
Abstract
Transient expression systems in mesophyll protoplasts have been utilised in many plant species as an indispensable tool for gene function analysis and efficacious genome editing constructs. However, such a system has not been developed in Cannabis due to the recalcitrant nature of the plant to tissue culture as well as its illegal status for many years. In this study, young expanding leaves from aseptic in vitro Cannabis explants were used for protoplast isolation. Factorial designs were used to optimise variables in viable protoplast isolation and transient expression of GFP, with a range analyses performed to determine, and quantify, significantly impacting variables. Viable protoplast yields as high as 5.7 × 106 were achieved with 2.5% (w/v) Cellulase R-10, 0.3% (w/v) Macerozyme R-10 and 0.7 M mannitol, incubated for 16 h. As indicated by the transient expression of GFP, efficiency reached 23.2% with 30 μg plasmid, 50% PEG, 1 × 106 protoplasts and a transfection duration of 20 min. Application of the optimised protocol for protoplast isolation was successfully evaluated on three subsequent unrelated genotypes to highlight the robustness and broad applicability of the developed technique.
Similar content being viewed by others

Introduction
Cannabis sativa L. (Cannabis) is a highly polymorphic, wind pollinated herb originating in China where evidence of its cultivation dates back to 4000 bc (Zuardi 2006). Recent interest in the medicinal properties of phytocannabinoids produced by Cannabis has led to increased legalisation around the world, along with a growing medicinal industry (ProCon.org 2021). Recently, chromosomal resolution of the Cannabis genome was published along with other genetic resources for identification of important cannabinoid biosynthesis genes (Grassa et al. 2018; Laverty et al. 2019). Next generation sequencing has also started to unravel the complexity of the Cannabis genome and transcriptome atlas (Braich et al. 2019). Progress in the genetic tools for manipulation and analysis of genes have prompted research into Cannabis, but currently, Cannabis remains a recalcitrant species to deliver biotechnology tools to. Transgenic hairy root cultures of Cannabis have been performed previously using agroinfiltration of vectors with the GUS reporter gene (Wahby et al. 2013). However, hairy root cultures only offer the accumulation of metabolites within the root structures (Gurunani et al. 2015) making this transformational technique unsuitable for biotechnological applications seeking phytocannabinoids, which accumulate in the female floral tissues. Transformation of hemp callus cultures with a foreign Escherichia coli gene, manA, has previously been successful using Agrobacterium tumefaciens with a success rate of 31% (Feeney and Punja 2003). PCR analysis of callus cultures confirmed stable gene integration with up to four T-DNA copies being integrated into the genome; however, plantlet regeneration was unsuccessful.
With the recent release of the chromosomal assembly and high-density linkage map of the Cannabis genome (Grassa et al. 2018; Laverty et al. 2019), identification of genes involved in cannabinoid biosynthesis is now relatively straightforward, with benchmark standards being developed for genetic engineering of these genes (Matchett-Oates et al. 2020.). However, to date, no procedures for the evaluation of genes through functional screening exist, due to the recalcitrant nature of Cannabis in vitro and the difficulty in obtaining transgenic explants (Feeney and Punja 2017). In addition, the illegal status of cannabis during the time in which biotechnology has emerged and has caused Cannabis to fall behind in genetic improvement studies. Genetic transformation is used for the study of gene function and genetic improvement in plants. Protocols for many non-model species, in which regeneration of transgenic plants has not been achieved, have been developed for screening genome editing constructs. To date, only brief protocol outlines for protoplast isolation have been reported (Jones 1979; Morimoto et al. 2007), with no protoplast transient expression protocols existing for Cannabis. Creating stable transformants is expensive and time consuming making this approach for large scale evaluation of cannabinoid biosynthesis genes limiting. Utilising protoplasts ability to transiently express DNA constructs has allowed for high-throughput transient screening of genes in Arabidopsis (Marion et al. 2008), gene-silencing in barley (Douchkov et al. 2005) and functional analysis of newly isolated genes in tobacco (Fischer and Hain 1995). Such studies are yet to be conducted on Cannabis making the need for an efficient transient expression system in protoplasts of high importance.
Protoplasts are osmotically fragile due to the lack of a cell wall from enzymatic digestion, allowing for the transfer of DNA constructs through the plasma membrane using common methods such as PEG-mediated transfection (Yoo et al. 2007), Agrobacterium infiltration (Clough and Bent 1998) and biolistic bombardment (Vain et al. 1993). Transient gene expression from plant protoplasts has widely been used to study cell death related processes (Chen et al. 2015), developmental studies (Sheen 2001), subcellular localisation of proteins (Su et al. 2010) and expression of foreign genes (Zhang et al. 2011). During isolation, optimum conditions such as osmotic balance, enzyme concentration and digestion time need to be established by optimising all variables contributing to yield and viability. The use of reporter genes in transient assays is often used due to the non-toxic nature, stability and ease of detection allowing for protein localisation and interactions studies (Leffel et al. 1997) making protoplasts an important tool in gene analysis.
The evaluation of transiently expressed shared terpene biosynthesis genes between Cannabis, and other species has been reported (Reed and Osbourn 2018; Smirnoff 2019). However, Cannabis contains a unique set of enzymes in the cannabinoid biosynthesis pathway, which synthesise phytocannabinoids that are only produced in this species. The elucidation of the cannabinoid biosynthesis genes in the draft Cannabis genome provided the first complete look into the multiple pathways involved (Van Bakel et al. 2011). Identification and characterisation of the first unique enzyme in the cannabinoid pathway, OLS, have evolved from identification of an unknown polyketide synthase (Raharjo et al. 2004). Initially characterised as a novel polyketide synthase-olivetol synthase (Taura et al. 2009), later, the mechanism for olivetol production requiring olivetol acid cyclase, a DABB protein (Gagne et al. 2012), was identified and correctly characterised. Similarly, identification and functional studies on the oxidocyclases, tetrahydrocannabinolic acid synthase (THCAS) and cannabidiolic acid synthase (CBDAS) have evolved as technological advancements into molecular cloning techniques, and DNA sequencing has improved. Tetrahydrocannabinol (THC), one of the major constituents in Cannabis, was originally presumed to be formed by the isomerisation of cannabidiolic acid (CBDA) (Shoyama et al. 1975). Protein sequencing and PCR cloning of tetrahydrocannabinolic acid (THCA) confirmed that THC was produced from cannabigerolic acid (CBGA) by THCAS (Sirikantaramas et al. 2004). Identification of CBDAS (Taura et al. 1996) occurred shortly after with a similar strategy used for gene identification (Taura et al. 2007).
Genetic engineering of Cannabis offers the opportunity to produce higher levels of cannabinoids with tailor-made chemical profiles for medicinal applications. However, the recalcitrant nature of Cannabis and the absence of methods to efficiently generate transgenic plants currently makes transient expression systems important to understand the molecular regulatory mechanisms responsible for cannabinoid biosynthesis, which remain largely unknown. Prior to this report, a transient expression system in Cannabis protoplasts did not exist, making the characterisation of unique gene function speculative. In this present study, an efficient protoplast isolation and transient expression system using Cannabis leaf mesophyll protoplasts are reported. Optimisation to obtain a high yield of viable protoplasts and PEG-mediated transfection of the protoplasts using fluorescent reporter genes is also reported. Furthermore, due to the outbreeding highly heterozygous nature of Cannabis, strain-dependent response variation is expected; this protoplast isolation and transient expression systems is shown to be suitable to many strains, highlighting the applicability for future protoplast and transient expression studies.
Materials and Methods
Plant Material and Expression Vectors
All research was performed under Medicinal Cannabis Research Licence (RL011/18) and Permit (RL01118P4) issued through the Department of Health (DoH), Office of Drug Control (ODC) Australia. A specific genotype (C. sativa) with high THC content (25%) was used for optimisation within this study. Aseptic plantlets were derived from apical meristems of mature vegetative mother plants, which were sterilised by a 1-min 80% ethanol wash, washed three times with sterile water, disinfected with 10% (v/v) sodium hypochlorite (White King, Melbourne, Australia) solution containing 4.5% active chlorine for 15 min and finally washed three times with sterile water. Apical meristems were cultured in root induction media containing ½ Murashige and Skoog salts and vitamins (MS; Murashige and Skoog 1962) (Duchefa Biochemie, Haarlem, The Netherlands), 1% sucrose (w/v) (Sigma-Aldrich, St. Louis, MO), 1% agar (w/v) (Duchefa Biochemie) and 1 mg L−1 Indole-3-butyric acid (IBA) (Sigma-Aldrich) adjusted to pH 5.7 prior to autoclaving at 121°C for 15 min. The apical meristems were cultured at 26°C under lighting of 74 μmol m−2 s−1 supplied by fluorescent lamps for 18 h per day. The green fluorescent protein (GFP) expression vector pDONR221-GFP was constructed from pDONR221 (Invitrogen, Carlsbad, CA) through BP Clonase reaction by the insertion of the attB1-CaMV35D-p_turboGFP(D)_AtuNos-t-attB2 cassette (Supplementary Information).
Protoplast Isolation, Purification and Quantification
Protoplasts were isolated from well rooted, 1- to 2 mo-old plantlets with young leaves cut into 0.5- to 1.0-mm-thin strips (Fig. 1a–d) and incubated in a Petri dish containing digestion media comprising of 1, 2 and 2.5% (w/v) Cellulase Onozuka R-10 (Yakult Honsha Co., Ltd., Tokyo, Japan); 0.3, 0.4 and 0.5% (w/v) Macerozyme R-10 (Yakult Honsha Co.); 0.3, 0.5 and 0.7 M mannitol (Sigma-Aldrich, St Louis, MO); 20 mM MES (Sigma-Aldrich, St Louis, MO); 20 mM KCl (Sigma-Aldrich, St Louis, MO); and 10 mM CaCl2 (Sigma-Aldrich), pH adjusted to 5.8 and filter-sterilised using a 0.22-μm filter (Sigma-Aldrich). Leaf strips were incubated in the dark at 28°C without agitation for between 8 and 24 h with each digestion replicated three times. Following digestion, the cellular suspension was mechanically filtrated through a 70-μm mesh (Corning, NY) into a sterile 50-mL polypropylene centrifuge tube (Corning, NY) and centrifuged at 700×g for 10 min. Following centrifugation, the resulting supernatant was discarded, and the pellet resuspended with 3 mL of W5 (5 mM glucose, 5 mM KCl, 10 mM MES, 125 mM CaCl2, 154 mM NaCl, pH 5.8), transferred to a 15-mL round bottomed tube and 3 mL 20% (w/v) sucrose added and centrifuged again. Protoplasts were collected from the interphase (Fig. 1e) into a fresh 15-mL round bottom tube and 3 mL W5 added and centrifuged again with the supernatant removed. Finally, the pellet was resuspended in 1 mL W5, and 100 μL of the resuspended protoplasts were diluted with 0.5 M Evans Blue, and approximately 50 to 200 protoplasts were counted using a haemocytometer under a light microscope. The viability of the protoplasts was calculated by (viable protoplasts/total number of protoplasts) × 100%.

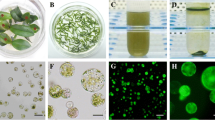
(a) In vitro 1- to 2-mo-old rooted explants grown on Murashige and Skoog medium containing 1 mg L−1 indole-3-butyric acid suitable for protoplast isolation. Scale bar = 2 cm. (b) Healthy Cannabis sativa L. leaves suitable for protoplast isolation. Scale bar = 2 cm. (c) Cannabis sativa L. leaves sliced into 0.5- to 1-mm-thin strips with a fresh razor blade and placed in media. Scale bar = 2 cm. (d) Cannabis sativa L. 16 h post digestion in the dark at 28°C without shaking. Scale bar = 2 cm. (e) Purified Cannabis sativa L. protoplast interphase after density gradient centrifugation.
PEG-Mediated Transfection
Isolated protoplasts were divided into aliquots of 5 × 105 or 1 × 106 and centrifuged at 700×g for 10 min with the supernatant removed. PEG4000 solutions (20%, 30%, 40% and 50% (w/v)) (Sigma-Aldrich) were prepared by dissolving in ddH20 containing 0.1 M Ca(NO3)2 4H2O and 0.4 M Mannitol (Sigma-Aldrich). A volume of 100 μL transformation buffer (15 mM MgCl2, 0.5 M Mannitol, 0.1% (w/v) MES, pH 5.7) was added to the protoplasts and gently mixed. Plasmid concentrations (5, 10 and 30 μg) in 60 μL of ddH2O were added to the protoplast solution and gently mixed followed by addition of 150 μL of warmed (42°C) PEG4000 solutions. The protoplast solution was mixed gently and incubated in the dark at room temperature (22 ± 1°C) for between 10 and 60 min. The transfection reaction was stopped by the addition of 5 mL W5 dropwise, followed by a further 5 mL in a gentle stream. The protoplast mixture was centrifuged once more at 700×g for 10 min, removing the supernatant and the addition of 150 μL W5 followed by incubation in the dark at room temperature for 48 h. The expression of GFP was observed under a fluorescence microscope (OLYMPUS CKX53, Tokyo, Japan) (excitation emission wavelengths 470 to 490 nm, 510 nm).
FACS Analysis
The protoplasts were analysed using InfluxTM FACS instrument (BD Biosciences, Franklin Lakes, NJ) fitted with a 200-μm nozzle using W5 buffer as a sheath fluid. The sheath pressure was set at 4 psi, and the sample pressure was set at 5 psi. A 466-nm Coherent Sapphire Solid state laser was used for excitation, and emission was measured using a 517/18 nm band-pass filter for GFP. The photomultiplier tube voltage was set at 16.41 V for forward scatter, 20.59 V for side scatter and 40.37 V for GFP. The threshold value for event detection was set at 0.3 on side scattering. For analysis, a gate was set using PEG-transfected control protoplasts. Frequency of GFP expressing cells was recorded, and data was processed using BD FACSTM Software v1.0.0.650 (BD Biosciences).
Statistical Analysis
Experimental data was statistically analysed using Minitab 19 Statistical Software (Version 19, State College, PA) and R Studio (Version 1.1.453, RStudio, Inc., Boston, MA).
Results
Mesophyll Protoplast Isolation From Cannabis Leaf Tissue
Initially, protoplasts were generated from rapidly expanding leaves from 1- to 2-mo-old in vitro plantlets that were used as the source material (Fig. 1a, b). Briefly, plantlets cultured at 26°C in ½ MS medium with 5 μM IBA were used for mesophyll protoplast isolation. One gram of young leaf material was cut into 0.5- to 1-mm strips and immediately transferred into digestion medium in the dark without agitation at 28°C for 8 to 24 h (Fig. 1a–d). Protoplasts were collected through a 70-μm nylon mesh, centrifuged and washed as previously described.
Single Factor Effect on Mesophyll Protoplast Isolation
To optimise mesophyll protoplast isolation, enzyme and mannitol concentrations and enzymatic digestion time were adjusted. Mesophyll protoplasts were isolated from young, expanding leaves with variable digestion parameters, outlined in Table 1. Total protoplast yields ranged from 1.2 × 106 to 9.2 × 106 per gram of fresh leaf weight with the highest yield obtained from treatment 7. Protoplast viability ranged from 39 to 79%, with the highest viability obtained from treatment 9. An L9 (34) orthogonal test (Table 1) was designed to identify statistically significant variables to optimise protoplast isolation, with a range analysis performed to predict the optimal combination of the variables. The range analysis of the means between the factors and levels (Table 2), calculated Delta scores (mean score of the highest range minus the lowest mean range) for enzymolysis time (R 2.21) and Cellulase R-10 (R 1.56) significantly influencing protoplast isolation, with Macerozyme R-10 (R 1.44) and mannitol (R 1.23) proving less significant. Analysis of each level calculated response (k1–3) from the range analysis (Table 2), the Duncans multiple range test (Duncans) assigned the best possible combination of each factor as A3:B1:C3:D2. The calculated theoretical best combination was already performed in treatment 7, with an average yield of 5.7 × 106 viable protoplasts.
Increasing Cellulase-R10 concentration significantly increased protoplast yield within multilevel factors (Fig. 3), from 1.31 (1%) to 2.86 (2.5%) × 106 viable protoplasts. This trend, of increasing concentrations greatly increasing yields, was not observed for the other variables, where applicable. Macerozyme R-10 effected protoplast yields significantly at 0.3% producing 2.93 × 106 viable protoplasts, whereas at 0.4% and 0.5%, this decreased to 1.50 × 106 and 2.14 × 106 viable protoplasts, respectively. A concentration of 0.7 M mannitol was more substantial in producing 2.89 × 106 viable protoplasts, whereas 0.3 M produced 2.01 × 106, which performed greater than 0.5 M, which produced 1.67 × 106 viable protoplasts (Table 3). Enzymolysis time of 16 h produced a two-fold increase in viable protoplasts, with 3.61 × 106 compared to a shorter digestion time of 8 h (1.58 × 106) or a longer digestion time of 24 h (1.39 × 106).
Transient Transfection of Cannabis Mesophyll Protoplasts
To establish the first transient expression system, pDONR221-GFP was used to study the effects of plasmid, PEG and protoplast concentration, including incubation time on transfection efficiency. The transfection efficiency reached 23.2% in treatment 12 (30 μg plasmid, 50% PEG (w/v), 20 min of incubation and 1 × 106 protoplasts), with the lowest recorded transfection rate of 5.78% in treatment 5 (10 μg plasmid, 20% PEG (w/v), 20 min incubation time and 1 × 106 protoplasts).
Single Factor Effect on Mesophyll Protoplast Transfection
Factors with multiple levels across variables (Table 3) were designed to optimise plasmid concentration, PEG concentration (w/v), transfection time and protoplast concentration variables in transfection efficiency. Mesophyll protoplasts were transfected using the PEG-mediated transfection protocol described previously (Fig. 2) with transfection efficiency ranging from 5.78 to 23.20% with the highest average transfection rate, 23.20%, obtained from treatment 12. Within multilevel factors, increasing plasmid concentration greatly increased protoplast transfection within efficiencies between 10 and 30 μg (11.11 to 17.29%). Increasing PEG concentration from 20 to 50% saw increases in transfection efficiency, with 50% achieving 16.23% on average, compared to 20% PEG (w/v), which saw 10.93% transfection efficiency. Incubation time of 30 min gave the highest average transient efficiency of 15.82%. Similarly, a 20-min incubation resulted in 16.61% transfection efficiency. Incubation of 10 and 60 min saw a reduction of transfection efficiency, 12.02% and 13.25%, respectively. Protoplast density of 5 × 105 achieved higher transient efficiencies on average compared to 1 × 106, with 14.63% and 11.72% efficiency, respectively. From the range analysis of the means (Table 4), the calculated Delta values of plasmid concentration (R 6.18%) and PEG concentration (R 5.3%) greatly influence transfection efficiency, with transfection time (R 3.8%) and protoplasts density (R 2.91%) proving less significant.

(a) Visualisation of Cannabis sativa L. protoplasts under brightfield microscopy. Scale bar = 200 μm. (b) Visualisation of Cannabis sativa L. protoplasts under fluorescence microscopy with GFP filter set. Scale bar = 50 μm. (c) Isolated protoplasts under fluorescence microscopy with GFP filter set. Scale bar = 200 μm. (d) Isolated Cannabis sativa L. protoplasts under fluorescence microscopy with GFP filter set. Scale bar = 100 μm.
Discussion
Protoplasts offer a versatile experimental system, with transient expression systems widely applied in Arabidopsis (Yoo et al. 2007), rice (Wang et al. 1988) and tobacco (Töpfer et al. 1988) to analyse gene expression and function and to deliver genetic improvements in plants. Crucial for the development of an efficient transient expression system for genome editing constructs is the routine generation of high-quality protoplasts and robust transfection protocols. Only two reports of isolated Cannabis protoplasts exist; however, the reports briefly discuss the methods for protoplast isolation with no data on protoplast yield and viability, or the data collection was not described in detail. Morimoto et al. (2007) digested Cannabis leaves with 1% Cellulase R-10, 0.2% Macerozyme R-10, 0.1% pectolyase Y-23 and 0.4 M mannitol at 30°C for 4 h with gentle agitation; however, no yield or viability data was given. Jones (1979) explored a range of enzyme combinations to produce protoplast from young and old leaf tissue, as well as callus, with concentrations between 1 × 103 and 1 × 105 reported. Protoplast isolation from the closely related species Humulus lupulus (hops) has previously been reported from cell suspension cultures. Several cell wall digesting enzyme mixtures were trialled with varying Cellulysin and Driselase concentrations in 0.4 M mannitol osmoticum, with the highest yield of protoplasts (9.3 to 9.9 × 106 per gFW) obtained from 2% Cellulysin and 1% Driselase with 99 to 100% viability reported (Furze et al. 1987). Heale et al. (Heale et al. 1989) reported leaf mesophyll isolation from the hop cultivar, Challenger, by firstly removing the epidermis followed by incubating in 4% Cellulase, 0.3% Macerozyme and 2% hemicellulose for 7 h on an orbital shaker protoplast yields between 4 and 7 × 105 mL−1 were achieved with viability ranging from 80 to 90% under optimal conditions. An efficient protocol for transient expression of LUC activity in rose (another member of the Order Rosales with Cannabis) leaves using Agrobacterium has also been reported (Lu et al. 2017). Recently, transient expression of β-glucuronidase (GUS) and GFP in a range of agroinfiltrated organs and tissues has been reported in Cannabis, with the protocol being optimised for hemp cultivars (Deguchi et al. 2020).
The development of an efficient protoplast transient expression system to screen genome editing constructs requires established protocols for the isolation of viable protoplasts and a competent transfection workflow, which until now have not been reported for Cannabis. With several factors affecting yield and viability of protoplasts and transfection efficiencies, this development of a routine system allows for significant advances to be made towards understanding and improving Cannabis cultivars. The method described here investigates the effect each variable has on protoplast isolation and transfection, with each being analysed for their significance.
To optimise protoplast isolation, Cellulase-R10, Macerozyme-R10, mannitol concentrations and digestion times were adjusted (Table 1). Under the optimum conditions, viable Cannabis leaf mesophyll protoplasts reached 5.7 × 106 protoplasts g FW−1 (Table 1). The increase in Cellulase concentration to 2.5% (w/v) showed significantly increased yields, 2.88 × 106, from the range analysis. Although 2.5% (w/v) Cellulase concentration was the highest tested, increasing levels past a saturation point has been shown to decrease protoplast yields in tobacco (Kuriakose et al. 2012) and Magnolia (Shen et al. 2017). The optimal Macerozyme concentration was determined to be 0.3% (w/v), whilst an increase to 0.4% saw more than a 50% drop in protoplast recovery to 1.50 × 106 calculated from the range analysis. For both enzymes, the increase in concentration at which viable protoplast yield decreases is presumably due to the influence those enzymes have on the membrane integrity. The application of the optimised protoplast isolation protocol to verify robustness on Cannbio-2, the alternative high THC strain and a high CBD strain produced a significant yield of viable protoplasts for subsequent transient expression experiments, demonstrating the robustness and versatility of the protocol described. The protocol produced similar protoplast viability, with Cannbio-2 averaging 79%, equalling to the best performing treatment 9, and the high CBD strain producing higher levels of viability with 82% (Table 5). Although viable protoplast yield from these cultivars is proportionately fewer, the protocol has been optimised for a specific high yielding THC strain, with the calculated optimal conditions proven to be an advisory starting point for further optimisation on any chosen cultivar (Table 5).
Protoplasts lack of cell walls that require a stabilized environment, which is controlled by the osmotic gradient for proper osmolarity to sustain viable protoplasts (Sain et al. 2017). Mannitol is frequently used for it is inert metabolically and slowly diffuses through the cellular membrane (Chawla 2011). Mannitol concentration of 0.7 M resulted in the highest yield, calculated by the range analysis, of 2.91 × 106, with lower concentrations of 0.3 M and 0.5 M yielding 2.02 × 106 and 1.67 × 106, respectively. Similar to enzyme concentration, increasing mannitol concentration, thus causing an imbalance in the osmoticum, decreases protoplast yield, as is seen with wheat (Jia et al. 2016) and pineapple (Priyadarshani et al. 2018). Enzymolysis time significantly affected viable protoplast yield, with digestions of 24-h yielding as low as 39% viability in treatment 8, which most certainly caused by over digestion of the cell walls. Comparatively, 8 and 16 h were determined to yield significantly higher levels of viability, with 16 h shown to result in a larger concentration of viable protoplasts (Fig. 3) and the best digestion time for the release of viable protoplasts without over digestion.

Effects of different factors on viable Cannabis sativa L. protoplast isolation yield. Bars represent standard errors (SE). Statistical significance was determined using a one-way ANOVA test (*p < 0.05, **p < 0.01).
The uptake of DNA through the plasma membrane of protoplasts for transient expression studies requires considerable concentrations of PEG acting in tandem with divalent cations (Maas and Werr 1989). With increasing concentrations of PEG, DNA hydration reduces causing structural changes and thus reducing the transfection efficiency (Saenger et al. 1986). In Cannabis protoplasts, transient expression efficiency at the highest investigated PEG concentration (50%) achieved 16.23% (Fig. 4), which is a significantly higher concentration of PEG than has been previously reported in rice (Page et al. 2019) or pea (Nicolaisen and Poulsen 1993). Results show that increasing plasmid concentration from 5 to 10 μg achieved similar transfection efficiencies, 11.11% and 11.13%, respectively (Table 3). These results are inconsistent with findings in pepper (Jeon et al. 2007), pineapple (Priyadarshani et al. 2018) and Phaseolus (Nanjareddy et al. 2016) in which there is an approximate doubling in relevant transient expression efficiencies between these two concentrations. This suggests that Cannabis protoplasts require higher concentrations of plasmid to achieve increased levels of expression. Intermediate exposure time to high concentrations on PEG was shown to increase transfection efficiencies (Fig. 4), with the optimal exposure time determined to be 30 min, achieving 15.82%. Increasing the incubation time to 60 min saw a sharp decrease in transfection efficiency, falling to 12.02%. The increased exposure to the high concentrations of PEG resulting in lower transfection efficiencies is expected due to DNA becoming less hydrated, with similar results found in carrot, rapeseed and soybean (Rasmussen and Rasmussen 1993). This protocol has been optimised for this particular cultivar (Fig. 4). This protocol provides a relevant starting point for optimisation regardless of genetics. This level of efficiency in transfection allows for cellular studies, including genome editing using CRISPR and ZFNs.

Effects of different factors on Cannabis sativa L. protoplast transfection efficiency. Bars represent standard errors (SE). Statistical significance was determined using a one-way ANOVA test (*p < 0.05).
Conclusion
The method described here is the first reported for the transient expression of heterologous genes in Cannabis protoplasts. The variables involved in protoplast isolation were verified on three cultivars with varying cannabinoid content for protocol robustness. The transfection protocol was optimised for a high THC yielding strain within mesophyll protoplasts. This method can be easily adapted for transient expression studies using CRISPR/Cas-9, protein-protein interaction or other investigations in Cannabis where a transient gene expression system is desired.
References
Braich S, Ballie R, Jewell L, Spangenberg G, Cogan N (2019) Generation of a comprehensive transcriptome atlas and transcriptome dynamics in medicinal cannabis. Sci Rep 9:1–12
Chawla HS (2011) Introduction to plant biotechnology, vol 3. Science Publishers, Enfield New Hampshire
Chen J, Yi Q, Song Q, Gu Y, Zhang J, Hu Y, Liu H, Liu Y, Yu G, Hang Y (2015) A highly efficient maize nucellus protoplast system for transient gene expression and studying programmed cell death-related processes. Plant Cell Rep 34:1239–1251
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Deguchi M, Bogush D, Weeden H, Spuhler Z, Potlakayala S, Kondo T, Zhang Z, Rudrabhatla S (2020) Establishment and optimization of a hemp (Cannabis sativa L.) agroinfiltration system for gene expression and silencing studies. Sci Rep 10:1–11
Douchkov D, Nowara D, Zierold U, Schweizer P (2005) A high-throughput gene-silencing system for the functional assessment of defense-related genes in barley epidermal cells. Mol Plant Microbe Interact 18:755–761
Feeney M, Punja ZK (2003) Tissue culture and Agrobacterium-mediated transformation of hemp (Cannabis sativa L.). In Vitro Cell Dev Biol - Plant 39:578–585
Feeney M, Punja ZK (2017) The role of Agrobacterium-mediated and other gene-transfer technologies in Cannabis research and product development, Chandra, S., Lata, H., ElSohly, M. (eds), Cannabis sativa L.-Botany and Biotechnology, vol 1, Springer, Cham. pp. 343–363
Fischer R, Hain R (1995) Tobacco protoplast transformation and use for functional analysis of newly isolated genes and gene constructs. In: Galbraith DW, Bourque DP, Bohnert HJ (eds) Methods in Cell Biology, vol 50. Elsvier, pp 401–410
Furze JM, Rhodes MJC, Robins RJ (1987) The use of agarose bead culture for the regeneration of single cell-derived colonies from protoplasts isolated from suspension cultures of Humulus lupulus. Plant Cell Tiss Org Cult 8:17–25
Gagne SJ, Stout J, Liu E, Boubakir Z, Clark S, Page J (2012) Identification of olivetolic acid cyclase from Cannabis sativa reveals a unique catalytic route to plant polyketides. Proc Natl Acad Sci 109:12811–12816
Grassa CJ, Wenger JP, Dabney C, Poplawski SG, Motley ST, Michael TP, Schwartz CJ, Weiblen GD (2018) A complete Cannabis chromosome assembly and adaptive admixture for elevated cannabidiol (CBD) content. bioRxiv:458083. https://doi.org/10.1101/458083
Gurunani SG, Gholse YN, Yeole MP, Chaple DR (2015) Hairy root culture: an experimental system for secondary metabolite production. Res J Pharm Tech 8:728–730
Heale JB, Legg T, Connell S (1989) Humulus lupulus L.(Hop): In vitro culture; Attempted production of bittering components and novel disease resistance. In: Bajaj YPS (ed) Medicinal and Aromatic Plants II, Biotechnology in Agriculture and Forestry, vol 7. Springer, Berlin, Heidelberg, pp 264–285
Jeon JM, Ahn NY, Son BH, Kim CY, Han C, Kim G, Gal SW, Lee S (2007) Efficient transient expression and transformation of PEG-mediated gene uptake into mesophyll protoplasts of pepper (Capsicum annuum L.). Plant Cell Tiss Org Cult 88:225–232
Jia X, Zhang X, Qu J, Han R (2016) Optimization conditions of wheat mesophyll protoplast isolation. Agri Sci 7:850–858
Jones RL (1979) Cell culture, protoplast isolation, and cell fusion of Cannabis Sativa L.: evaluation of chilling preventative chemicals and quality control of bananas in the tropics, University of Houston Central Campus, M. Sc. Thesis, DOI: 10.13140/RG.2.2.19104.43527
Kuriakose B, Du Toit ES, Jordaan A (2012) Transient gene expression assays in rose tissues using a Bio-Rad Helios hand-held gene gun. S Afr J Bot 78:307–311
Laverty KU, Stout J, Sullivan MJ, Shah H, Gill N, Holbrook L, Deikus G, Sebra R, Hughes TR, Page JE, van Bakel H (2019) A physical and genetic map of Cannabis sativa identifies extensive rearrangements at the THC/CBD acid synthase loci. Genome Res 29:146–156
Leffel SM, Mabon SA, Stewart CN Jr (1997) Applications of green fluorescent protein in plants. Biotechniques 23:912–918
Lu J, Bai M, Ren H, Liu J, Wang C (2017) An efficient transient expression system for gene function analysis in rose. Plant Method 13:116
Maas C, Werr W (1989) Mechanism and optimized conditions for PEG mediated DNA transfection into plant protoplasts. Plant Cell Rep 8:148–151
Marion J, Bach L, Bellec Y, Meyer C, Gissot L, Faure J (2008) Systematic analysis of protein subcellular localization and interaction using high-throughput transient transformation of Arabidopsis seedlings. Plant J 56:169–179
Matchett-Oates L, Braich S, Spangenberg G, Rochfort S, Cogan N (2020) In silico enablinginformed design for genome editing in medicinal cannabis, gene families and variant characterisation. Plant Methods. IN REVIEW
Morimoto S, Tanaka Y, Sasaki K, Tanaka H, Fukamizu T, Shpyama Y, Shoyama Y, Taura F (2007) Identification and characterization of cannabinoids that induce cell death through mitochondrial permeability transition in Cannabis leaf cells. J Biol Chem 282:20739–20751
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nanjareddy K, Arthikala M, Blanco L, Arellano E, Lara M (2016) Protoplast isolation, transient transformation of leaf mesophyll protoplasts and improved Agrobacterium-mediated leaf disc infiltration of Phaseolus vulgaris: tools for rapid gene expression analysis. BMC Biotechnol 16:53
Nicolaisen M, Poulsen GB (1993) Optimization of polyethylene glycol mediated transient gene expression in pea protoplasts. Plant Cell Tiss Org Cult 35:93–97
Page MT, Parry MAJ, Carmo-Silva E (2019) A high-throughput transient expression system for rice. Plant Cell Environ 42:2057–2064
Priyadarshani S, Hu B, Li W, Ali H, Jia H, Zhao L, Ojolo SP, Azam SM, Xiong J, Yan M, Rahman ZU, Wu Q, Qin Y (2018) Simple protoplast isolation system for gene expression and protein interaction studies in pineapple (Ananas comosus L.). Plant Method 14:95
ProCon.org (2021) Legal Medical Marijuana States and DC. Available at: https://medicalmarijuana.procon.org/legal-medical-marijuana-states-and-dc/ (Accessed: 24 March 2021).
Raharjo TJ, Cheng W, Verberne MC, Peltenburg-Looman AMG, Linthorst HJM, Verpoorte R (2004) Cloning and over-expression of a cDNA encoding a polyketide synthase from Cannabis sativa. Plant Physiol and Biochem 42:291–297
Rasmussen JO, Rasmussen OS (1993) PEG mediated DNA uptake and transient GUS expression in carrot, rapeseed and soybean protoplasts. Plant Sci 89:199–207
Reed J, Osbourn A (2018) Engineering terpenoid production through transient expression in Nicotiana benthamiana. Plant Cell Rep 37:1431–1441
Saenger W, Hunter WN, Kennard O (1986) DNA conformation is determined by economics in the hydration of phosphate groups. Nature 324:385–388
Sain PM, Rodríguez S, Disalvo EA (2017) Effect of osmotic stress on the surface properties of protoplasts as measured by merocyanine 540. Soc Plant Res 30:75–80
Sheen J (2001) Signal transduction in maize and Arabidopsis mesophyll protoplasts. Plant Physiol 127:1466–1475
Shen Y, Meng D, McGrouther K, Zhang J, Cheng L (2017) Efficient isolation of Magnolia protoplasts and the application to subcellular localization of MdeHSF1. Plant Method 13:44
Shoyama Y, Yagi M, Nishioka I, Yamauchi T (1975) Biosynthesis of cannabinoid acids. Phytochem 14:2189–2192
Sirikantaramas S, Morimoto S, Shoyama Y, Ishikawa Y, Wada Y, Shoyama Y, Taura F (2004) The gene controlling marijuana psychoactivity molecular cloning and heterologous expression of Delta1-tetrahydrocannabinolic-acid synthase from Cannabis sativa. J Biol Chem 279:39767–39774
Smirnoff N (2019) Engineering of metabolic pathways using synthetic enzyme complexes. Plant Physiol 179:918–928
Su C, Wang Y, Hsieh T, Lu C, Tseng T, Yu S (2010) A novel MYBS3-dependent pathway confers cold tolerance in rice. Plant Physiol 153:145–158
Taura F, Morimoto S, Shoyama Y (1996) Purification and characterization of cannabidiolic-acid synthase from Cannabis sativa L. Biochemical analysis of a novel enzyme that catalyzes the oxidocyclization of cannabigerolic acid to cannabidiolic acid. J Bio Chem 271:17411–17416
Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y, Morimoto S (2007) Cannabidiolic-acid synthase, the chemotype-determining enzyme in the fiber-type Cannabis sativa. FEBS Lett 581:2929–2934
Taura F, Tanaka S, Taguchi C, Fukamizu T, Tanaka H, Shoyama Y, Morimoto S (2009) Characterization of olivetol synthase, a polyketide synthase putatively involved in cannabinoid biosynthetic pathway. FEBS Lett 583:2061–2066
Töpfer R, Pröls M, Schell J, Steinbiß HH (1988) Transient gene expression in tobacco protoplasts: II. Comparison of the reporter gene systems for CAT, NPT II, and GUS. Plant Cell Rep 7:225–228
Vain P, McMullen MD, Finer JJ (1993) Osmotic treatment enhances particle bombardment-mediated transient and stable transformation of maize. Plant Cell Rep 12:84–88
Van Bakel H, Stout J, Cote A, Tallon C, Sharpe A, Hughes T, Page J (2011) The draft genome and transcriptome of Cannabis sativa. Genome Biol 12:R102
Wahby I, Caba JM, Ligero F (2013) Agrobacterium infection of hemp (Cannabis sativa L.): establishment of hairy root cultures. J Plant Interact 8:312–320
Wang YC, Klein TM, Fromm M, Cao J, Sanford JC, Wu R (1988) Transient expression of foreign genes in rice, wheat and soybean cells following particle bombardment. Plant Mol Biol 11:433–439
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Prot 2:1565–1572
Zhang Y, Su J, Duan S, Ao Y, Dai J, Liu J, Wang P, Li Y, Liu B, Feng D, Wang J, Wang H (2011) A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Method 7:30–38
Zuardi AW (2006) History of cannabis as a medicine: a review. Braz J Psychiatry 28:153–157
Acknowledgements
Authors acknowledge Canopy Growth Corporation for access to plant material to evaluate the protocols, where applicable. The authors also wish to acknowledge Navdeep Kaur for FACS operation and optimisation.
Funding
Funding was provided by Agriculture Victoria Services and Agriculture Victoria Research.
Author information
Authors and Affiliations
Contributions
LMO and EM in protocol development and optimisation. LMO assembled the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Editor: Ted Klein
Supplementary information
ESM 1
(DOCX 211 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matchett-Oates, L., Mohamaden, E., Spangenberg, G.C. et al. Development of a robust transient expression screening system in protoplasts of Cannabis. In Vitro Cell.Dev.Biol.-Plant 57, 1040–1050 (2021). https://doi.org/10.1007/s11627-021-10178-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-021-10178-0