
Abstract
Protein methylation, a post-translational modification (PTM), is observed in a wide variety of cell types from prokaryotes to eukaryotes. With recent and rapid advancements in epigenetic research, the importance of protein methylation has been highlighted. The methylation of histone proteins that contributes to the epigenetic histone code is not only dynamic but is also finely controlled by histone methyltransferases and demethylases, which are essential for the transcriptional regulation of genes. In addition, many nonhistone proteins are methylated, and these modifications govern a variety of cellular functions, including RNA processing, translation, signal transduction, DNA damage response, and the cell cycle. Recently, the importance of protein arginine methylation, especially in cell cycle regulation and DNA repair processes, has been noted. Since the dysregulation of protein arginine methylation is closely associated with cancer development, protein arginine methyltransferases (PRMTs) have garnered significant interest as novel targets for anticancer drug development. Indeed, several PRMT inhibitors are in phase 1/2 clinical trials. In this review, we discuss the biological functions of PRMTs in cancer and the current development status of PRMT inhibitors in cancer therapy.
Similar content being viewed by others
Introduction
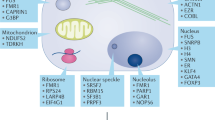
Since the discovery of arginine residue methylation on histone proteins1, protein arginine methylation has been emphasized as an indispensable post-translational modification (PTM) and an epigenetic regulation mechanism2,3. Arginine methylation is catalyzed by a family of enzymes called protein arginine methyltransferases (PRMTs), and nine PRMTs have been identified in mammals to date (Fig. 1a)2,4,5. All PRMTs share four conserved sequence motifs (I, post-I, II, and III) and one THW loop, which compose the S-adenosyl-L-methionine (AdoMet) binding pocket in the tertiary structure6,7. PRMTs transfer a methyl group from the AdoMet molecule to the guanidino group of the arginine residue in substrate proteins8. There are three types of methyl arginine (Fig. 1b): ω-NG-monomethyl arginine (MMA), ω-NG,NG-asymmetric dimethyl arginine (ADMA), and ω-NG,N’G-symmetric dimethyl arginine (SDMA)8. PRMTs are classified into three subgroups based on the type of methyl arginine they produce: Type I PRMTs (PRMT1, 2, 3, 4, 6, and 8) generate MMA and ADMA, Type II PRMTs (PRMT5 and 9) produce MMA and SDMA, and Type III PRMT (PRMT7) produces only MMA7,9.

a The mammalian PRMT family. Nine PRMTs were identified, and these have unique signatures (dark blue lines) with high sequence similarity (a, Motif I: VLD/EVGXGXG; b, Post-I: V/IXG/AXD/E; c, Motif II: F/I/VDI/L/K; d, Motif III: LR/KXXG; e, THW loop). Their enzymatic types and cellular localization are shown. b Types of arginine methylation. The arginine residue has two equivalent nitrogen atoms in its guanidino group. Types I, II, and III PRMTs generate monomethyl arginine (MMA) marks. The subsequent generation of asymmetric dimethyl arginine (ADMA) is catalyzed by type I enzymes (PRMT1, PRMT2, PRMT3, CARM1, PRMT6, and PRMT8), and symmetric dimethyl arginine (SDMA) is produced by type II enzymes (PRMT5 and PRMT9). PRMT7, a type III enzyme, generates only MMA.
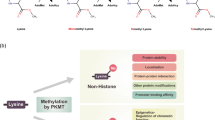
The arginine residue consists of a guanidino group on its side chain, which is protonated and positively charged at physiological pH3,5. The guanidino group forms multiple hydrogen bonds that bind with other interacting proteins or cofactors2,5. Although the methylated arginine residue retains its positive charge, the ability to form hydrogen bonds is reduced, probably affecting the protein-protein interaction. In addition, arginine methylation is very stable compared to that of other PTMs, and hence, its kinetics are less dynamic2,5. PRMTs are associated with many essential cellular processes, including transcription, splicing, translation, signal transduction, DNA damage and repair, and cell cycle regulation (Fig. 2)2,3,4, and the knockout phenotypes of some PRMTs show embryonic or perinatal lethality2,10,11,12, indicating the significance of PRMTs in maintaining functional homeostasis in biological systems. Tissue-specific deletion studies of PRMTs strongly support the supposition that they are involved in cancer and metabolic, immune, neurodegenerative, and muscular disorders4,13,14. Since the dysregulation of PRMTs has been closely associated with cancer development2,15,16, the use of PRMTs as novel targets for anticancer drug development is rapidly increasing. Recent studies have revealed considerable advances in the identification of clinically relevant PRMT inhibitors17,18. Here, we focus on the biological functions of PRMTs in cancer and the therapeutic potential of PRMT inhibitors.

Protein arginine methylation is observed in both histones and nonhistone proteins, which contribute to diverse cellular responses for maintaining cellular homeostasis in biological systems. The expression and activity of PRMTs are regulated by developmental and pathogenic processes, genetic mutations, and various environmental factors.
Biological functions of protein arginine methylation
As histone proteins tightly regulate gene transcription through various PTMs, including acetylation, lysine methylation, phosphorylation, ubiquitination, and SUMOylation19,20, early studies of PRMTs have also focused on their epigenetic functions. PRMTs synthesize methyl arginine on nucleosomes after being recruited into chromatin remodeling complexes, and these methylated arginine residues not only serve as key epigenetic marks but also engage in crosstalk with other epigenetic marks21,22. These orchestrated epigenetic modifications are recognized by epigenetic reader proteins, leading to the recruitment of activating/repressing transcriptional machinery. The histone modifications generated by PRMTs and their roles are summarized in Table 1. The methylation status of an arginine residue in histones can determine whether the transcription process is activated or suppressed. For example, H4R3me2a, a modification generated by PRMT1/PRMT3, acts as a mark of activated transcription, whereas H4R3me2s, generated by PRMT5, functions as a repression mark, implying that there is a sophisticated and competitive mechanism between PRMTs for regulating the transcription process. In addition to histone proteins, various proteins involved in transcription, such as transcription factors, coactivators, and corepressors, are also methylated by PRMTs (Table 1 and Fig. 2)23. Hence, PRMTs also contribute to the precise regulation of the transcription process. A number of RNA-binding proteins (RBPs) have RG/RGG-rich motifs that have been established as representative consensus sequences of PRMTs24,25. Indeed, theoretical insights and proteomic analysis revealed that several RBPs are methylated by PRMTs and that these modifications are essential for mRNA splicing, RNA localization, and translation processes26,27,28. In addition to gene expression regulation, the functions of PRMTs are extended to various cellular processes, including cell signaling, cell cycle regulation, and the DNA damage response (DDR)2,3. Methylation of arginine residues in signal receptors and their downstream mediators determines the amplitude or duration of signal transduction, contributing to the regulation of cell proliferation, survival, differentiation, and metabolism. Although all of these functions are critical for maintaining cellular homeostasis and normal cell growth, we highlight the biological roles of PRMTs in both cell cycle regulation and the DDR, which are the key pathways that are dysregulated in the hallmarks of cancer.
Regulation of the cell cycle through arginine methylation
Cell cycle progression is precisely orchestrated by the cooperation of various signaling pathways and post-translational modifications29,30. Arginine methylation is implicated in the cell cycle via gene expression regulation or the direct methylation of cell-cycle-related regulators (Fig. 3).

The cell cycle is mainly regulated by phase-specific oscillation of cyclin-dependent kinase (CDK)-cyclin complexes. The expression of several cyclins (Cyclin E, Cyclin D1, etc.) and CDKs is epigenetically regulated by PRMTs (not shown). CDK4 is directly methylated by PRMT1, which inhibits binding with Cyclin D and blocks cell cycle progression. In contrast, methylation of E2F1 by either PRMT1 or PRMT5 results in cell progression from G1 to S phase. Several CKIs (CDK inhibitors), such as p16, p21, and p27, are directly methylated by PRMTs to regulate their binding with CDK-cyclin complexes or their cellular localization. During mitosis, PRMT6-mediated H3R2me2a recruits Aurora B kinase into chromosomes along with CPC components, enabling H3S10 phosphorylation. Another CPC component, INCENP, is also methylated by PRMT1, which promotes its interaction with Aurora B kinase. Together, the activities of PRMT1 and PRMT6 during M phase are required for chromosome condensation and proper segregation.
PRMT1 methylates cyclin-dependent kinase 4 (CDK4), a key regulator of the G1-S transition, at four residues (Arg55/73/82/163) located near the Cyclin D3-binding area31. These multiple arginine methylations disrupt the formation of the CDK4-Cyclin D3 complex and advance cell cycle progression, promoting pre-B-cell differentiation and inhibiting leukemogenesis. The inner centromere protein (INCENP), a component of the chromosomal passenger complex (CPC), is methylated by PRMT1 at the Arg887 residue located in the Aurora kinase B (AURKB)-binding region32. The methylation of Arg887 in INCENP facilitates its interaction with AURKB, thereby augmenting AURKB activity and contributing to the enhancement of chromosome alignment and segregation during mitosis in cancer cells. PRMT1 also regulates chromosome alignment via arginine methylation of ubiquitin-associated protein 2-like (UBAP2L)33. PRMT1 directly interacts with and methylates UBAP2L on its N-terminal RGG/RG motif, and its methylation is essential for the proper alignment and accurate distribution of chromosomes in metaphase.
As a transcriptional coactivator, CARM1 (coactivator-associated arginine methyltransferase 1, also known as PRMT4) participates in cell cycle progression by regulating the expression of genes associated with the cell cycle. With the p160 coactivator member ACTR/SRC3/AIB1, CARM1 acts as a coactivator of Cyclin E (CCNE1) transcription in an E2F1-dependent manner34. CARM1 recruited to the CCNE1 promoter increases the levels of H3R17me2a and H3R26me2a, resulting in transcriptional activation of CCNE1 and subsequent cellular entry into S phase. E2F1 is a target for transcriptional regulation of CARM1 for cell cycle regulation35. Upon estrogen stimulation, CARM1 is recruited to the E2F1 promoter with ERα in an oncogenic coactivator AIB1-dependent manner and then induces the H3R17me2a modification at the E2F1 promoter. The epigenetic upregulation of CCNE1 and E2F1 mediated by CARM1 is associated with the development of breast cancer.
PRMT5 activity is primarily implicated in G1 progression and the G1-S transition. PRMT5 overexpression accelerates cell cycle progression by increasing the expression of cell cycle regulators, including CDK4, CDK6, Cyclin D1, Cyclin D2, Cyclin E1, and phospho-Rb36. In addition, upregulation of PRMT5 activates PI3K, AKT, mTOR/eIF4E, and NF-κB signaling, contributing to the proliferation of cancer cells36. PRMT5 epigenetically suppresses RBL2, a member of the retinoblastoma tumor suppressor family, and indirectly enhances RB1 phosphorylation, resulting in the activation of the polycomb repressor complex PRC2 and Cyclin D137. The upregulation of the expression of PRC2 and Cyclin D1 facilitates cell cycle progression and cell survival via activation of cyclin D1-CDK4/6 signaling and suppression of proapoptotic target genes of PRC2. As an alternative mechanism for Cyclin D1 upregulation mediated by PRMT5, the regulation of tumor suppressor miRNA expression by PRMT5 was recently studied, and the results were published38. PRMT5 epigenetically suppresses the expression of several tumor suppressor miRNAs, such as miR33b, miR96, and miR503, which bind to and target the mRNA corresponding to Cyclin D1 and/or c-Myc. In aggressive B-cell lymphoma, upregulated expression of PRMT5 leads to a decrease in the levels of these miRNAs and an increase in Cyclin D1 and c-Myc expression. PRMT5 directly interacts with CDK4, impeding the competitive interaction between CDK4 and p1639. This PRMT5-CDK4 complex promotes the activation of CDK4-pRb-E2F-mediated transcription and, in turn, the cell cycle progression of hepatocarcinoma cells.
PRMT6 regulates the cell cycle via epigenetic repression of cell cycle-related regulators, such as p21WAF1, p27KIP1, and p1840,41,42. The H3R2me2a modification mediated by PRMT6 transcriptionally turns off these genes and, in turn, induces abnormal bypass of the cell cycle. This outcome demonstrates the oncogenic function of PRMT6. The PRMT6-mediated H3R2me2a modification is essential for the recruitment of CPC to chromosome arms during mitosis43. AURKB preferentially binds to H3R2me2a and phosphorylates H3S10, which recruits the CPC complex to chromosome arms for precise chromosome condensation and segregation during mitosis.
Regulation of the DNA damage response through arginine methylation
One of the important biological roles of arginine methylation is the regulation of DNA damage signaling and DNA repair processes. Several DDR regulators have been identified as substrates for PRMTs, and their methylated arginine residues modulate their functions, stability, DNA-binding affinity, and interaction with other proteins (Table 1).
The double-strand break repair protein MRE11, a component of the MRE11–RAD50-NBS1 (MRN) complex, is methylated by PRMT1 in its C-terminal glycine-arginine rich (GAR) domain44,45, which is critical for its exonuclease activity and allows the recruitment of MRE11 to the damaged DNA site44. PRMT1-mediated MRE11 methylation is implicated in the S-phase DNA damage checkpoint, ATR/CHK1 signaling, and the recruitment of replication protein A (RPA) and RAD51 to DNA lesions45,46, suggesting that arginine methylation in the GAR domain is required for the normal functioning of MRE11 in response to DNA damage stress and repair signaling. PRMT1 methylates arginine residues in the GAR motif in 53BP1 (p53-binding protein 1), a key regulator of the nonhomologous end-joining (NHEJ) repair process47,48. Asymmetrically dimethylated 53BP1 enhances DNA-binding activity without affecting its oligomerization. During the repair of DNA single-strand breaks and single-base lesions, DNA polymerase β (pol β) plays an indispensable role in the DNA base excision repair (BER) pathway49. PRMT1 methylates the Arg137 residue of pol β, which interferes with the binding of proliferating cell nuclear antigen (PCNA) without affecting its polymerase or dRP-lyase activities50. Another DNA repair regulator, Flap endonuclease 1 (FEN1), is methylated by PRMT1 at Arg19251. This methylation suppresses its phosphorylation at Ser187, promoting its interaction with PCNA and its localization to damaged DNA foci. Furthermore, upregulation of PRMT1 correlates with high expression of FEN1 in lung cancer due to stabilization of the FEN1 protein via PRMT1-mediated arginine methylation52.
The tumor suppressor BRCA1, a key regulator of the HR repair process, is directly or indirectly regulated by arginine methylation. PRMT1 interacts with and methylates the 504–802 region of BRCA1 to consolidate its target promoters53. The methylation status of the 504–802 region determines the binding preference of BRCA1 for SP1 or STAT1. Furthermore, methylation of the Arg754 residue of CBP/p300 by CARM1 is preferentially recognized by the BRCT domain of BRCA1, which is critical for the recruitment of BRCA1 to the p53-binding region of the p21WAF1 promoter54.
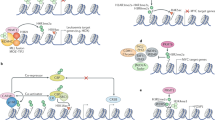
Many studies have revealed that the activity of PRMT5 is crucial in the DDR. PRMT5 methylates three arginine residues (Arg172/174/175) of Rad955. This process is required for the activation of Chk1 signaling and, in turn, S/M and G2/M cell cycle checkpoints. In the DDR, the p53 transcription factor is a major determinant of cell survival or apoptosis56. PRMT5 interacts with and methylates p53 at Arg333/335/337 residues, affecting the promoter specificity of p53 associated with apoptosis or cell cycle arrest57. Moreover, the translation of p53 is regulated by PRMT5 upon DNA damage via the expression of the translation initiation factor eIF4E58. PRMT5-mediated Arg111/113 methylation of E2F1 negatively regulates its protein stability59. Under stress caused by DNA damage, E2F1 methylation by PRMT5 is reduced, and consequently, the protein levels of E2F1 are elevated, which contributes to the induction of apoptosis. Upregulation of PRMT5 in cancer downregulates the apoptotic activity of E2F1, contributing to tumorigenesis. Krüppel-like factor 4 (KLF4) is also methylated by PRMT5 at Arg374/376/377 residues. This methylation inhibits VHL-mediated ubiquitination, thereby increasing the protein stability of KLF460. Stress induced by DNA damage increases PRMT5 protein levels and subsequently facilitates the methylation and accumulation of KLF4, modulating the cell cycle and survival of cancer cells. Consistent with these outcomes, aberrant accumulation of PRMT5 and subsequent KLF4 methylation/accumulation correlate with poor prognosis in breast cancer. PRMT5 plays a role in homologous recombination (HR)-mediated DNA repair through arginine methylation of the TIP60 complex61. PRMT5 methylates the Arg205 residue of RUVBL1, a cofactor of the TIP60 complex, which promotes TIP60/KAT5-dependent chromatin acetylation and subsequent 53BP1 removal from double-strand break sites. Moreover, the loss of PRMT5 leads to aberrant splicing of DNA repair regulators, including TIP60/KAT5 histone acetyltransferase (HAT) and KMT5C/SUV4-20H2 lysine methyltransferase62. A decrease in TIP60α expression by aberrant splicing of TIP60 results in the reduction in TIP60-mediated chromatin acetylation and, in turn, defects in HR. In addition to HR, PRMT5 is implicated in the NHEJ pathway via arginine methylation of 53BP163. Hwang et al. found that the GAR motif of 53BP1 is competitively methylated by PRMT1 and PRMT5. While asymmetric dimethylation of 53BP1 by PRMT1 affects its DNA-binding activity, symmetric dimethylation by PRMT5 regulates its protein stability. Inhibition or deletion of PRMT5 leads to a decrease in 53BP1 protein levels and defects in the NHEJ process (Fig. 4).

Under DNA double-strand breaks, the MRE11–RAD50–NBS1 complex is recruited into the DNA lesion and activates ATM/CHK2 kinase signaling. PRMT1-mediated MRE11 methylation is essential for exonuclease activity and localization to DNA. There are two main repair pathways, homologous recombination (HR) and nonhomologous end-joining (NHEJ). 53BP1, a major regulator of NHEJ, is competitively methylated by PRMT1 and PRMT5 in the GAR motif. PRMT1-mediated methylation of 53BP1 promotes DNA binding (not shown), and PRMT5-mediated methylation increases the stability of 53BP1, which contributes to NHEJ repair. BRCA1, a well-established key regulator of HR, is methylated by PRMT1, but its role is unknown. Arginine methylation of RUVBL1 (a cofactor of the TIP60 complex) by PRMT5 facilitates TIP60α-dependent histone H4 Lys16 acetylation (H4K16ac), which blocks 53BP1 recruitment to reinforce HR.
The roles of PRMTs in cancer
With the accumulation of studies on the biological and pathological roles of protein arginine methylation, evidence for a direct link between PRMTs and cancer is emerging. In this section, we discuss the roles of PRMTs in cancer and the molecular mechanisms of each isoform (Table 2).
Type I PRMTs
PRMT1
PRMT1 is the most predominant enzyme in the PRMT family, and its activity is responsible for more than 90% of the overall arginine methylation in mammalian cells64. The dysregulation of PRMT1 expression and its pathological mechanisms in various human carcinomas are summarized in Table 2. For instance, EZH2 (enhancer of zeste homolog 2) is asymmetrically dimethylated at Arg342 by PRMT165, which leads to an increase in EZH2 levels because TNF receptor associated factor 6 (TRAF6)-mediated ubiquitination is interrupted. Upregulation of EZH2 by Arg342 methylation consequently reduces the expression of EZH2 target genes such as HOXA10, DAB2IP, HOXA9, and HOXA7, promoting breast cancer cell migration and metastasis. Indeed, the expression levels of PRMT1 and the methylation levels of the Arg342 residue of EZH2 correlate with poor clinical outcomes in breast cancer patients, suggesting the utility of PRMT1 as a diagnostic marker and therapeutic target for cancer. In colorectal cancer patients, PRMT1-mediated Arg198/200 methylation of EGFR is correlated with tumor growth, a high recurrence rate after cetuximab treatment, and reduced overall survival66. PRMT1 methylates GLI1 at the Arg597 residue, which enhances its transcriptional activity67. In pancreatic ductal adenocarcinoma, increased PRMT1 expression correlates with GLI1 expression and leads to SMO-independent GLI1 activation, thereby mediating its oncogenic functions. PRMT1-mediated Twist1 methylation is involved in the regulation of the epithelial-mesenchymal transition (EMT) in lung cancer cells68. The Twist1 transcription factor, known as an E-cadherin repressor, is methylated by PRMT1 at the Arg34 residue to promote its repressive activity. Upregulation of PRMT1 expression in lung cancer is linked to a decrease in E-cadherin and an increase in N-cadherin levels, which stimulates cell migration, invasion, and metastasis.
PRMT2
The role of PRMT2 in cancer remains controversial. In breast cancer cells, PRMT2 is recruited to the AP-1-binding site of the CCND1 promoter and ERα binding is simultaneously suppressed69. Depletion of PRMT2 expression leads to an increase in estrogen-induced CCND1 expression and promotion of cell proliferation and colony formation, indicating That PRMT2 has tumor-suppressive activity. In contrast, an oncogenic function of PRMT2 in glioblastoma has been reported70. PRMT2 expression is elevated in glioblastoma and is correlated with tumor grade. The PRMT2-mediated H3R8me2a modification is implicated in the activation of the oncogenic transcriptome, leading to the enhancement of GBM cell growth and tumorigenesis.
CARM1
The role of CARM1 in cancer is still debated. CARM1 positively regulates the transcription of CCNE1 via H3R17 and H3R26 methylation in collaboration with E2Fs and ACTR34. In high-grade breast tumors, the mRNA levels of CARM1 and ACTR are elevated, indicating an oncogenic role of CARM1 in breast cancer. CARM1-mediated BAF155 methylation promotes cancer cell migration and metastasis71. The chromatin remodeling factor BAF155 (BRG1-associated factor 155) is methylated by CARM1 at Arg1604, which modulates the chromatin association patterns of BAF155. Arg1604 methylation of BAF155 facilitates cell migration and metastasis and correlates with breast cancer progression, malignancy, and recurrence-free survival. Arginine methylation of pyruvate kinase 2 (PKM2) by CARM1 is implicated in tumorigenesis via modulation of energy metabolism72. The Arg445/447/455 residues of PKM2 are methylated by CARM1. This methylation does not affect PKM2 enzymatic activity and is involved in regulating mitochondrial respiration in cancer cells. PKM2 methylation leads to decreased Ca2+ uptake and diminished mitochondrial membrane potential, causing an increase in cell proliferation, migration, and metastasis. Recently, the oncogenic function of CARM1 in CBP/P300-mutated lymphomas was well characterized73. Inhibition of CARM1 activity slows diffuse large B-cell lymphoma (DLBCL) growth, which is positively correlated with CBP/P300-mutation status, indicating that the CBP/P300 mutation in cancer creates a vulnerability to targeting CARM1 activity.
In contrast to these oncogenic functions, several reports have described the role of CARM1 as a tumor suppressor. As a coactivator of the estrogen receptor ERα, CARM1 regulates estrogen-dependent breast cancer cell proliferation and differentiation74. CARM1 suppresses estradiol (E2)-dependent cell cycle progression and proliferation of breast cancer cells via modulation of the ERα-mediated transcription of proteins, especially p21WAF1, p27KIP1, Cyclin G2, MAZ, KRTAP10.12, and GATA-3. In ER-positive breast cancers, the expression level of CARM1 is positively correlated with ERα levels and inversely correlated with tumor grade, suggesting that CARM1 is a biomarker of well-differentiated breast cancer cells. In addition, CARM1 activity contributes to the sensitization of cancer cells to chemotherapy drugs via arginine methylation of RNA polymerase II mediator complex subunit 12 (MED12)75. CARM1 interacts with and methylates MED12 at Arg1862/1912 located in the C-terminal proline-glutamine-leucine-rich (PQL) domain. ChIP-seq analysis revealed that arginine methylation of MED12 enhances chromatin association with target genes, especially p21WAF1, resulting in suppression of p21WAF1 transcription. Methylation of MED12 renders cancer cells sensitive to chemotherapy drugs under in vitro and in vivo conditions, and higher levels of MED12 and CARM1 correlate with a better response to chemotherapy drugs.
PRMT6
PRMT6 demonstrates oncogenic activity by inducing the addition of the epigenetic repressive H3R2me2a mark on tumor suppressor genes, such as p21WAF1 and p16INK4A, which facilitates cell proliferation and prevents senescence76. In addition, PRMT6-mediated H3R2me2a impedes the recruitment of UHRF1 (an accessory factor of DNMT1) onto chromatin, leading to DNA hypomethylation77. Indeed, PRMT6 expression inversely correlates with global DNA methylation in many human cancer cells, and PRMT6 depletion or inhibition restores DNA methylation. These observations demonstrate the potential of targeting PRMT6 for cancer therapy.
Type II PRMT
PRMT5
A major type II enzyme, PRMT5, is emerging as the most promising target for a range of solid and blood cancers. Overexpression or dysregulation of PRMT5 has been observed in various cancer types, including breast78,79, lung80,81, ovarian82, prostate83, colorectal84, gastric85,86, liver87,88, pancreatic89,90, head and neck91, bladder92, lymphoma93,94, melanoma95, and glioma96. Epigenetically, PRMT5 associates with BRG1- and hBRM-based hSWI/SNF chromatin remodeling complexes and induces H3R8me2s and H4R3me2s modifications, repressing the transcription of tumor suppressor genes such as suppressor of tumorigenicity 7 (ST7) and nonmetastatic 23 (NM23)97. The PRMT5-pICln (but not the MEP50) complex is recruited to the proximal region of the androgen receptor (AR) promoter and mediates symmetric dimethylation of H4R3, which acts as an epigenetic activation modification98. An increase in AR expression mediated by PRMT5 promotes the growth of castration-resistant prostate cancer cells. The epigenetic regulation of cancer-specific miRNA expression by PRMT5 is critical for tumor growth, progression, and metastasis. The PRMT5-mediated H4R3me2s modification silences miR-29b expression, resulting in increased levels of Sp1 and FLT3. This increase leads to the growth of cancer cells in acute myeloid leukemia94. In addition, overexpression of PRMT5 in lung cancer enriches the epigenetic repressive mark H4R3me2s on the promoter of the miR-99 family and subsequently suppresses the expression of member miRs99. Reduced expression of miR-99 family members increases the expression of fibroblast growth factor receptor 3 (FGFR3) and, in turn, activates ERK1/2 and AKT signaling, promoting lung cancer cell migration and invasion. PRMT5 also contributes to carcinogenesis via the arginine methylation of several oncoproteins and tumor suppressors (Table 2). For instance, programmed cell death 4 (PDCD4), a tumor suppressor, is methylated at Arg110 by PRMT578. High expression of PDCD4 alone correlates with better outcomes for breast cancer patients. However, patients with both high PDCD4 and PRMT5 demonstrate poor prognoses, suggesting that arginine methylation of PDCD4 by PRMT5 decreases the ability of PDCD4 to suppress cancer cell growth. Indeed, Arg110 methylation of PDCD4 by PRMT5 modulates PDCD4 subcellular translocalization from the nucleus to the cytoplasm and facilitates its interaction with eIF4A in the cytoplasm, leading to enhanced cancer cell viability100.
Recently, several reports have been published on the correlation between S-methyl-5′-thioadenosine phosphorylase (MTAP) gene deletion and susceptibility to PRMT5 action, which is worth considering101,102,103. The chromosome 9p21 (chr9p21) locus, which encodes the CDKN2A gene, is homozygously deleted in approximately 15% of all human cancers, with frequent codeletion of the MTAP gene, in 80–90% of tumors, along with CDKN2A deletion. Due to the intracellular accumulation of methylthioadenosine (MTA), an endogenous PRMT5 antagonist, MTAP deletion renders cancer cells sensitive to PRMT5. Based on this rationale, the combination of a PRMT1 inhibitor and PRMT5 inhibitor synergistically inhibits the proliferation of cancer cells with MTAP deletion104.
Type III PRMT
PRMT7
PRMT7 is prominently overexpressed in malignant breast tumors and is associated with the EMT105. In PRMT7-overexpressing cells, enriched H4R3me2s at the E-cadherin promoter antagonizes the H3K4me3 epigenetic modification and, in turn, represses the transcription of E-cadherin during the EMT. The recruitment of PRMT7 to the E-cadherin promoter depends on the YY1 transcription factor, and the PRMT7-YY1-HDAC3 ternary complex acts as a transcriptional repressor of E-cadherin. In the EMT, SHANK2 (scaffolding protein SH3 and multiple ankyrin repeat domain 2) is symmetrically dimethylated at Arg240 by PRMT7, which activates endosomal FAK/cortactin signaling, contributing to cancer cell invasion, metastasis, and malignancy106. As mentioned above, since PRMT7 is a type III PRMT that can only deposit MMA, it is unclear how the SDMA mark is enriched. Perhaps the following possibilities should be considered: the H4R3me1 mark becomes a substrate for PRMT5, or there is an auxiliary factor that can convert PRMT7 into a type II enzyme.
PRMT inhibitors constitute a novel class of anticancer drugs
As described above, PRMTs regulate various cellular processes, including transcription, mRNA splicing, translation, DNA damage/repair response, and the cell cycle. Since they are closely associated with cancer and tumorigenesis, PRMTs have recently emerged as molecular targets for anticancer drug development and play essential roles in cancer research17,107. As a result, enormous efforts have been undertaken to develop effective and selective PRMT inhibitors. Although many candidates are still in the preclinical stage, some inhibitors have entered clinical trials. In Table 3, we summarize the main features of the inhibitors that have been developed thus far and briefly discuss them below.
Type I PRMT inhibitors
Most PRMT inhibitors target type I not type II PRMTs. The first discovered PRMT inhibitor was AMI-1 (2004), which inhibits type I PRMTs108. Despite its usefulness, more specific and potent inhibitors needed to be developed. This requirement was partially fulfilled by the development of allantodapsone with specificity for PRMT1109. MS023 also contributed to the available potent inhibitors by inhibiting type I PRMT at concentrations much lower than those of AMI-1110. The in vitro working concentration was reduced from the micromolar to nanomolar range. Finally, GSK3368715 was developed and entered a phase 1 clinical trial in 2018104. GSK3368715 is being examined for its use as a treatment for refractory diffuse large B-cell lymphoma and select solid tumors with MTAP deficiency (http://clinicaltrials.gov/ct2/show/NCT03666988). Since the loss of MTAP leads to the accumulation of MTA, an endogenous PRMT5 inhibitor, GSK3368715 can be effective in MTAP-null cancer cells by mediating the blockage of the compensatory relationship between ADMA and SDMA107. This finding also suggests that combination therapy with type I PRMT inhibitors and PRMT5 inhibitors may demonstrate synergistic effects111.
In contrast to PRMT1 inhibitors that bind mainly to the substrate-binding pocket, a substrate and AdoMet noncompetitive inhibitor, SGC707, has been developed;112 this is the first allosteric PRMT3 inhibitor to have been developed. Although SGC707 is insufficient for therapeutic purposes, it is a good reference to encourage the development of other allosteric inhibitors113. As efforts to develop type I PRMT inhibitors continued, several CARM1 inhibitors, such as EZM2302 and TP-064, were also identified 2114,115. They have demonstrated remarkable efficacy under in vivo and in vitro conditions. MS049 is a dual inhibitor of CARM1 and PRMT6116. EPZ020411 is a representative PRMT6 inhibitor that can also inhibit PRMT1, PRMT8, and other PRMTs but has a high affinity for PRMT6117. Similarly, many compounds have been developed to inhibit PRMT6, such as GMS, which have an effect in the nanomolar range but lack selectivity118. Recently developed SGC6870 is a highly selective inhibitor of PRMT6119.
Type II PRMT inhibitors
Despite many expectations for and investments into the development of type I PRMT inhibitors, only a few satisfactory outcomes have been observed. However, the development of PRMT5 inhibitors has been more successful. This outcome is not surprising, as PRMT5 plays an essential role in cancer stem cell survival, mRNA splicing, and DNA repair processes61,120,121. Thus, PRMT5 inhibitors can be useful for treating cancer in mono- or combination therapy with DNA-damaging agents. EPZ015666 was the first PRMT5 inhibitor;122 similar compounds have also been developed123,124. Among these inhibitors, GSK3326595 has entered phase 1/2 clinical trials (http://clinicaltrials.gov/ct2/show/NCT02783300 and http://clinicaltrials.gov/ct2/show/NCT03614728). These inhibitors bind at the substrate-binding pocket. This binding is enhanced via AdoMet, which competes with MTA101. Therefore, EPZ015666 is less effective in MTAP-null cancer cells. Hence, PRMT5 inhibitors that bind at the AdoMet-binding pocket have been developed. LLY-283 and JNJ-64619178 are representative examples125,126. Specifically, JNJ-64619178 has entered Phase 1 clinical trials and is being examined for use in the treatment of advanced solid tumors, non-Hodgkin lymphoma, and lower-risk myelodysplastic syndromes (http://clinicaltrials.gov/ct2/show/NCT03573310). In 2019, PF-06939999 (http://clinicaltrials.gov/ct2/show/NCT0385427), PRT543 (http://clinicaltrials.gov/ct2/show/NCT03886831), and PRT811 (http://clinicaltrials.gov/ct2/show/NCT04089449) also entered phase 1 clinical trials.
Interest in PRMT5 inhibitors has been increasing, for several reasons for this: PRMT5 inhibitors have successfully entered clinical trials, and the relationship between MTAP loss and PRMT5 activity has been demonstrated103. PRMT5 has a unique characteristic that requires MEP50 to serve as its complex partner127. Considering these observations, scientists of recent studies have suggested the development of allosteric PRMT5 inhibitors that stabilize MTA or enhance the formation of the PRMT5-MTA complex and inhibitors that disrupt the formation of the PRMT5-MEP50 complex3. These suggestion are interesting. MS4322, the first developed PRMT5 degrader, is an example of the application of proteolysis targeting chimera (PROTAC)128. PROTAC is a technology employed to degrade a specific target protein in a proteasome-dependent manner by recruiting the E3 ubiquitin ligase129. Since it demonstrates broad applications and has enabled the resistance to small-molecule inhibitors to be overcome, PROTAC technology has been frequently used for novel drug discovery and development130. MS4322 forms a link between the structure of EPZ015666 and the von Hippel-Lindau E3 ubiquitin ligase ligand, and hence, MS4322 effectively and selectively inhibits PRMT5. This discovery is meaningful, as it confirmed the possibility that a therapeutic PRMT degrader can be developed.
Type III PRMT inhibitors
PRMT7 was identified in 2004 and is associated with metastasis and DNA damage131. Although PRMT7 is considered a potential target for treating breast cancer105, studies on the development of PRMT7 inhibitors are still limited. DS-437 was developed as a dual inhibitor of PRMT5 and PRMT7132. Recently, SGC3027 was developed as the first PRMT7 inhibitor133. It is a prodrug that can be converted to the active form: SGC8158.
Future perspectives
Protein arginine methylation, as reviewed in this paper, plays an essential role in maintaining biological homeostasis4. Dysregulation of arginine methylation is observed not only in cancer cells but also in various tumors (Table 2). Hence, the development of anticancer drugs targeting PRMTs has gained traction (Table 3). The fact that PRMT inhibitors are included in multiple clinical trials may be sufficient to fuel research examining arginine methylation. However, several issues still need to be addressed to better understand the roles of arginine methylation and successfully develop its inhibitor: (1) novel PRMT substrates need to be identified and characterized, (2) a regulatory mechanism for arginine methylation needs to be found, and (3) isoform-specific inhibitors need to be developed.
PRMTs govern diverse cellular processes, including transcription, signaling pathways, splicing, cell cycle progression, and DNA damage and repair processes, via methylation of a variety of substrate proteins, as summarized in Table 1. Research on arginine methylation is still in the early stages. Many proteins can be methylated at arginine residues, and this modification regulates various cellular responses. Therefore, one of the obvious future goals is to clarify the downstream pathways by identifying novel substrates of PRMTs, which will guide help us to understand the mechanisms of various diseases, including cancer, and establish treatment strategies. Next, the regulatory mechanism of arginine methylation should be more clearly understood. In contrast to other PTMs, arginine methylation is known to be quite stable2,5. Arginine methylation appears to be a nondynamic and static reaction, as the presence of dedicated arginine demethylase that enables cycles of methylation and demethylation has not been identified. As several reports have shown that the levels of arginine methylation change dynamically depending on the cellular environment134,135, we are confident that a arginine demethylase will soon be discovered. In addition, the regulatory mechanisms of PRMT activity in cellular systems are poorly understood. PRMT5 is active only when it interacts with MEP50136,137. In some cases, the enzyme activity of PRMTs can be regulated by other PTMs, such as phosphorylation. PRMT5 can be phosphorylated at tyrosine residues by JAK2-V617F or Src family kinases, which leads to a decrease in its methyltransferase activity63,138. In contrast, the phosphorylation of PRMT5 Thr80 by RhoA-activated kinase increases its methyltransferase activity139. However, since these regulatory mechanisms are observed under limited and special circumstances, the detailed regulatory mechanism of arginine methylation is a concept that must be explored. Finally, developing PRMT inhibitors as novel anticancer drugs requires a careful approach. PRMTs, especially PRMT1 and PRMT5, are considered promising targets for the development of anticancer drugs because of their pro-oncogenic functions2,3,18. However, since arginine methylation is an essential response to normal cell growth and homeostasis maintenance3,4,5, nonselective and indiscriminate inhibition of PRMTs is likely to lead to undesirable effects. Therefore, it is necessary to select and target tumors that are relatively vulnerable to PRMT inhibition. From this perspective, it is quite remarkable that the lack of the MTAP gene increases sensitivity to PRMT5 inhibitors101,102,103,104 and that CBP/P300 mutation creates vulnerability to CARM1 inhibitors73. In addition, the development of isoform-specific inhibitors will ensure successful cancer treatment.
In summary, systematic approaches to arginine methylation, including the issues discussed above, will not only provide a better understanding of biological phenomena but also lead to the development of a novel class of anticancer drugs.
References
Paik, W. K. & Kim, S. Enzymatic methylation of protein fractions from calf thymus nuclei. Biochem. Biophys. Res. Commun. 29, 14–20 (1967).
Yang, Y. & Bedford, M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 (2013).
Guccione, E. & Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 20, 642–657 (2019).
Blanc, R. S. & Richard, S. Arginine methylation: the coming of age. Mol. Cell 65, 8–24 (2017).
Bedford, M. T. & Clarke, S. G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 (2009).
Schapira, M. & Ferreira de Freitas, R. Structural biology and chemistry of protein arginine methyltransferases. MedChemComm 5, 1779–1788 (2014).
Tewary, S. K., Zheng, Y. G. & Ho, M. C. Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level. Cell Mol. Life Sci. 76, 2917–2932 (2019).
Lee, H. W., Kim, S. & Paik, W. K. S-adenosylmethionine: protein-arginine methyltransferase. Purification and mechanism of the enzyme. Biochemistry 16, 78–85 (1977).
Zurita-Lopez, C. I., Sandberg, T., Kelly, R. & Clarke, S. G. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J. Biol. Chem. 287, 7859–7870 (2012).
Pawlak, M. R., Scherer, C. A., Chen, J., Roshon, M. J. & Ruley, H. E. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell Biol. 20, 4859–4869 (2000).
Yadav, N. et al. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc. Natl Acad. Sci. USA 100, 6464–6468 (2003).
Tee, W. W. et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 24, 2772–2777 (2010).
vanLieshout, T. L. & Ljubicic, V. The emergence of protein arginine methyltransferases in skeletal muscle and metabolic disease. Am. J. Physiol. Endocrinol. Metab. 317, E1070–E1080 (2019).
Hashimoto, M. et al. Severe hypomyelination and developmental defects are caused in mice lacking protein arginine methyltransferase 1 (PRMT1) in the central nervous system. J. Biol. Chem. 291, 2237–2245 (2016).
Yoshimatsu, M. et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 128, 562–573 (2011).
Shailesh, H., Zakaria, Z. Z., Baiocchi, R. & Sif, S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 9, 36705–36718 (2018).
Smith, E. et al. Recent advances in targeting protein arginine methyltransferase enzymes in cancer therapy. Expert. Opin. Ther. Targets 22, 527–545 (2018).
Jarrold, J. & Davies, C. C. PRMTs and arginine methylation: cancer’s best-kept secret? Trends Mol. Med. 25, 993–1009 (2019).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Zhao, Z. & Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 20, 245 (2019).
Litt, M., Qiu, Y. & Huang, S. Histone arginine methylations: their roles in chromatin dynamics and transcriptional regulation. Biosci. Rep. 29, 131–141 (2009).
Huang, S., Litt, M. & Felsenfeld, G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 19, 1885–1893 (2005).
Lee, Y. H. & Stallcup, M. R. Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol. Endocrinol. 23, 425–433 (2009).
Liu, Q. & Dreyfuss, G. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol. Cell Biol. 15, 2800–2808 (1995).
Blackwell, E. & Ceman, S. Arginine methylation of RNA-binding proteins regulates cell function and differentiation. Mol. Reprod. Dev. 79, 163–175 (2012).
Brahms, H. et al. The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J. Biol. Chem. 275, 17122–17129 (2000).
Boisvert, F. M., Cote, J., Boulanger, M. C. & Richard, S. A proteomic analysis of arginine-methylated protein complexes. Mol. Cell Proteomics 2, 1319–1330 (2003).
Hebert, M. D., Shpargel, K. B., Ospina, J. K., Tucker, K. E. & Matera, A. G. Coilin methylation regulates nuclear body formation. Dev. Cell 3, 329–337 (2002).
Vermeulen, K., Van Bockstaele, D. R. & Berneman, Z. N. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36, 131–149 (2003).
Satyanarayana, A. & Kaldis, P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28, 2925–2939 (2009).
Dolezal, E. et al. The BTG2-PRMT1 module limits pre-B cell expansion by regulating the CDK4-Cyclin-D3 complex. Nat. Immunol. 18, 911–920 (2017).
Deng, X. et al. PRMT1 promotes mitosis of cancer cells through arginine methylation of INCENP. Oncotarget 6, 35173–35182 (2015).
Maeda, M. et al. Arginine methylation of ubiquitin-associated protein 2-like is required for the accurate distribution of chromosomes. FASEB J. 30, 312–323 (2016).
El Messaoudi, S. et al. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc. Natl Acad. Sci. USA 103, 13351–13356 (2006).
Frietze, S., Lupien, M., Silver, P. A. & Brown, M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res. 68, 301–306 (2008).
Wei, T. Y. et al. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 103, 1640–1650 (2012).
Chung, J. et al. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) silencing. J. Biol. Chem. 288, 35534–35547 (2013).
Karkhanis, V. et al. Protein arginine methyltransferase 5 represses tumor suppressor miRNAs that down-regulate CYCLIN D1 and c-MYC expression in aggressive B-cell lymphoma. J. Biol. Chem. 295, 1165–1180 (2020).
Yang, H. et al. PRMT5 competitively binds to CDK4 to promote G1-S transition upon glucose induction in hepatocellular carcinoma. Oncotarget 7, 72131–72147 (2016).
Phalke, S. et al. p53-Independent regulation of p21Waf1/Cip1 expression and senescence by PRMT6. Nucleic Acids Res. 40, 9534–9542 (2012).
Kleinschmidt, M. A., de Graaf, P., van Teeffelen, H. A. & Timmers, H. T. Cell cycle regulation by the PRMT6 arginine methyltransferase through repression of cyclin-dependent kinase inhibitors. PLoS ONE 7, e41446 (2012).
Tang, J., Meng, Q., Shi, R. & Xu, Y. PRMT6 serves an oncogenic role in lung adenocarcinoma via regulating p18. Mol. Med. Rep. 22, 3161–3172 (2020).
Kim, S. et al. PRMT6-mediated H3R2me2a guides Aurora B to chromosome arms for proper chromosome segregation. Nat. Commun. 11, 612 (2020).
Boisvert, F. M., Hendzel, M. J., Masson, J. Y. & Richard, S. Methylation of MRE11 regulates its nuclear compartmentalization. Cell Cycle 4, 981–989 (2005).
Boisvert, F. M., Dery, U., Masson, J. Y. & Richard, S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev. 19, 671–676 (2005).
Yu, Z. et al. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 22, 305–320 (2012).
Boisvert, F. M., Rhie, A., Richard, S. & Doherty, A. J. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle 4, 1834–1841 (2005).
Adams, M. M. et al. 53BP1 oligomerization is independent of its methylation by PRMT1. Cell Cycle 4, 1854–1861 (2005).
Krokan, H. E. & Bjoras, M. Base excision repair. Cold Spring Harb Perspect. Biol. 5, a012583 (2013).
El-Andaloussi, N. et al. Methylation of DNA polymerase beta by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. FASEB J. 21, 26–34 (2007).
Guo, Z. et al. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 6, 766–773 (2010).
He, L. et al. PRMT1 is critical to FEN1 expression and drug resistance in lung cancer cells. DNA Repair (Amst) 95, 102953 (2020).
Guendel, I. et al. Methylation of the tumor suppressor protein, BRCA1, influences its transcriptional cofactor function. PLoS ONE 5, e11379 (2010).
Lee, Y. H., Bedford, M. T. & Stallcup, M. R. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev. 25, 176–188 (2011).
He, W. et al. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res. 39, 4719–4727 (2011).
Hafner, A., Bulyk, M. L., Jambhekar, A. & Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210 (2019).
Jansson, M. et al. Arginine methylation regulates the p53 response. Nat. Cell Biol. 10, 1431–1439 (2008).
Scoumanne, A., Zhang, J. & Chen, X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 37, 4965–4976 (2009).
Cho, E. C. et al. Arginine methylation controls growth regulation by E2F-1. EMBO J. 31, 1785–1797 (2012).
Hu, D. et al. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 6, 8419 (2015).
Clarke, T. L. et al. PRMT5-dependent methylation of the TIP60 coactivator RUVBL1 is a key regulator of homologous recombination. Mol. Cell 65, 900–916 e907 (2017).
Hamard, P. J. et al. PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Rep. 24, 2643–2657 (2018).
Hwang, J. W. et al. PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun. Biol. 3, 428 (2020).
Tang, J. et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275, 7723–7730 (2000).
Li, Z. et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. 27, 3226–3242 (2020).
Liao, H. W. et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Invest. 125, 4529–4543 (2015).
Wang, Y. et al. Oncogenic functions of Gli1 in pancreatic adenocarcinoma are supported by its PRMT1-mediated methylation. Cancer Res. 76, 7049–7058 (2016).
Avasarala, S. et al. PRMT1 is a novel regulator of epithelial-mesenchymal-transition in non-small cell lung cancer. J. Biol. Chem. 290, 13479–13489 (2015).
Zhong, J. et al. Nuclear loss of protein arginine N-methyltransferase 2 in breast carcinoma is associated with tumor grade and overexpression of cyclin D1 protein. Oncogene 33, 5546–5558 (2014).
Dong, F. et al. PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma. Nat. Commun. 9, 4552 (2018).
Wang, L. et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25, 21–36 (2014).
Liu, F. et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 19, 1358–1370 (2017).
Veazey, K. J. et al. CARM1 inhibition reduces histone acetyltransferase activity causing synthetic lethality in CREBBP/EP300-mutated lymphomas. Leukemia 34, 3269–3285 (2020).
Al-Dhaheri, M. et al. CARM1 is an important determinant of ERalpha-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer Res. 71, 2118–2128 (2011).
Wang, L. et al. MED12 methylation by CARM1 sensitizes human breast cancer cells to chemotherapy drugs. Sci. Adv. 1, e1500463 (2015).
Stein, C., Riedl, S., Ruthnick, D., Notzold, R. R. & Bauer, U. M. The arginine methyltransferase PRMT6 regulates cell proliferation and senescence through transcriptional repression of tumor suppressor genes. Nucleic Acids Res. 40, 9522–9533 (2012).
Veland, N. et al. The arginine methyltransferase PRMT6 regulates DNA methylation and contributes to global DNA hypomethylation in cancer. Cell Rep. 21, 3390–3397 (2017).
Powers, M. A., Fay, M. M., Factor, R. E., Welm, A. L. & Ullman, K. S. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 71, 5579–5587 (2011).
Chiang, K. et al. PRMT5 is a critical regulator of breast cancer stem cell function via histone methylation and FOXP1 expression. Cell Rep. 21, 3498–3513 (2017).
Gu, Z. et al. Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem. J. 446, 235–241 (2012).
Shilo, K. et al. Cellular localization of protein arginine methyltransferase-5 correlates with grade of lung tumors. Diagn. Pathol. 8, 201 (2013).
Bao, X. et al. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 61, 206–217 (2013).
Deng, X. et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 36, 1223–1231 (2017).
Hartley, A. V. et al. PRMT5-mediated methylation of YBX1 regulates NF-kappaB activity in colorectal cancer. Sci. Rep. 10, 15934 (2020).
Liu, X. et al. Protein arginine methyltransferase 5-mediated epigenetic silencing of IRX1 contributes to tumorigenicity and metastasis of gastric cancer. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2835–2844 (2018).
Kanda, M. et al. Protein arginine methyltransferase 5 is associated with malignant phenotype and peritoneal metastasis in gastric cancer. Int. J. Oncol. 49, 1195–1202 (2016).
Jeon, J. Y. et al. Protein arginine methyltransferase 5 is implicated in the aggressiveness of human hepatocellular carcinoma and controls the invasive activity of cancer cells. Oncol. Rep. 40, 536–544 (2018).
Jiang, H. et al. PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 7, 869–882 (2018).
Qin, Y. et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal. 17, 30 (2019).
Ge, L. et al. PRMT5 promotes epithelial-mesenchymal transition via EGFR-beta-catenin axis in pancreatic cancer cells. J. Cell Mol. Med. 24, 1969–1979 (2020).
Fan, Z. et al. Targeting methyltransferase PRMT5 retards the carcinogenesis and metastasis of HNSCC via epigenetically inhibiting Twist1 transcription. Neoplasia 22, 617–629 (2020).
Hu, G., Wang, X., Han, Y. & Wang, P. Protein arginine methyltransferase 5 promotes bladder cancer growth through inhibiting NF-kB dependent apoptosis. EXCLI J. 17, 1157–1166 (2018).
Zhu, F. et al. PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia 33, 2898–2911 (2019).
Tarighat, S. S. et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30, 789–799 (2016).
Tamiya, H. et al. SHARPIN-mediated regulation of protein arginine methyltransferase 5 controls melanoma growth. J. Clin. Invest. 128, 517–530 (2018).
Yan, F. et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 74, 1752–1765 (2014).
Pal, S., Vishwanath, S. N., Erdjument-Bromage, H., Tempst, P. & Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol. 24, 9630–9645 (2004).
Beketova, E. et al. Protein arginine methyltransferase 5 promotes pICln-dependent androgen receptor transcription in castration-resistant prostate cancer. Cancer Res. 80, 4904–4917 (2020).
Jing, P. et al. Protein arginine methyltransferase 5 promotes lung cancer metastasis via the epigenetic regulation of miR-99 family/FGFR3 signaling. Cancer Lett. 427, 38–48 (2018).
Fay, M. M., Clegg, J. M., Uchida, K. A., Powers, M. A. & Ullman, K. S. Enhanced arginine methylation of programmed cell death 4 protein during nutrient deprivation promotes tumor cell viability. J. Biol. Chem. 289, 17541–17552 (2014).
Marjon, K. et al. MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep. 15, 574–587 (2016).
Kryukov, G. V. et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 (2016).
Mavrakis, K. J. et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213 (2016).
Fedoriw, A. et al. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell 36, 100–114 e125 (2019).
Yao, R. et al. PRMT7 induces epithelial-to-mesenchymal transition and promotes metastasis in breast cancer. Cancer Res. 74, 5656–5667 (2014).
Liu, Y. et al. Arginine methylation of SHANK2 by PRMT7 promotes human breast cancer metastasis through activating endosomal FAK signalling. elife https://doi.org/10.7554/eLife.57617 (2020).
Fong, J. Y. et al. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 36, 194–209 e199 (2019).
Hu, H. et al. Exploration of cyanine compounds as selective inhibitors of protein arginine methyltransferases: synthesis and biological evaluation. J. Med. Chem. 58, 1228–1243 (2015).
Spannhoff, A. et al. Target-based approach to inhibitors of histone arginine methyltransferases. J. Med. Chem. 50, 2319–2325 (2007).
Eram, M. S. et al. A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 11, 772–781 (2016).
Gao, G. et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 47, 5038–5048 (2019).
Siarheyeva, A. et al. An allosteric inhibitor of protein arginine methyltransferase 3. Structure 20, 1425–1435 (2012).
Kaniskan, H. U. et al. A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew. Chem. Int. Ed. Engl. 54, 5166–5170 (2015).
Nakayama, K. et al. TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 9, 18480–18493 (2018).
Drew, A. E. et al. Identification of a CARM1 inhibitor with potent in vitro and in vivo activity in preclinical models of multiple myeloma. Sci. Rep. 7, 17993 (2017).
Shen, Y. et al. Discovery of a potent, selective, and cell-active dual inhibitor of protein arginine methyltransferase 4 and protein arginine methyltransferase 6. J. Med. Chem. 59, 9124–9139 (2016).
Mitchell, L. H. et al. Aryl pyrazoles as potent inhibitors of arginine methyltransferases: identification of the first PRMT6 tool compound. ACS Med. Chem. Lett. 6, 655–659 (2015).
Wu, H. et al. Structural basis of arginine asymmetrical dimethylation by PRMT6. Biochem. J. 473, 3049–3063 (2016).
Shen, Y. et al. Discovery of a first-in-class protein arginine methyltransferase 6 (PRMT6) covalent inhibitor. J. Med. Chem. 63, 5477–5487 (2020).
Jin, Y. et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Invest. 126, 3961–3980 (2016).
Koh, C. M. et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100 (2015).
Chan-Penebre, E. et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 11, 432–437 (2015).
Duncan, K. W. et al. Structure and property guided design in the identification of PRMT5 tool compound EPZ015666. ACS Med. Chem. Lett. 7, 162–166 (2016).
Gerhart, S. V. et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 8, 9711 (2018).
Brehmer, D. et al. In Proc. American Association for Cancer Research Annual Meeting 2017. DDT02-04 (American Association for Cancer Research, 2017).
Bonday, Z. Q. et al. LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med. Chem. Lett. 9, 612–617 (2018).
Antonysamy, S. et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl Acad. Sci. USA 109, 17960–17965 (2012).
Shen, Y. et al. Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem. 63, 9977–9989 (2020).
Sun, X. et al. PROTACs: great opportunities for academia and industry. Signal. Transduct. Target Ther. 4, 64 (2019).
Gao, H., Sun, X. & Rao, Y. PROTAC technology: opportunities and challenges. ACS Med. Chem. Lett. 11, 237–240 (2020).
Karkhanis, V. et al. Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase delta catalytic subunit gene, POLD1. J. Biol. Chem. 287, 29801–29814 (2012).
Nagai, Y. et al. PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185(erbB2/neu) tumor immunotherapy. Front. Immunol. 10, 174 (2019).
Szewczyk, M. M. et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat. Commun. 11, 2396 (2020).
Katsuno, Y. et al. Arginine methylation of SMAD7 by PRMT1 in TGF-beta-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J. Biol. Chem. 293, 13059–13072 (2018).
Xu, J. et al. Arginine methylation initiates BMP-induced Smad signaling. Mol. Cell 51, 5–19 (2013).
Stopa, N., Krebs, J. E. & Shechter, D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol. Life Sci. 72, 2041–2059 (2015).
Karkhanis, V., Hu, Y. J., Baiocchi, R. A., Imbalzano, A. N. & Sif, S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 36, 633–641 (2011).
Liu, F. et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 19, 283–294 (2011).
Sipos, A. et al. Myosin phosphatase and RhoA-activated kinase modulate arginine methylation by the regulation of protein arginine methyltransferase 5 in hepatocellular carcinoma cells. Sci. Rep. 7, 40590 (2017).
Wang, H. et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293, 853–857 (2001).
Strahl, B. D. et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 11, 996–1000 (2001).
Min, Z. et al. Asymmetrical methyltransferase PRMT3 regulates human mesenchymal stem cell osteogenesis via miR-3648. Cell Death Dis. 10, 581 (2019).
Zhao, Q. et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 16, 304–311 (2009).
Cesaro, E. et al. The Kruppel-like zinc finger protein ZNF224 recruits the arginine methyltransferase PRMT5 on the transcriptional repressor complex of the aldolase A gene. J. Biol. Chem. 284, 32321–32330 (2009).
Fabbrizio, E. et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645 (2002).
Hyllus, D. et al. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 21, 3369–3380 (2007).
Guccione, E. et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449, 933–937 (2007).
Blythe, S. A., Cha, S. W., Tadjuidje, E., Heasman, J. & Klein, P. S. beta-Catenin primes organizer gene expression by recruiting a histone H3 arginine 8 methyltransferase, Prmt2. Dev. Cell 19, 220–231 (2010).
Dacwag, C. S., Ohkawa, Y., Pal, S., Sif, S. & Imbalzano, A. N. The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol. Cell Biol. 27, 384–394 (2007).
Schurter, B. T. et al. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 40, 5747–5756 (2001).
Ma, H. et al. Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr. Biol. 11, 1981–1985 (2001).
Daujat, S. et al. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr. Biol. 12, 2090–2097 (2002).
Casadio, F. et al. H3R42me2a is a histone modification with positive transcriptional effects. Proc. Natl Acad. Sci. USA 110, 14894–14899 (2013).
Waldmann, T. et al. Methylation of H2AR29 is a novel repressive PRMT6 target. Epigenet. Chromatin 4, 11 (2011).
Mowen, K. A. et al. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 104, 731–741 (2001).
Liu, L. M. et al. Methylation of C/EBPalpha by PRMT1 inhibits its tumor-suppressive function in breast cancer. Cancer Res. 79, 2865–2877 (2019).
Zhao, X. et al. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 22, 640–653 (2008).
Yamagata, K. et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231 (2008).
Liu, Q., Zhang, X. L., Cheng, M. B. & Zhang, Y. PRMT1 activates myogenin transcription via MyoD arginine methylation at R121. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 194442 (2019).
Liu, X. et al. Methylation of arginine by PRMT1 regulates Nrf2 transcriptional activity during the antioxidative response. Biochim. Biophys. Acta. 1863, 2093–2103 (2016).
Reintjes, A. et al. Asymmetric arginine dimethylation of RelA provides a repressive mark to modulate TNFalpha/NF-kappaB response. Proc. Natl Acad. Sci. USA 113, 4326–4331 (2016).
Xu, W. et al. A transcriptional switch mediated by cofactor methylation. Science 294, 2507–2511 (2001).
Chevillard-Briet, M., Trouche, D. & Vandel, L. Control of CBP co-activating activity by arginine methylation. EMBO J. 21, 5457–5466 (2002).
Lee, Y. H., Coonrod, S. A., Kraus, W. L., Jelinek, M. A. & Stallcup, M. R. Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc. Natl Acad. Sci. USA 102, 3611–3616 (2005).
Ceschin, D. G. et al. Methylation specifies distinct estrogen-induced binding site repertoires of CBP to chromatin. Genes Dev. 25, 1132–1146 (2011).
Zhao, H. Y., Zhang, Y. J., Dai, H., Zhang, Y. & Shen, Y. F. CARM1 mediates modulation of Sox2. PLoS ONE 6, e27026 (2011).
Liu, Y., Li, J., Shang, Y., Guo, Y. & Li, Z. CARM1 contributes to skeletal muscle wasting by mediating FoxO3 activity and promoting myofiber autophagy. Exp. Cell Res. 374, 198–209 (2019).
Ito, T. et al. Arginine methyltransferase CARM1/PRMT4 regulates endochondral ossification. BMC Dev. Biol. 9, 47 (2009).
Wei, H. et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc. Natl Acad. Sci. USA 110, 13516–13521 (2013).
Harris, D. P., Chandrasekharan, U. M., Bandyopadhyay, S., Willard, B. & DiCorleto, P. E. PRMT5-mediated methylation of NF-kappaB p65 at Arg174 is required for endothelial CXCL11 gene induction in response to TNF-alpha and IFN-gamma costimulation. PLoS ONE 11, e0148905 (2016).
Abe, Y., Suzuki, Y., Kawamura, K. & Tanaka, N. MEP50/PRMT5-mediated methylation activates GLI1 in Hedgehog signalling through inhibition of ubiquitination by the ITCH/NUMB complex. Commun. Biol. 2, 23 (2019).
Bandyopadhyay, S. et al. HOXA9 methylation by PRMT5 is essential for endothelial cell expression of leukocyte adhesion molecules. Mol. Cell Biol. 32, 1202–1213 (2012).
Vuong, T. A. et al. PRMT7 methylates and suppresses GLI2 binding to SUFU thereby promoting its activation. Cell Death Differ. 27, 15–28 (2020).
Meister, G. et al. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 11, 1990–1994 (2001).
Friesen, W. J., Massenet, S., Paushkin, S., Wyce, A. & Dreyfuss, G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 7, 1111–1117 (2001).
Friesen, W. J. et al. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell Biol. 21, 8289–8300 (2001).
Miranda, T. B. et al. Spliceosome Sm proteins D1, D3, and B/B’ are asymmetrically dimethylated at arginine residues in the nucleus. Biochem. Biophys. Res. Commun. 323, 382–387 (2004).
Cheng, D., Cote, J., Shaaban, S. & Bedford, M. T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 25, 71–83 (2007).
Brahms, H., Meheus, L., de Brabandere, V., Fischer, U. & Luhrmann, R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B’ and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA 7, 1531–1542 (2001).
Arribas-Layton, M., Dennis, J., Bennett, E. J., Damgaard, C. K. & Lykke-Andersen, J. The C-terminal RGG domain of human Lsm4 promotes processing body formation stimulated by arginine dimethylation. Mol. Cell Biol. 36, 2226–2235 (2016).
Boisvert, F. M. et al. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol. 159, 957–969 (2002).
Whitehead, S. E. et al. Determinants of the interaction of the spinal muscular atrophy disease protein SMN with the dimethylarginine-modified box H/ACA small nucleolar ribonucleoprotein GAR1. J. Biol. Chem. 277, 48087–48093 (2002).
Nichols, R. C. et al. The RGG domain in hnRNP A2 affects subcellular localization. Exp. Cell Res. 256, 522–532 (2000).
Passos, D. O., Quaresma, A. J. & Kobarg, J. The methylation of the C-terminal region of hnRNPQ (NSAP1) is important for its nuclear localization. Biochem. Biophys. Res. Commun. 346, 517–525 (2006).
Ostareck-Lederer, A. et al. Asymmetric arginine dimethylation of heterogeneous nuclear ribonucleoprotein K by protein-arginine methyltransferase 1 inhibits its interaction with c-Src. J. Biol. Chem. 281, 11115–11125 (2006).
Zhang, L. et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. elife https://doi.org/10.7554/eLife.07938 (2015).
Tadesse, H., Deschenes-Furry, J., Boisvenue, S. & Cote, J. KH-type splicing regulatory protein interacts with survival motor neuron protein and is misregulated in spinal muscular atrophy. Hum. Mol. Genet. 17, 506–524 (2008).
Rengasamy, M. et al. The PRMT5/WDR77 complex regulates alternative splicing through ZNF326 in breast cancer. Nucleic Acids Res. 45, 11106–11120 (2017).
Yang, Y. et al. PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun. 6, 6428 (2015).
Hadjikyriacou, A., Yang, Y., Espejo, A., Bedford, M. T. & Clarke, S. G. Unique features of human protein arginine methyltransferase 9 (PRMT9) and its substrate RNA splicing factor SF3B2. J. Biol. Chem. 290, 16723–16743 (2015).
Thandapani, P. et al. Aven recognition of RNA G-quadruplexes regulates translation of the mixed lineage leukemia protooncogenes. elife https://doi.org/10.7554/eLife.06234 (2015).
Huang, L., Wang, Z., Narayanan, N. & Yang, Y. Arginine methylation of the C-terminus RGG motif promotes TOP3B topoisomerase activity and stress granule localization. Nucleic Acids Res. 46, 3061–3074 (2018).
Shin, H. S. et al. Arginine methylation of ribosomal protein S3 affects ribosome assembly. Biochem. Biophys. Res. Commun. 385, 273–278 (2009).
Swiercz, R., Cheng, D., Kim, D. & Bedford, M. T. Ribosomal protein rpS2 is hypomethylated in PRMT3-deficient mice. J. Biol. Chem. 282, 16917–16923 (2007).
Choi, S., Jung, C. R., Kim, J. Y. & Im, D. S. PRMT3 inhibits ubiquitination of ribosomal protein S2 and together forms an active enzyme complex. Biochim. Biophys. Acta. 1780, 1062–1069 (2008).
Lee, J. & Bedford, M. T. PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep. 3, 268–273 (2002).
Gao, G., Dhar, S. & Bedford, M. T. PRMT5 regulates IRES-dependent translation via methylation of hnRNP A1. Nucleic Acids Res. 45, 4359–4369 (2017).
Ren, J. et al. Methylation of ribosomal protein S10 by protein-arginine methyltransferase 5 regulates ribosome biogenesis. J. Biol. Chem. 285, 12695–12705 (2010).
Haghandish, N. et al. PRMT7 methylates eukaryotic translation initiation factor 2alpha and regulates its role in stress granule formation. Mol. Biol. Cell 30, 778–793 (2019).
Hsu, J. M. et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 13, 174–181 (2011).
Andreu-Perez, P. et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci. Signal. 4, ra58 (2011).
Chan, L. H. et al. PRMT6 regulates RAS/RAF binding and MEK/ERK-mediated cancer stemness activities in hepatocellular carcinoma through CRAF methylation. Cell Rep. 25, 690–701 e698 (2018).
Jeong, H. J. et al. Prmt7 promotes myoblast differentiation via methylation of p38MAPK on arginine residue 70. Cell Death Differ. 27, 573–586 (2020).
Cho, J. H. et al. Arginine methylation-dependent regulation of ASK1 signaling by PRMT1. Cell Death Differ. 19, 859–870 (2012).
Chen, M. et al. Cross-talk between Arg methylation and Ser phosphorylation modulates apoptosis signal-regulating kinase 1 activation in endothelial cells. Mol. Biol. Cell 27, 1358–1366 (2016).
Zheng, S. et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell 52, 37–51 (2013).
Choucair, A. et al. The arginine methyltransferase PRMT1 regulates IGF-1 signaling in breast cancer. Oncogene 38, 4015–4027 (2019).
Gao, Y. et al. The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci. Rep. 6, 19874 (2016).
Wang, L. et al. Methylation of HSP70 orchestrates its binding to and stabilization of BCL2 mRNA and renders pancreatic cancer cells resistant to therapeutics. Cancer Res. 80, 4500–4513 (2020).
Ryu, J. W. et al. Novel prognostic marker PRMT1 regulates cell growth via downregulation of CDKN1A in HCC. Oncotarget 8, 115444–115455 (2017).
Li, L., Zhang, Z., Ma, T. & Huo, R. PRMT1 regulates tumor growth and metastasis of human melanoma via targeting ALCAM. Mol. Med. Rep. 14, 521–528 (2016).
Chuang, C. Y. et al. PRMT1 expression is elevated in head and neck cancer and inhibition of protein arginine methylation by adenosine dialdehyde or PRMT1 knockdown downregulates proliferation and migration of oral cancer cells. Oncol. Rep. 38, 1115–1123 (2017).
Zhou, W., Yue, H., Li, C., Chen, H. & Yuan, Y. Protein arginine methyltransferase 1 promoted the growth and migration of cancer cells in esophageal squamous cell carcinoma. Tumour Biol. 37, 2613–2619 (2016).
Zhong, J. et al. Identification and characterization of novel spliced variants of PRMT2 in breast carcinoma. FEBS J. 279, 316–335 (2012).
Hsu, M. C. et al. Protein arginine methyltransferase 3-induced metabolic reprogramming is a vulnerable target of pancreatic cancer. J. Hematol. Oncol. 12, 79 (2019).
Liu, J. et al. Arginine methylation-dependent LSD1 stability promotes invasion and metastasis of breast cancer. EMBO Rep. 21, e48597 (2020).
Ou, C. Y. et al. A coactivator role of CARM1 in the dysregulation of beta-catenin activity in colorectal cancer cell growth and gene expression. Mol. Cancer Res. 9, 660–670 (2011).
Wang, Y. P. et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol. Cell 64, 673–687 (2016).
Zhong, X. Y. et al. CARM1 methylates GAPDH to regulate glucose metabolism and is suppressed in liver cancer. Cell Rep. 24, 3207–3223 (2018).
Karakashev, S. et al. CARM1-expressing ovarian cancer depends on the histone methyltransferase EZH2 activity. Nat. Commun. 9, 631 (2018).
Vu, L. P. et al. PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep. 5, 1625–1638 (2013).
Greenblatt, S. M. et al. CARM1 is essential for myeloid leukemogenesis but dispensable for normal hematopoiesis. Cancer Cell 33, 1111–1127 e1115 (2018).
Chung, J., Karkhanis, V., Baiocchi, R. A. & Sif, S. Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/beta-catenin and AKT/GSK3beta proliferative signaling. J. Biol. Chem. 294, 7692–7710 (2019).
Wang, L., Pal, S. & Sif, S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell Biol. 28, 6262–6277 (2008).
Radzisheuskaya, A. et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat. Struct. Mol. Biol. 26, 999–1012 (2019).
Wang, Z. et al. PRMT5 determines the sensitivity to chemotherapeutics by governing stemness in breast cancer. Breast Cancer Res. Treat. 168, 531–542 (2018).
Yang, F. et al. Proliferative role of TRAF4 in breast cancer by upregulating PRMT5 nuclear expression. Tumour Biol. 36, 5901–5911 (2015).
Zhang, S., Ma, Y., Hu, X., Zheng, Y. & Chen, X. Targeting PRMT5/Akt signalling axis prevents human lung cancer cell growth. J. Cell Mol. Med. 23, 1333–1342 (2019).
Fu, T., Lv, X., Kong, Q. & Yuan, C. A novel SHARPIN-PRMT5-H3R2me1 axis is essential for lung cancer cell invasion. Oncotarget 8, 54809–54820 (2017).
Zakrzewicz, D. et al. Protein arginine methyltransferase 5 mediates enolase-1 cell surface trafficking in human lung adenocarcinoma cells. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1816–1827 (2018).
Mounir, Z. et al. ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the Androgen Receptor. elife https://doi.org/10.7554/eLife.13964 (2016).
Liu, M. et al. PRMT5-dependent transcriptional repression of c-Myc target genes promotes gastric cancer progression. Theranostics 10, 4437–4452 (2020).
AbuHammad, S. et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl Acad. Sci. USA 116, 17990–18000 (2019).
Okuno, K. et al. Asymmetric dimethylation at histone H3 arginine 2 by PRMT6 in gastric cancer progression. Carcinogenesis 40, 15–26 (2019).
Jiang, N. et al. PRMT6 promotes endometrial cancer via AKT/mTOR signaling and indicates poor prognosis. Int. J. Biochem. Cell Biol. 120, 105681 (2020).
Avasarala, S. et al. PRMT6 promotes lung tumor progression via the alternate activation of tumor-associated macrophages. Mol. Cancer Res. 18, 166–178 (2020).
Baldwin, R. M. et al. Protein arginine methyltransferase 7 promotes breast cancer cell invasion through the induction of MMP9 expression. Oncotarget 6, 3013–3032 (2015).
Cheng, D. et al. PRMT7 contributes to the metastasis phenotype in human non-small-cell lung cancer cells possibly through the interaction with HSPA5 and EEF2. Onco Targets Ther. 11, 4869–4876 (2018).
Liu, F. et al. PRMT7 promotes the growth of renal cell carcinoma through modulating the beta-catenin/C-MYC axis. Int. J. Biochem. Cell Biol. 120, 105686 (2020).
Jiang, H. et al. PRMT9 promotes hepatocellular carcinoma invasion and metastasis via activating PI3K/Akt/GSK-3beta/Snail signaling. Cancer Sci. 109, 1414–1427 (2018).
Cheng, D. et al. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 279, 23892–23899 (2004).
Mcalpine, I. J. et al. In Proceedings of the American Association for Cancer Research Annual Meeting 2018. 4857 (American Association for Cancer Research, 2018).
Jain, K. & Clarke, S. G. PRMT7 as a unique member of the protein arginine methyltransferase family: a review. Arch. Biochem. Biophys. 665, 36–45 (2019).
Yan, L. et al. Diamidine compounds for selective inhibition of protein arginine methyltransferase 1. J Med. Chem. 57, 2611–2622 (2014).
Smil, D. et al. Discovery of a dual PRMT5-PRMT7 inhibitor. ACS Med. Chem. Lett. 6, 408–412 (2015).
van Haren, M. J. & Martin, N. I. PRMT Inhibitors Vol. 33 159–196 (Springer, 2019).
Acknowledgements
This study was supported by grants from the National Research Foundation of Korea (NRF) and the Korean government (MSIP) (NRF-2018R1A2B2005646, NRF-2021R1A2C2013613, and NRF-2018R1D1A1A02051170)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hwang, J.W., Cho, Y., Bae, GU. et al. Protein arginine methyltransferases: promising targets for cancer therapy. Exp Mol Med 53, 788–808 (2021). https://doi.org/10.1038/s12276-021-00613-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00613-y
This article is cited by
-
PRMT3 methylates HIF-1α to enhance the vascular calcification induced by chronic kidney disease
Molecular Medicine (2024)
-
ALS-linked FUS R521C disrupts arginine methylation of UBAP2L and stress granule dynamics
Journal of Analytical Science and Technology (2023)
-
Automated imaging and identification of proteoforms directly from ovarian cancer tissue
Nature Communications (2023)
-
Targeting epigenetic regulators to overcome drug resistance in cancers
Signal Transduction and Targeted Therapy (2023)
-
Histone modifications in drug-resistant cancers: From a cancer stem cell and immune evasion perspective
Experimental & Molecular Medicine (2023)
