1974;1:431–7.
Turner P, Bayliss PFC, Ghose K. Clinical pharmacoolgy of viloxazine (Vivalan). J Int Med Res. 1975;3:41–7.
Meldrum B, Anlezark G, Adam H, Greenwood D. Anticonvulsant and proconvulsant properties of viloxazine hydrochloride: pharmacological and pharmacokinetic studies in rodents and the epileptic baboon. Psychopharmacology. 1982;76(3):212–7.
Edwards JG, Glen-Bott M. Does viloxazine have epileptogenic properties? J Neurol Neurosurg Psychiatry. 1984;47:960–4.
Pisani F, Fazio A, Oteri G, Perucca E, Russo M, Trio R, et al. Carbamazepine-viloxazine interaction in patients with epilepsy. J Neurol Neurosurg Psychiatry. 1986;49:1142–5.
Murphy JE. Vivalan: drug profile. J Int Med Res. 1975;3:112–5.
Březinová V, Adam K, Chapman K, Oswald I, Thomson J. Viloxazine, sleep, and subjective feelings. Psychopharmacology. 1977;55:121–8.
Kirby M, Turner P. Some preliminary observations on ICI 58 834, a new psychotropic agent, in man. Br J Clin Pharmacol. 1973;1(2):169.
Biederman J, Spencer T. Attention-deficit/hyperactivity disorder (ADHD) as a noradrenergic disorder. Biol Psychiatry. 1999;46:1234–42.
Banerjee E, Nandagopal K. Does serotonin deficit mediate susceptibility to ADHD? Neurochem Int. 2015;82:52–68. https://doi.org/10.1016/j.neuint.2015.02.001.
Oades R. The role of the serotonin system in ADHD. Expert Rev Neurother. 2007;7:1357–74.
Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann Pharmacother. 2014;48(2):209–25. https://doi.org/10.1177/1060028013510699.
Viggiano D, Ruocco LA, Arcieri S, Sadile AG. Involvement of norepinephrine in the control of activity and attentive processes in animal models of attention deficit hyperactivity disorder. Neural Plast. 2004;11(1–2):133–49.
Johnson JK, Liranso T, Saylor K, Tulloch G, Adewole T, Schwabe S, et al. A phase II double-blind, placebo-controlled, efficacy and safety study of SPN-812 (extended-release viloxazine) in children with ADHD. J Atten Disord. 2020;24(2):348–58.
Catala-Lopez F, Hutton B, Nunez-Beltran A, Page MJ, Ridao M, Macias Saint-Gerons D, et al. The pharmacological and non-pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: a systematic review with network meta-analyses of randomised trials. PLoS ONE. 2017;12(7):e0180355. https://doi.org/10.1371/journal.pone.0180355.
Cortese S. Pharmacologic treatment of attention deficit-hyperactivity disorder. N Engl J Med. 2020;383(11):1050–6.
Biederman J, Spencer T, Wilens T. Evidence-based pharmacotherapy for attention-deficit hyperactivity disorder. Int J Neuropsychopharmacol. 2004;7(1):77–97. https://doi.org/10.1017/S1461145703003973.
Biederman J, Wilens T, Mick E, Milberger S, Spencer TJ, Faraone SV. Psychoactive substance use disorders in adults with attention deficit hyperactivity disorder (ADHD): effects of ADHD and psychiatric comorbidity. Am J Psychiatry. 1995;152:1652–8.
Wilens T. Impact of ADHD and its treatment on substance abuse in adults. J Clin Psychiatry. 2004;65:38–45.
Soutullo C, DelBello MP, Ochsner JE, McElroy SL, Taylor SA, Strakowski SM, et al. Severity of bipolarity in hospitalized manic adolescents with history of stimulant or antidepressant treatment. J Affect Disord. 2002;70:323–7.
Corkum P, Moldofsky H, Hogg-Johnson S, Humphries T, Tannock R. Sleep problems in children with attention-deficit/hyperactivity disorder: impact of subtype, comorbidity, and stimulant medication. J Am Acad Child Adolesc Psychiatry. 1999;38(10):1285–93.
Briars L, Todd T. A review of pharmacological management of attention-deficit-hyperactivity disorder. J Pediatr Pharmacol Ther. 2016;21(3):192–206.
Mydayis (mixed salts of a single-entity amphetamine product) extended-release [package insert]. Lexington: Shire US, Inc.; 2019.
Adhansia XR (methylphenidate hydrochloride) extended-release [package insert]. Stamford: Purdue Pharma L.P.; 2019.
Spencer T, Biederman J, Wilens T, Harding M, O’Donnell D, Griffin S. Pharmacotherapy of attention-deficit hyperactivity disorder across the life cycle. J Am Acad Child Adolesc Psychiatry. 1996;35(4):409–32. https://doi.org/10.1097/00004583-199604000-00008.
Treuer T, Gau SS, Mendez L, Montgomery W, Monk JA, Altin M, et al. A systematic review of combination therapy with stimulants and atomoxetine for attention-deficit/hyperactivity disorder, including patient characteristics, treatment strategies, effectiveness, and tolerability. J Child Adolesc Psychopharmacol. 2013;23(3):179–93. https://doi.org/10.1089/cap.2012.0093.
Molife C, Bernauer MJ, Farr AM, Haynes VS, Kelsey D. Combination therapy patterns and predictors of ADHD in commercially insured and Medicaid populations. Postgrad Med. 2012;124(5):7–22. https://doi.org/10.3810/pgm.2012.09.2586.
Kollins SH, Jain R, Brams M, Segal S, Findling RL, Wigal SB, et al. Clonidine extended-release tablets as add-on therapy to psychostimulants in children and adolescents with ADHD. Pediatrics. 2011;127(6):e1406–13. https://doi.org/10.1542/peds.2010-1260.
Adler LA, Reingold LS, Morrill MS, Wilens TE. Combination pharmacotherapy for adult ADHD. Curr Psychiatry Rep. 2006;8:409–15.
Faraone SV, Glatt SJ. A comparison of the efficacy of medications for adult attention-deficit/hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry. 2010;71(6):754–63. https://doi.org/10.4088/JCP.08m04902pur.
Martinez-Raga J, Knecht C, Szerman N, Martinez MI. Risk of serious cardiovascular problems with medications for attention-deficit hyperactivity disorder. CNS Drugs. 2013;27(1):15–30.
Sayer GR, McGough JJ, Levitt J, Cowen J, Sturm A, Castelo E, et al. Acute and long-term cardiovascular effects of stimulant, guanfacine, and combination therapy for attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2016;26(10):88–98.
Schelleman H, Bilker WB, Kimmel SE, Daniel GW, Newcomb C, Guevara JP, et al. Amphetamines, atomoxetine and the risk of serious cardiovascular events in adults. PLoS ONE. 2013;8(1):e52991.
Henderson TA, Hartman K. Aggression, mania, and hypomania induction associated with atomoxetine. Pediatrics. 2004;114(3):895–6.
Horrigan JP, Barnhill LJ. Guanfacine and secondary mania in children. J Affect Disord. 1999;54(3):309–14.
Bangs ME, Tauscher-Wisniewski S, Polzer J, Zhang S, Acharya N, Desaiah D, et al. Meta-analysis of suicide-related behavior events in patients treated with atomoxetine. J Am Acad Child Adolesc Psychiatry. 2008;47(2):209–18.
Greenwood DT. Viloxazine and neurotransmitter function. Adv Biochem Psychopharmacol. 1982;31:287–300.
Acknowledgements
Editorial assistance (proofreading, formatting, and medical writing support) was provided by IMPRINT Science (New York, NY, USA), and funded by Supernus Pharmaceuticals, Inc. We thank Rick Nullmeier for the research support, and Alisa Kosheleff for her help with the writing and editing of the article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This review was funded by Supernus Pharmaceuticals, Inc. The open access fee was provided by Supernus Pharmaceuticals, Inc.
Conflict of interest
Azmi F. Nasser, Stefan Schwabe, Chungping Yu, Jennie Garcia-Olivares, and Welton O’Neal are employees of Supernus Pharmaceuticals, Inc. At the time of this work, Shawn A. Candler was an employee of Supernus Pharmaceuticals, Inc. Robert L. Findling receives or has received research support, acted as a consultant and/or has received honoraria from Acadia, Adamas, Aevi, Afecta, Akili, Alkermes, Allergan, American Academy of Child & Adolescent Psychiatry, American Psychiatric Press, Arbor, Axsome, Daiichi-Sankyo, Emelex, Gedeon Richter, Genentech, Idorsia, Intra-Cellular Therapies, Kempharm, Luminopia, Lundbeck, MedAvante-ProPhase, Merck, MJH Life Sciences, National Institutes of Health, Neurim, Otsuka, PaxMedica, PCORI, Pfizer, Physicians Postgraduate Press, Q BioMed, Receptor Life Sciences, Roche, Sage, Signant Health, Sunovion, Supernus Pharmaceuticals, Syneos, Syneurx, Takeda, Teva, Tris, and Validus. In the past year, Jeffrey H. Newcorn is/has been an advisor and/or consultant for Adlon Therapeutics, Arbor, Corium, Eisai, Medice, Myriad Neuroscience, NLS, OnDosis, Rhodes, Shire/Takeda, and Supernus. He has received research support from the National Institute on Drug Abuse, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Otsuka, Shire, and Supernus. He also has received speaker fees from Shire/Takeda for disease-state presentations, and served as a consultant for the US National Football League.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author contributions
Conceptualization: RLF, JHN, SAC. Literature search: RLF, SAC, AFN, SS, CY, JGO, WON, JHN. Original draft preparation: RLF, SAC, AFN, SS, CY, JGO, WON, JHN. Reviewing and editing: RLF, JHN, SAC.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Findling, R.L., Candler, S.A., Nasser, A.F. et al. Viloxazine in the Management of CNS Disorders: A Historical Overview and Current Status. CNS Drugs 35, 643–653 (2021). https://doi.org/10.1007/s40263-021-00825-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00825-w