
Abstract
Disease resistance is essential for reliable maize production. In a long-term tug-of-war between maize and its pathogenic microbes, naturally occurring resistance genes gradually accumulate and play a key role in protecting maize from various destructive diseases. Recently, significant progress has been made in deciphering the genetic basis of disease resistance in maize. Enhancing disease resistance can now be explored at the molecular level, from marker-assisted selection to genomic selection, transgenesis technique, and genome editing. In view of the continuing accumulation of cloned resistance genes and in-depth understanding of their resistance mechanisms, coupled with rapid progress of biotechnology, it is expected that the large-scale commercial application of molecular breeding of resistant maize varieties will soon become a reality.
Similar content being viewed by others


Introduction
Maize (Zea mays L.) is one of the most important crops for food, feed, and fuel production worldwide. The global demand for maize continues to grow because of the increasing need for feed and industrial raw materials. In recent years, due to the ever-shrinking genetic diversity of maize varieties, continuous cropping, returning straw to the field, high-fertility management, high-density planting, and extreme climatic events, maize diseases are becoming more and more serious (Duan et al. 2019; Sun et al. 2020). Annual yield losses caused by maize diseases (excluding viral diseases) are estimated to account for 4–14% of the global harvest (https://portal.nifa.usda.gov/web/crisprojectpages/1008502-genetic-architecture-of-disease-resistance-in-maize.html). Since the arable land area of maize will not increase significantly, it will be crucial to ensure stable maize yields and high kernel quality by reducing disease severity in this crop.
In the long history of maize cultivation, disease epidemics in maize is changing dynamically, as some diseases increase or decrease in importance relative to other diseases. Disease prevalence in maize is closely correlated with pathogen resources, cultivated varieties, weather conditions, farming systems, and agricultural ecology (Yang et al. 2017a). The outbreak of southern corn leaf blight (SCLB) in the USA in 1970 was due to the wide deployment of susceptible cultivars with T-type male-sterile cytoplasm (cms-T), which suffered from the new Bipolaris maydis race T (Ullstrup 1972). The recent occurrence of northern corn leaf blight (NCLB) in North China was due to the widespread cultivation of the susceptible variety XianYu335 (Pu 2013). The high temperature and humidity in the Huang-Huai-Hai plain are conducive to the outbreak of stalk rot and ear rot diseases (Duan et al. 2019). In mechanical harvesting, maize plants are left in the field for a longer period of time for dehydration, which will undoubtedly increase the severity of stalk/ear rot diseases. Moreover, the failure to remove rotted ears during mechanical harvesting further reduces kernel quality (Silva et al. 2017; Holland et al. 2020).
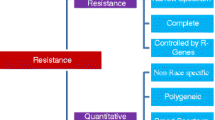
The plant immune response is a highly complex, tightly regulated, multi-layered process that can be roughly divided into qualitative disease resistance and quantitative disease resistance (QDR) (Poland et al. 2009; Kou and Wang 2010). Resistance genes (R-gene) underlying qualitative resistance tend to provide complete or near-complete resistance and are therefore also known as major genes (Nelson et al. 2018). QDR confers an incomplete or partial resistance and is controlled by multiple small-effect genes (Niks et al. 2015). Although single R-genes are often non-durable, when used in combination with QDR genes, they can effectively promote crop protection against pathogens (Palloix et al. 2009). Therefore, combining multiple R-genes and/or QDRs into a single genome is the optimal choice for breeding varieties with strong and durable disease resistance.
As early as 1992, a major gene Hm1 was isolated in maize by transposon-tagging method, which confers resistance to Cochliobolus carbonum race 1 (Johal and Briggs 1992). Notably, Hm1 is also the first resistance gene identified in any plant species. With the same transposon-tagging approach, another major gene Rp1-D was isolated in 1999, which confers resistance against common leaf rust (Collins et al. 1999). During the maize growth period, however, the most devastating diseases are caused by necrotrophic or hemibiotrophic pathogens, and the resistance to such diseases mainly depends on QDR genes (Yang et al. 2017a). It turns out that cloning of the QDR gene is much more difficult than the major gene. It is not until recently that there have been reports of successful cloning of QDR genes (Zuo et al. 2015; Hurni et al. 2015; Yang et al. 2017b; Wang et al. 2017; Liu et al. 2017; Leng et al. 2017; Li et al. 2019; Ye et al. 2019; Yang et al. 2021; Liu et al. 2020a).
In this review, we summarize recent advances in functional genomics on maize disease resistance, describe the current works on molecular breeding, and predict the potential development in the future. Several similar reviews may help to better understand the maize disease resistance (Poland et al. 2009; Kou and Wang 2010; St Clair 2010; Zhang et al. 2013; Niks et al. 2015; Krattinger and Keller 2016; Ali and Yan 2012; Yang et al. 2017a).
Functional genomics of disease resistance in maize
Inheritance of resistance to fungal diseases in maize
Most maize diseases are caused by pathogenic fungi (Table 1). These diseases cause significant economic losses due to reduced yield/quality and the increasing input cost for disease control. Foliar disease, smut, and stem/ear rot are among the most serious fungal diseases of maize (Azra and Hussain 2019).
Foliar fungal diseases of cereals are usually associated with reduced photosynthetic area, chlorosis, and premature leaf senescence (Fig. 1a–e), which result in incomplete grain filling and reduced grain yields (Zheng et al. 2018). A recent global survey highlighted several foliar fungal diseases that significantly reduce maize yields in Africa, Asia, and the Americas (Savary et al. 2019).

The phenotypes of major maize diseases. a NCLB mainly damages leaves and forms local lesions that progress until necrosis occurs. Lesions may coalesce, blighting the entire leaf. b GLS begins as small, regular, elongated necrotic spots. Lesions grow parallel to the veins. The growth is limited by adjacent veins, so the final lesion shape is rectangular. c SCLB mainly infects maize leaves. Lesions are initially small and diamond shaped, then become elongated as they mature. Under severe disease pressure, lesions may coalesce, blighting the entire leaf. d Common rust has small, powdery pustules over both surfaces of the leaves. Pustules are brown in early stages of infection; later, the epidermis ruptures and the lesions turn black as the plant matures. e Southern rust has small circular, pinhead-shaped pustules. Pustules are smaller, lighter in color, and more circular than those of common rust. Southern rust also presents on both leaf surfaces. f BLSB develops on leaves, sheaths, and husks. The symptoms are characteristic concentric spots that cover large areas of infected leaves and husks. g Head smut spreads systemically into the ear and tassel. The most conspicuous symptom is masses of black spores in the individual male florets and the ear. h Common smut is easily identified by white tumor-like galls which can develop in all aerial organs of maize. i Gibberella stalk rot may look similar to Fusarium stalk rot. Symptoms of stalk rot include rotting of the roots, crown, and lower internodes. Corn infected with Gibberella has pink to reddish coloration of the pith and vascular strands. j Fusarium ear rot is the most common fungal disease found on corn ears. Mold may be white, pink, or salmon-colored. Infected kernels may turn tan or brown. k SCMV infection causes characteristic chlorosis, here shown in a greenhouse-grown seedling. The new leaves of susceptible plants show yellow and green stripes. l MRDD-infected plants are usually dwarfed and severely stunted, with dark-green leaves, shortened internodes, and waxy enations on the abaxial surfaces of upper leaves
Northern corn leaf blight (NCLB) (Fig. 1a) causes the yield loss of > 1% globally (Savary et al. 2019). In the northern USA, NCLB was the most prominent corn disease in 2015 (Mueller et al. 2016). In Jilin Province of China, the NCLB outbreak in 2012 caused a substantial yield loss (Liu et al. 2013). Qualitative resistance conveyed by Ht genes results in distinct phenotypes in response to infection by avirulent races of Exserohilum turcicum. The Ht1 gene, identified from the popcorn cultivar Ladyfinger and the field corn inbred line GE440, confers a chlorotic-lesion reaction that reduces sporulation and lesion size (Hooker 1963). The Ht2 and Ht3 genes also confer chlorotic-lesion-mediated resistance (Hooker 1977; Hurni et al. 2015). The Htn1 locus was originally introgressed into modern maize cultivars from Mexican landrace Pepitilla in the 1970s (Gevers 1975). ZmWAK-RLK1 is the causal gene at Htn1, which encodes an unusual innate immunity receptor with an extracellular wall-associated kinase domain (Hurni et al. 2015), and the fungal resistance correlates with reduced benzoxazinoid content (Yang et al. 2019b). The latest research showed that Ht2 and Ht3 are identical and allelic to Htn1. The difference between the ZmWAK-RLK1 variants encoded by Htn1 and Ht2/Ht3 lies in multiple amino acid polymorphisms, which particularly affect the putative extracellular domain (Yang et al. 2021). The recessive gene ht4, identified in a line derived from the maize synthetic BS19, confers a chlorotic halo reaction to infection by E. turcicum (Carson 1995). Loci affecting quantitative resistance to NCLB have been mapped on all 10 maize chromosomes (Welz and Geiger 2000). ZmREM6.3, the causal gene of qNLB1.02B73, was identified by combing fine mapping, expression analysis, and mutant evaluations. ZmREM6.3 appears to have a specific effect on NCLB symptom development (Jamann et al. 2016).
Gray leaf spot (GLS) (Fig. 1b) is the second most serious foliar disease of maize worldwide (Savary et al. 2019). GLS resistance is a typical quantitative trait controlled by multiple resistance QTLs (Menkir and Ayodele 2005). ZmCCoAOMT2 was confirmed to be the causal gene at QTL qMdr9.02 conferring resistance to Cercospora zeae-maydis GLS. This gene encodes caffeoyl-CoA O-methyltransferase, an enzyme involved in the phenylpropanoid pathway and lignin production (Yang et al. 2017b). Another C. zeae-maydis GLS resistance QTL, Qgls8, was mapped to a ~ 130-kb region on chromosome 8 (Zhang et al. 2017b). Two major resistance QTLs against Cercospora zeina GLS, qRgls1 and qRgls2, were identified and fine-mapped to 1.4-Mb and 1-Mb regions on chromosomes 8 and 5, respectively (Zhang et al. 2012b; Xu et al. 2014). To date, more than 100 QTLs for GLS resistance have been detected (Du et al. 2020).
Southern corn leaf blight (SCLB) (Fig. 1c), once a major threat to global maize production, has declined to a relatively low level due to the use of resistant cultivars. A major recessive SCLB resistance locus, rhm1, was mapped to an 8.56-kb region on chromosome 6 with only one candidate gene, encoding the lysine histidine transporter 1 (LHT1) (Zhao et al. 2012b). To date, many QTL analyses of maize resistance to SCLB have been conducted, revealing numerous resistance QTLs that primarily exhibit additive or partially dominant or epistatic effects (Carson et al. 2004; Balint-Kurti and Carson 2006; Balint-Kurti et al. 2007; Balint-Kurti et al. 2008; Zwonitzer et al. 2009; Kaur et al. 2019). ZmCCoAOMT2, the causal gene of the QTL qMdr9.02, also confers quantitative resistance to SCLB (Yang et al. 2017b). Differences in ZmCCoAOMT2-mediated SCLB resistance are attributed to allelic variations at both the gene expression and amino acid sequence levels, which lead to differences in the levels of metabolites (e.g., lignin) in the phenylpropanoid pathway and programmed cell death (Yang et al. 2017b).
Common rust (Fig. 1d) is an important foliar disease that is widely distributed in tropical, subtropical, temperate, and highland areas (Vivek et al. 2009; Wright et al. 2014), and causes up to 49% yield losses in susceptible genotypes (Groth et al. 1983). The rp1 complex, a cluster of resistance genes, is located on the short arm of chromosome 10 (Hulbert 1997). Sixteen different genes were identified in the rp1 cluster by examining their responses to an extensive collection of rust biotypes, and fourteen of which were given the Rp1 designation (Rp1-A to Rp1-N) (Hooker 1969; Hulbert 1997). Among them, Rp1-D, encoding a typical resistance protein with nucleotide-binding and leucine-rich repeat (NB-LRR) domains, confers race-specific resistance to the disease (Collins et al. 1999). Since many Puccinia sorghi races that are virulent on Rp1-D have been found throughout North America (Pataky and Tracy 1999; Pate et al. 2000; Pataky et al. 2000), it is important to combine multiple R-genes with QDRs to generate maize varieties with durable resistance to common rust (Yang et al. 2017a). With genome-wide association studies (GWAS), 25 resistance QTLs were identified and distributed on chromosomes 1, 3, 5, 6, 8, and 10 (Zheng et al. 2018).
Southern rust (Fig. 1e) is generally more harmful to corn than common rust due to its ability to develop and spread rapidly under favorable conditions. To date, at least 18 race-specific resistance genes have been identified, and most have been widely used in commercial maize varieties, such as Rpp1-11 (Storey and Howland 1957; Ullstrup 1965; Brewbaker et al. 2011), Rpp25 (Zhao et al. 2013), RppQ (Chen et al. 2004; Zhou et al. 2007), RppD (Zhang et al. 2010), RppC (Yao et al. 2013), RppS313 (Wang et al. 2019a), RppS (Wu et al. 2015), and RppCML496 (Lv et al. 2020). Like other plant pathogenic microbes, Puccinia polysora is notorious for its rapid mutation to overcome maize resistance. For example, Rpp9 once provided effective resistance to SCR in the southern USA, but it has since been overcome by a new race of P. polysora (Brewbaker et al. 2011). The resistance QTLs have been identified and mapped on chromosomes 3 and 4 (Holland et al. 1998); 3, 4, and 9 (Jiang et al. 1999); 4, 8, 9, and 10 (Jines et al. 2007); 6 (Brewbaker et al. 2011); and 1, 2, 5, 6, 9, and 10 (Wanlayaporn et al. 2013). Plant-specific remorins are important for plant responses to microbial infections and plant signaling processes. Overexpressing the remorin gene ZmREM1.3 enhanced resistance to southern rust in maize (Wang et al. 2019c).
Banded leaf and sheath blight (BLSB) (Fig. 1f) is a widespread soil-borne fungal disease of both maize and rice in South and Southeast Asia (Zhao et al. 2006; Chen et al. 2013; Li et al. 2019). The F-box gene ZmFBL41 was identified as a causal gene conferring quantitative resistance to BLSB (Li et al. 2019). The activity of ZmFBL41 was evaluated in the transposon-insertion line zmfbl41 selected from the maize UniformMu resource. The zmfbl41 line exhibited weaker disease symptoms than the wild type (W22) following Rhizoctonia solani infection. Two amino acid substitutions in ZmFBL41 prevented its interaction with ZmCAD (the final enzyme in the monolignol biosynthetic pathway). This resulted in inhibited ZmCAD degradation, leading to lignin accumulation and limiting lesion expansion (Li et al. 2019).
Head smut (Fig. 1g) and common smut (Fig. 1h) are both soil-borne diseases of maize and pose serious threats to maize production. Many head smut resistance QTLs have been identified across all 10 chromosomes (Lübberstedt et al. 1999; Chen et al. 2008; Li et al. 2015). A major dominant QTL qHSR1 on the long arm of chromosome 2 reduced the disease incidence by ~ 25% (Chen et al. 2008). ZmWAK is the causal resistance gene at qHSR1 and encodes a cell wall–associated kinase (WAK), composing of a cytoplasmic serine/threonine kinase domain, a calcium-binding epidermal growth factor (EGF_CA) domain, and an extracellular galacturonan-binding (GUB) domain (Zuo et al. 2015). ZmWAK spans the plasma membrane and functions as a receptor-like kinase that may perceive and transduce extracellular signals. ZmWAK is highly expressed in the mesocotyls of maize seedlings, where it represses the growth of hyphae towards aboveground plant tissues, resulting in a significant decrease in the pathogen amount in floral organs, thereby reducing the disease severity (Zuo et al. 2015). Common smut, caused by Ustilago maydis, can be easily identified by the formation of tumor-like galls in all aerial organs of maize plants, which results in stunted growth and yield losses (Martínez-Espinoza et al. 2002; Tanaka et al. 2020). In recent years, great progress has been made in the study of the pathogenic mechanism of U. maydis and its interaction with plants (Ma et al. 2018; Tanaka et al. 2020; Zuo et al. 2019). The disease resistance loci are distributed on all 10 maize chromosomes; however, none of them have been identified (Pataky 1995; Lübberstedt et al. 1998; Ding et al. 2008).
With the development of agricultural mechanization, stalk rot (Fig. 1i) and ear rot (Fig. 1j) have attracted much attention, and prompted genetic studies on them. Two QTLs, the major qRfg1 and the minor qRfg2, were identified in the resistant inbred line 1145 (Yang et al. 2010; Zhang et al. 2012a). The ZmCCT gene containing a CCT domain is the causal gene at qRfg1 (Wang et al. 2017). The insertion or deletion of a CACTA-like transposon in the ZmCCT promoter causes differential histone modification and DNA methylation to regulate maize resistance to stalk rot (Wang et al. 2017). Without the transposon insertion, ZmCCT is in the “primed” state, allowing plants to respond quickly to pathogen challenge and mount defense responses. By contrast, ZmCCT with the transposon insertion is in the “silent” state, eliciting little or no defense response to pathogen invasion (Wang et al. 2017). ZmAuxRP1 is the causal gene at the minor QTL, qRfg2, that responds quickly to pathogen challenge with a rapid yet transient reduction in its expression, leading to arrested root growth but enhanced resistance to Gibberella stalk rot (Ye et al. 2019). ZmAuxRP1 promotes the biosynthesis of indole-3-acetic acid (IAA), while suppressing the formation of benzoxazinoid defense compounds (BXs). The concerted interplay between IAA and BXs helps maintain the growth-defense balance in a timely and efficient manner to optimize plant fitness (Ye et al. 2019). Interestingly, ZmAuxRP1 increases the resistance to Fusarium ear rot as well, suggesting that the same mechanism is used for resistance to both stalk rot and ear rot (Ye et al. 2019).
Rcg1 is a major QTL associated with resistance to Anthracnose stalk rot (ASR) caused by the fungus Colletotrichum graminicola (Jung et al. 1994). Rcg1 was identified in the inbred line MP305 by fine mapping, followed by mutant analysis. Rcg1 harbors an NB-LRR disease resistance gene that delays the occurrence of Anthracnose stalk rot, causing the disease to have little impact on plant yield (Frey et al. 2011).
LOX (lipoxygenase) genes are thought to be involved in plant susceptibility to fungal invasion and mycotoxin production (Christensen et al. 2014; Maschietto et al. 2015). Maize mutants with a defect in the 9-LOX gene ZmLOX3 show reduced levels of several 9-LOX-derived fatty acid hydroperoxides. The kernels of lox3 mutants show greatly reduced ear rot symptoms, including drastically reduced conidiation of F. verticillioides and reduced production of the mycotoxin fumonisin B1 (Gao et al. 2007; Gao et al. 2009). By contrast, infection by F. verticillioides is suppressed by the maize 9-LOX gene ZmLOX12 (Christensen et al. 2014). These observations suggest that a specific plant 9-LOX isoform is required for fungal pathogenesis, including disease development and spore and mycotoxin production (Lanubile et al. 2017).
Inheritance of resistance to viral diseases in maize
At least ten viruses cause significant agronomic losses in maize globally (Table 1) (White 1999). The incidence and severity of viral diseases are increasing, and new viral diseases continue to emerge. Maize dwarf mosaic disease (MDMD) is prevalent worldwide, especially in the USA, Europe, and the Huang-Huai-Hai plain in China. This disease seriously affects the yield and quality of maize (Fuchs and Gruntzig 1995). Maize lethal necrosis (MLN), a complex viral disease, is emerging as a serious threat to maize production (Boddupalli et al. 2020). MLN is caused by maize chlorotic mottle virus (MCMV; genus Machlomovirus in the Tombusviridae) in combination with one of several viruses from the Potyviridae, such as sugarcane mosaic virus (SCMV), maize dwarf mosaic virus (MDMV), Johnsongrass mosaic virus (JMV), and wheat streak mosaic virus (WSMV) (Redinbaugh and Stewart 2018; Boddupalli et al. 2020). MLN causes irreversible damage that kills maize plants before they reach maturity (Yang et al. 2017a). During 2012–2013, the estimated maize yield losses due to MLN were 23–100% in affected counties of Kenya (De Groote et al. 2016; Batchelor et al. 2020). Maize rough dwarf disease (MRDD) (Fig. 1l) poses a grave threat to maize production worldwide (Dovas et al. 2004; Achon et al. 2015). MRDD is caused by viruses in the Fijivirus genus in the Reoviridae family (Zhang et al. 2001; Liu et al. 2020a). In China, outbreaks of MRDD mainly occur in the Huang-Huai-Hai plain (Chen et al. 2015; Xu et al. 2020). Yield losses caused by MRDD range from 20 to 30% to as high as 100% in severely infected fields (Xu et al. 2020).
To date, only three viral disease resistance genes have been identified and validated, including ZmTrxh (Liu et al. 2017) and ZmABP1 (Leng et al. 2017) against SCMV, and ZmGDIα against MRDD (Liu et al. 2020a). ZmTrxh and ZmABP1 are the causal genes of the major QTLs Scmv1 and Scmv2, respectively (Xia et al. 1999; Xu et al. 1999), which function epistatically to confer complete resistance to SCMV (Xing et al. 2006). ZmTrxh encodes an atypical h-type thioredoxin, and its expression level is closely correlated with SCMV resistance (Tao et al. 2013a; Liu et al. 2017). ZmTrxh is dispersed in the cytoplasm to repress SCMV accumulation without eliciting salicylic acid- and/or jasmonic acid-mediated defense responses (Liu et al. 2017). ZmABP1 encodes an auxin-binding protein, and its expression level is closely associated with disease resistance, indicating that ZmABP1 cis-regulatory elements play a key role in SCMV resistance (Leng et al. 2017). ZmABP1 mainly functions during later stages of viral infection and thus adds a second tier of resistance to the immediate response mediated by ZmTrxh (Leng et al. 2017).
The major quantitative QTL qMrdd1 is proved to be associated with the ZmGDIα locus, which provides maize with recessive resistance to rough dwarf disease (MRDD) (Tao et al. 2013b; Liu et al. 2020a). ZmGDIα encodes a Rab GDP dissociation inhibitor alpha (RabGDIα), which is required for vesicle trafficking. The wild-type ZmGDIα is the dominant susceptible allele, and its splicing mutant ZmGDIα-hel is the recessive resistant allele. ZmGDIα-hel was generated when a helitron transposon inserted into its intron 10, inducing alternative splicing that replaces the wild-type exon 10 with a helitron-derived exon 10. ZmGDIα-hel reduces the disease severity index of MRDD by ~ 30% (Liu et al. 2020a). The viral protein P7-1 binds tightly to exon 10 and the C-terminal region of the wild-type ZmGDIα to recruit it for viral infection. The helitron-derived exon 10 weakens the binding of P7-1 to ZmGDIα-hel, resulting in quantitative resistance to MRDD (Liu et al. 2020a).
Inheritance of resistance to bacterial diseases in maize
Under favorable environmental conditions, such as protected cultivation, bacterial pathogens can cause tremendous crop losses (Table 1). Since its discovery in 1969, Goss’s bacterial wilt and leaf blight has emerged as an important disease of maize that causes more than 40% yield losses in susceptible maize hybrids (Carson 1991). In recent years, this disease has re-emerged and spread throughout all major corn-growing regions in the USA and Canada (Soliman et al. 2018) and caused 12.7 million tons of yield losses in maize between 2012 and 2015 (Mueller et al. 2016). Linkage mapping using three recombinant inbred line populations identified 19 QTLs (Singh et al. 2016). The effect size of each QTL was small, and none contributed > 6% of the total phenotypic variation (Singh et al. 2016).
Bacterial stalk rot, caused by Dickeya zeae, is an economically important disease that reduces crops yield by 21 to 98.8% (Kumar et al. 2017). This disease occurs in America, Canada, India, and Africa and is a major disease in tropical and subtropical maize planting areas. There are many other bacterial diseases in maize, such as bacterial leaf streak of corn (caused by Pseudomonas andropogonis) (Vidaver and Carlson 1978) and bacterial wilt of corn (caused by Pantoea stewartii) (EPPO 2006). Due to increasing global temperature and humidity, bacterial diseases pose a serious threat to the security of maize production. Unfortunately, there are few genetic studies about bacterial diseases in maize, so it is necessary to screen for various resistance sources and pay more attention to dig the resistance genes.
Inheritance of resistance to oomycete diseases in maize
Downy mildew (DM) diseases are caused by various fungal species in several genera of Oomycetes. This major group of diseases affects many crops, including maize and sorghum. With a recombinant inbred line (RIL) from the cross between B73 (susceptible) and Ki11 (resistant), seven QTLs were identified for three DM strains, located on chromosomes 2, 3, 6, and 9. The major QTL on chromosome 2 could explain 12.95% of the total phenotypic variation (Kim et al. 2020).
Pythium produces a white, rapidly growing mycelium, which can infect maize and cause a variety of diseases (Agrios 2005). Pythium stalk rot, caused by Pythium aphanidermatum and Pythium inflatum, is a serious disease that impairs maize production (Duan et al. 2019). Two independently inherited dominant genes, RpiQI319-1 and RpiQI319-2, confer resistance of Pythium stalk rot in maize (Song et al. 2015). Infection with P. aphanidermatum can also cause root rot, seedling blight, and seed rot (Wang and Duan 2020).
Molecular breeding of disease-resistant maize
Most inbred lines used in current commercial maize production are far from ideal in terms of disease resistance. For instance, very few elite inbred lines with resistance to head smut, stalk rot, and ear rot are available in China (Wang et al. 2014a; Duan et al. 2015). Several inbred lines with resistance against common rust and southern rust in China are highly susceptible to NCLB, SCLB, CLS, and GLS (Wang et al. 2014a). As known, the traditional breeding of disease-resistant varieties mainly depends on the breeder’s experiences in phenotypic selection. This is a time-consuming, inefficient process and highly dependent on environmental conditions. With the availability of elite disease resistance genes and their tagged molecular markers, the combination of traditional breeding and marker-assisted selection (MAS) has proven to be very efficient for developing elite resistant lines for maize production. Genomic selection (GS), transgenesis technique, and genome editing are all promising approaches as well. Combining these methods with doubled haploid (DH) technology could greatly accelerate the molecular breeding process in maize (Fig. 2).

Scheme for molecular breeding of disease-resistant maize. The collection of various excellent germplasm resources can provide sources for cloning and identification of disease resistance genes. The disease resistance genes can be used for MAS and transgenic disease resistance breeding. GS can speed up resistance breeding programs in maize. Combining gene editing with DH technology can quickly generate disease-resistant materials without transgenic components. Combining various strategies is an excellent way to greatly accelerate the maize breeding process. MAS, maker-assisted selection; DH, doubled haploid; WT, wild type
Sources of elite naturally occurring resistance genes
Natural germplasm resources, also known as genetic resources, show extensive genetic diversity in terms of disease resistance. Most disease resistance genes are present in tropical inbred lines, likely because high temperatures and high humidity favor the occurrence and maintenance of resistance genes. QTLs conferring resistance to GLS were identified in the highly resistant maize line Y32, derived from the tropical germplasm Suwan1 (Zhang et al. 2012b; Xu et al. 2014). The hybrid P78599, containing mixed ETO germplasm from South America and Suwan1 from Thailand, shows high resistance to most maize diseases. For instance, the stalk-rot disease resistance genes ZmCCT and ZmAuxRP1 were isolated from P78599-derived inbred line 1145 (Wang et al. 2017; Ye et al. 2019). Teosinte, the progenitor of maize, is another important genetic resource for maize improvement, as maize has lost a great deal of genetic variation compared to teosinte due to domestication and breeding bottlenecks (Tenaillon et al. 2004). A resistance QTL derived from teosinte conferred resistance to GLS, highlighting the potential value of teosinte for maize breeding programs (Zhang et al. 2017b). Furthermore, some important resistance alleles are rare in maize germplasms. For instance, ZmCCT was identified solely in inbred lines bred from the P78599 hybrid (Yang et al. 2013; Wang et al. 2017; Li et al. 2017). The ZmGDIα-hel allele against MRDD was detected in only 36 lines among the more than 1000 lines tested (Liu et al. 2020a). Thus, it is very necessary to conduct large-scale collection and evaluation of maize germplasm before identifying importantly rare natural resistance genes and using them in resistant breeding program.
Marker-assisted selection
MAS is a powerful tool to reduce maize diseases by using natural resistance genes. The introduction of the head smut resistance QTL qHSR1 via marker-assisted backcrossing significantly enhanced disease resistance of 10 inbred lines (Zhao et al. 2012a). ZmWAK, the causal gene at qHSR1, improves both head smut resistance and yield-related traits (Konlasuk et al. 2015; Zuo et al. 2015). ZmWAK has been widely used in the head smut resistance breeding program via MAS to improve the local Chinese susceptible germplasm Tongsipingtou and to develop a number of elite inbred lines (such as Ji853R and Chang7-2R) and resistant maize varieties (e.g., Jidan558) (Zhao et al. 2012a).
ZmCCT and ZmAuxRP1 are the causal factors for resistance to stalk rot (Wang et al. 2017; Ye et al. 2019). Both ZmCCT and ZmAuxRP1 have pleiotropic effects: ZmCCT is associated with photoperiod sensitivity, and ZmAuxRP1 is involved in root growth (Yang et al. 2013; Wang et al. 2017; Ye et al. 2019). Nine resistant ZmCCT haplotypes were introduced into seven elite inbred lines via MAS. The elite haplotype H5, selected from American inbred line GEMS14, exhibited enhanced resistance to stalk rot and less sensitivity to photoperiod (Li et al. 2017). Notably, inbred lines and hybrids carrying H5 also show stable stalk-rot resistance, little or no photosensitivity, and improved agronomic traits (such as yield and yield-related components). The H5 haplotype has been widely used for MAS in the stalk-rot resistance breeding programs in China and is expected to greatly alleviate the severity of stalk rot (Lanubile et al. 2017). Other stalk-rot resistance QTLs, such as qRfg3 (Ma et al. 2017), Rpi1 (Yang et al. 2005), and RpiQI319-1/2 (Song et al. 2015), have been identified in maize that will also be useful for reducing stalk rot diseases. If a resistance gene with pleiotropic effects is selected for resistant breeding program, it is necessary to simultaneously evaluate its resistance performance and influence on other traits in multiple genetic backgrounds.
Frequently, an inbred line bearing a single resistance QTL is not enough to achieve high-resistance performance. Hence, pyramiding of various resistance genes is an effective way to reduce disease severity. The introgression of Scmv1 and Scmv2 into the susceptible line F7 via MAS produced a near-isogenic line (NIL) with almost complete resistance to SCMV (Xing et al. 2006). Similarly, a maize NIL containing the qMdr9.02 locus with multiple disease resistance genes generated via MAS showed resistance to two important foliar diseases: SCLB and GLS (Yang et al. 2017b). MAS combined with phenotypic selection is a highly efficient, low-cost method that has greatly enhanced resistance breeding programs in maize (Yousef and Juvik 2001; Asea et al. 2012). However, MAS relies on the availability of markers linked to genes/dQTLs, which usually take a long time to identify by linkage or association mapping, especially for small-effect resistance QTLs. Because of this, several other strategies are currently used in disease resistance breeding programs.
Genomic selection
Genomic selection (GS) is conducted by combining genotypic (markers) and phenotypic data in a training population to estimate the breeding values of lines that have been genotyped but not phenotyped in a testing population (Meuwissen et al. 2001). GS has been successfully used in both animal and plant breeding programs, as it substantially increases the rate of genetic gain (Meuwissen et al. 2001). The prediction accuracy of GS is influenced by many factors, including trait heritability, prediction model, population size and structure (relationship between the training and testing populations), number of markers, and genotype × environment (GE) interactions. GS uses all markers to predict the breeding value of individuals in the testing population, and thus has a greater predictive power compared to approaches that use only markers with significant effects (Massman et al. 2013). Combining two different heterotic groups in a single training set can lead to significantly more accurate prediction for both heterotic groups, and more importantly, this approach saves available resources by avoiding the need to establish a training set of sufficient size for each heterotic group (Technow et al. 2013).
Although GS in maize is currently focused on grain yield, drought tolerance, and kernel zinc and oil content, it shows promise for use in disease-resistant corn breeding programs, as the prediction accuracy for resistance to NCLB reached ~ 0.7 (Technow et al. 2013; Beyene et al. 2015; Vivek et al. 2017; Guo et al. 2020; Mageto et al. 2020; Hao et al. 2019). For MLN resistance, GS gave a promising result despite being highly influenced by the number of markers, training population size, and population relevancy (Sitonik et al. 2019; Nyaga et al. 2019). The average accuracy ranges from 0.46 to 0.86 for the MLN disease severity and 0.46 to 0.87 for the MLN area under disease progress curve (Sitonik et al. 2019). GS also showed moderate-to-high accuracy in predicting Fusarium ear rot resistance, in which the maximum prediction accuracy was 0.46 for Fusarium ear rot and 0.67 for fumonisin (Liu et al. 2020b; Kuki et al. 2020; Holland et al. 2020). The prediction accuracy could be greatly elevated if using improved training population. For instance, GS generally showed low-to-moderate prediction accuracy of 0.29 to 0.56 for GLS resistance, which could be elevated to 0.77 or even 0.84 when increasing the diversity of the training set (Kibe et al. 2020). In any case, when trying to breed a resistant hybrid to a specific disease, both parental lines should be sufficiently resistant.
Transgenesis technique
Transgenic methods are useful for breeding disease-resistant maize. These techniques involve the direct introduction or modification of a target gene of interest using biotechnology (Christou 2013). Almost all disease resistance genes in maize function normally in the resulting transgenic lines, indicating that these techniques will be highly valuable for maize resistance breeding programs. More importantly, transgenic techniques can break the reproductive isolation between plant species to allow the introduction of resistance genes from other plant species. For instance, the expression of the durable wheat disease resistance gene Lr34 in maize conferred resistance to common rust and NCLB (Sucher et al. 2017). Maize containing the Rxo1 locus showed a strong hypersensitive response to a non-host bacterial pathogen (Zhao et al. 2004b; Zhao et al. 2004a). Pyramiding of different plant defense response genes and anti-apoptosis genes via genetic transformation conferred resistance to sheath blight disease and SCLB in maize (Zhu et al. 2018). In addition to resistance genes from plants, genes from fungi and viruses are also valuable in maize disease resistance breeding programs.
Functional analysis of resistance genes cloned from maize indicates that resistance performance is often closely associated with the expression of resistance gene. Thus, it is likely that disease-resistant maize could be bred by overexpressing or knocking down a gene of interest. However, the increased expression of some pleiotropic resistance genes can have negative effects on other traits. For example, in addition to conferring stalk rot resistance, overexpressing ZmCCT delayed flowering time in maize (Yang et al. 2013; Wang et al. 2017). The adverse effects of overexpression could be avoided by expressing a resistance gene under the control of a pathogen-inducible promoter. Although no such study has been reported in maize, this strategy has been highly successful in rice (Helliwell et al. 2013; Liu et al. 2019).
RNA interference (RNAi) induces post-transcriptional gene silencing via the expression of double-stranded RNA (dsRNA) or hairpin RNA (hpRNA). RNAi is a highly efficient method for controlling viral diseases. Expressing hpRNA derived from the capsid protein genes of MDMV and SCMV significantly enhanced maize resistance to MDMV and SCMV, respectively (Zhang et al. 2011; Gan et al. 2014). MLN, a viral disease caused by co-infection with several viruses, is destructive to maize production in Africa (Redinbaugh and Stewart 2018), suggesting that RNAi may be useful for controlling MLN.
However, transgenic approaches are not as widely used as MAS in breeding of disease-resistant maize. This is likely due to the shortage of available resistance genes and the restrictions imposed on the cultivation of genetically modified maize in many countries. Therefore, innovative transgene-free techniques have been developed that are more acceptable to disease-resistant maize breeding programs.
Genome editing by CRISPR/CAS9
Significant progress has been made in the field of genome editing, from zinc finger nucleases (ZFNs) to transcription activator-like effector nucleases (TALENs) to clustered regularly interspaced short palindromic repeats (CRISPR)–associated protein (CRISPR/Cas) (Carroll 2014; Yin et al. 2017; Adli 2018; Gao 2021). Most genome editing technologies involve the creation of double-strand breaks (DSBs) to trigger DNA repair mechanisms (Carroll 2014; Adli 2018). DSBs are mainly repaired in one of the following two ways: error-prone non-homologous end-joining (NHEJ), which creates small insertions and/or deletions (indels), and error-free homology-directed repair (HDR), which results in the insertion or replacement of homologous DNA (Carroll 2014). The NHEJ-mediated introduction of indels can disrupt the target gene’s function if they occur in the coding region or alter the gene expression if they occur in the cis-regulatory region. The HDR pathway requires the use of donor homologous DNA to introduce precise insertions or substitutions (Adli 2018).
The CRISPR/Cas9 system has revolutionized the genome editing due to its simplicity, flexibility, consistency, and high efficiency and has thus become the most powerful tool for genetic analysis and crop improvement (Hua et al. 2019; Zhu et al. 2020; Gao 2021). In the past few years, the CRISPR/Cas9 system has been successfully used for plant disease control (Langner et al. 2018; Chen et al. 2019; Mao et al. 2019). Moreover, once the genome has been edited, the transgenic cassette can be eliminated by selfing or hybridization (Hua et al. 2019).
Replacement/knock-in with dominant or partially dominant resistance genes
The major R-genes Hm1 and Rp1-D act in a dominant manner (Johal and Briggs 1992; Collins et al. 1999). A number of QDR genes also act in a dominant or partially dominant manner, such as ZmWAK (Zuo et al. 2015), Htn1 (Hurni et al. 2015), ZmCCoAOMT2 (Yang et al. 2017b), and ZmAuxRP1 (Ye et al. 2019). Such dominant (or partially dominant) R or QDR genes could be used to replace their weak or null counterparts by CRISPR/Cas9. Alternatively, these genes could be inserted into (or even stacked into) the maize genome by CRISPR/Cas9-mediated knock-in. The introduction of natural resistance genes in this manner would have many advantages, such as the lack of linkage drag, little or no fitness penalty, and stronger resistance due to the presence of multiple copies (Luo et al. 2016).
Due to the low efficiency of HDR, there are few successful examples of the replacement or knock-in of genes in plants. One of them was the improvement of drought tolerance of maize (Shi et al. 2017). Under drought-stress conditions, plants overexpressing ARGOS8 showed reduced sensitivity to ethylene and increased grain yield. However, the abundance of endogenous ARGOS8 transcript is relatively low in most maize inbred lines (Shi et al. 2015). The same research team used CRISPR/Cas9 technology to knock-in the GOS2 promoter to replace the original ARGOS8 promoter, leading to the production of plants with high levels of chimeric ARGOS8 transcripts and enhanced drought tolerance (Shi et al. 2017). Recently, a high-frequency and selectable marker-free intra-genomic gene targeting (GT) was reported in maize, in which a heat shock–inducible Cas9 was used to simultaneously generate double-strand breaks at the target locus and release the donor template from pre-integrated T-DNA, generating up to 4.7% targeted insertion in T0 plants (Barone et al. 2020). This gene targeting opens up a new way to use the CRISPR-Cas9 system to repair endogenous defective alleles, a technique with great potential for improving disease resistance in maize.
Modifying the cis-regulatory elements of resistance genes
Gene expression is regulated at both the transcriptional and post-transcriptional levels. The former mainly depends on regulatory elements in the promoter region, while the latter includes pre-RNA splicing, mRNA modification, mRNA transport, and mRNA degradation (Pramanik et al. 2020). Cis-regulatory elements are readily accessible targets for CRISPR/Cas9 (Swinnen et al. 2016). The promoter of the citrus canker susceptibility gene CsLOB1 contains the pathogen’s effector binding element. When they were edited by CRISPR/Cas9, the resultant plants showed enhanced resistance to citrus canker (Peng et al. 2017). Similarity, mutations of the promoters of SWEET11, SWEET13, and SWEET14 in rice conferred robust, broad-spectrum resistance to Xanthomonas oryzae pv. oryzae (Oliva et al. 2019; Xu et al. 2019).
The pleotropic gene ZmCCT confers quantitative resistance to Gibberella stalk rot and delays flowering time under long-day conditions (Yang et al. 2013; Wang et al. 2017). Thus, we reasoned that deleting the photosensitive elements in the ZmCCT promoter region would create an artificial allele with reduced photosensitivity but the same level of stalk rot resistance. We recently used CRISPR/Cas9 to systematically delete the photosensitive elements in the ZmCCT promoter to create ZmCCT variants with the aim to select an artificial ZmCCT allele to meet the requirement (unpublished data).
Inactivation of host susceptibility factors
Host susceptibility (S) factors can be exploited by pathogenic microbes to facilitate their proliferation. Disabling these key links between plants and pathogens might provide the host with broad-spectrum, durable disease resistance (Langner et al. 2018; Zaidi et al. 2018). A classic example of the use of CRISPR/Cas9 to improve plant disease resistance involves the S gene MLO, which is conserved throughout monocots and dicots. Two teams successfully edited MLO in different species (wheat and tomato) through CRISPR/Cas9, and the edited mlo gene improved resistance to powdery mildew in both species (Wang et al. 2014b; Nekrasov et al. 2017) (Wang et al. 2014b; Nekrasov et al. 2017). Similarly, OsERF922, encoding the negative regulator of rice blast resistance (Liu et al. 2012), was successfully knocked out by CRISPR/Cas9, thereby increasing resistance to rice blast (Wang et al. 2016). In maize resistance to BLSB, ZmFBL41 is a negative regulator, and the transposon-insertion line zmfbl41 improved maize resistance to BLSB (Li et al. 2019). This indicates that direct knockout of Zmfbl41 via CRISPR/Cas9 technology can also enhance the BLSB resistance.
Since its appearance, CRISPR/Cas9 technology has been extensively exploited to meet various demands. Among them, base editing is an ideal solution for nucleotide conversion. By fusing a CRISPR-Cas9 variant with cytidine deaminase (or adenosine deaminase), base editing allows for the direct transition of C·G to T·A (or A·T to G·C) at the target site without the need of DSBs (Shimatani et al. 2017; Zong et al. 2017; Zong et al. 2018; Li et al. 2018; Chen et al. 2019; Lin et al. 2020; Gao 2021). DSB-free base editing can be used to introduce a stop codon at a specific position, thus avoiding the side effects of DSBs (Billon et al. 2017). Given that disease resistance resulting from the knockout of an S gene is often accompanied by fitness costs, base editing could greatly reduce changes to the target S protein, thereby minimizing fitness costs (Zaidi et al. 2018). For instance, ZmGDIα-hel is the recessive resistance gene against RBSDV (Liu et al. 2020a). If the key amino acids in ZmGDIα that bind to the viral P7-1 protein are identified, then base editing can be used to modify ZmGDIα to disrupt the interaction between ZmGDIα and P7-1, thereby generating stronger resistance ZmGDIα alleles.
Combining genome editing and double-haploid technology
The removal of the CRISPR/Cas9 cassette requires several generations. Double-haploid (DH) technology is a powerful tool to promote the breeding efficiency by reducing the need for multiple generation selection (Ren et al. 2017). Combining CRISPR/Cas9 with DH technology represents an excellent way to accelerate maize breeding. Using roughly similar methods, the Haploid Induction Editing (HI-Edit) technique (Kelliher et al. 2019) and Haploid-Inducer Mediated Genome Editing (IMGE) system (Wang et al. 2019b), two different teams obtained edited haploids without the CRISPR/Cas9 cassette in a single step. Using the HI-Edit technique, 4.8–8.8% of the resulting plants were shown to lack the CRISPR/Cas9 cassette and contain the edited inducer gene matl (Kelliher et al. 2019). Similarly, using the IMGE system, the editing efficiency of ZmLG1 in haploids was estimated to be ~ 4.1%, and all zmlg1-haploids were Cas9-free (Wang et al. 2019b).
Perspectives
It is a daunting task to clone a QDR gene, especially a small-effect QDR gene (Yang et al. 2012). Thus far, only a few QDR genes have been cloned, and many more QDR genes remain to be identified (Mackay et al. 2009; Yang et al. 2017a). In the long run, however, it will be crucial to clone all resistance genes and understand their resistance mechanisms. This is because (1) only if resistance genes and related markers are available, can we replace susceptibility genes with resistance genes with less or no genetic drag; (2) the availability of a natural resistance gene allows for the identification of other downstream resistance-related genes in the same defense pathway; (3) all resistance genes could be modified via gene editing to create a series of artificial alleles for breeding of potentially resistant varieties.
Since most maize QDR genes only contribute a small genetic effect to help reduce disease severity (Holland 2007; Mackay et al. 2009), a lot of time and efforts are required to complete gene discovery by traditional map-based cloning strategy. In an attempt to accelerate gene discovery process, various resources must be utilized, such as diverse genetic stocks, various biological techniques, big data analysis, and bioinformatics tools. Sequential fine mapping based on recombinant-derived progeny is highly effective for narrowing down small-effect QDR loci (Yang et al. 2012; Ye et al. 2019). Online access to ever-increasing maize genome sequences is very helpful for identifying candidate resistance genes (Schnable et al. 2009; Springer et al. 2018; Sun et al. 2018; Yang et al. 2019a). Omic analysis, such as transcriptomics and metabolomics, play key roles in identifying candidate resistance genes and understanding their mechanisms (Zhang et al. 2017a; Yang et al. 2019b; Ye et al. 2019; Yao et al. 2020). Various Mu-/EMS-induced mutation libraries allow us to quickly examine the resistance performance of the candidate gene (Lu et al. 2018; Liang et al. 2019). Transgenic techniques, coupled with more powerful genome editing tools, can be used to accurately identify function of a candidate resistance gene (Christou 2013; Adli 2018).
Most sequence changes between resistant and susceptible alleles are related to transposable elements, i.e., the presence/absence variations resulted from transposon insertions, such as ZmCCT (Wang et al. 2017) and ZmGDIα (Liu et al. 2020a), or residual sequences caused by frequent transposon insertion/deletion activities, such as ZmWAK (Zuo et al. 2015), ZmTrxh (Liu et al. 2017), and ZmABP1 (Leng et al. 2017). Given that transposable elements account for approximately 85% of the whole maize genome (Schnable et al. 2009), it is conceivable that one transposon or another will be activated by biotic stresses to create genetic variants for natural selection. Only those alleles with enhanced disease resistance and no negative effect on agronomic traits are prone to be selected and preserved in maize.
Genome editing opens up infinite possibilities to edit a target gene based on a human’s blueprint. If a resistance gene comes from the loss-of-function of a susceptibility gene, i.e., the so-called recessive resistance gene (usually found in viral resistance), the simplest way is to disrupt or delete the susceptibility gene by gene editing to create an artificial resistance allele. Alternatively, key nucleotides related to disease susceptibility need to be identified and modified to generate resistance alleles while maintaining the other functions. If a resistance gene is dominant/semi-dominant over the susceptibility gene due to the gene expression level, the cis-regulatory region could be modified by inserting a strongly induced promoter or increasing the copy number of resistance gene to achieve stronger resistance. On the other hand, if protein structure is essential for disease resistance, the key residues/peptides to perceive pathogen effectors should be pinpointed in an attempt to generate stronger resistance alleles. Furthermore, all genes involved in the defense pathway are the potential targets for gene editing to increase resistance.
In short, only by discovering enough resistance genes and understanding their molecular mechanisms, coupled with advanced biotechnology, can we achieve the goal of breeding super maize varieties with high disease resistance and ideal agronomic traits.
Data Availability
Not applicable.
Change history
14 September 2021
A Correction to this paper has been published: https://doi.org/10.1007/s11032-021-01250-z
References
Achon MA, Serrano L, Sabaté J, Porta C (2015) Understanding the epidemiological factors that intensify the incidence of maize rough dwarf disease in Spain. Ann Appl Biol:166. https://doi.org/10.1111/aab.12184
Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9(1):1911. https://doi.org/10.1038/s41467-018-04252-2
Agrios G (2005) Plant Pathology (Fifth Edition). chapter eleven-Plant diseases caused by fungi. Academic Press, San Diego. https://doi.org/10.1016/B978-0-08-047378-9.50017-8
Ali F, Yan J (2012) Disease resistance in maize and the role of molecular breeding in defending against global threat. J Integr Plant Biol 54(3):134–151. https://doi.org/10.1111/j.1744-7909.2012.01105.x
Asea G, Vivek BS, Lipps PE, Pratt RC (2012) Genetic gain and cost efficiency of marker-assisted selection of maize for improved resistance to multiple foliar pathogens. Mol Breed 29(2):515–527. https://doi.org/10.1007/s11032-011-9568-8
Azra A, Hussain S (2019) Screening of maize genotypes against southern corn leaf blight (Bipolaris maydis) under artificial epiphytotic conditions. Sarhad J Agric 35. https://doi.org/10.17582/journal.sja/2019/35.4.1122.1128
Balint-Kurti PJ, Carson ML (2006) Analysis of quantitative trait Loci for resistance to southern leaf blight in juvenile maize. Phytopathology 96(3):221–225. https://doi.org/10.1094/phyto-96-0221
Balint-Kurti PJ, Zwonitzer JC, Wisser RJ, Carson ML, Oropeza-Rosas MA, Holland JB, Szalma SJ (2007) Precise mapping of quantitative trait loci for resistance to southern leaf blight, caused by Cochliobolus heterostrophus race O, and flowering time using advanced intercross maize lines. Genetics 176(1):645–657. https://doi.org/10.1534/genetics.106.067892
Balint-Kurti PJ, Zwonitzer JC, Pè ME, Pea G, Lee M, Cardinal AJ (2008) Identification of quantitative trait loci for resistance to southern leaf blight and days to anthesis in two maize recombinant inbred line populations. Phytopathology 98(3):315–320. https://doi.org/10.1094/phyto-98-3-0315
Barone P, Wu E, Lenderts B, Anand A, Gordon-Kamm W, Svitashev S, Kumar S (2020) Efficient gene targeting in maize using inducible CRISPR-Cas9 and marker-free donor template. Mol Plant 13(8):1219–1227. https://doi.org/10.1016/j.molp.2020.06.008
Batchelor WD, Suresh LM, Zhen X, Beyene Y, Wilson M, Kruseman G, Prasanna B (2020) Simulation of maize lethal necrosis (MLN) damage using the CERES-maize model. Agronomy 10(5):710
Beyene Y, Semagn K, Mugo S, Tarekegne A, Babu R, Meisel B, Sehabiague P, Makumbi D, Magorokosho C, Oikeh S, Gakunga J, Vargas M, Olsen M, Prasanna BM, Banziger M, Crossa J (2015) Genetic gains in grain yield through genomic selection in eight bi-parental maize populations under drought stress. Crop Sci 55(1):154–163. https://doi.org/10.2135/cropsci2014.07.0460
Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, Ciccia A (2017) CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol Cell 67(6):1068–1079.e1064. https://doi.org/10.1016/j.molcel.2017.08.008
Boddupalli P, Suresh LM, Mwatuni F, Beyene Y, Makumbi D, Gowda M, Olsen M, Hodson D, Worku M, Mezzalama M, Molnar T, Dhugga KS, Wangai A, Gichuru L, Angwenyi S, Alemayehu Y, Grønbech Hansen J, Lassen P (2020) Maize lethal necrosis (MLN): Efforts toward containing the spread and impact of a devastating transboundary disease in sub-Saharan Africa. Virus Res 282:197943. https://doi.org/10.1016/j.virusres.2020.197943
Brewbaker JL, Kim SK, So YS, Logroño M, Moon HG, Ming R, Lu XW, Josue AD (2011) General resistance in maize to southern rust (Puccinia polysora Underw.). Crop Sci 51(4):1393–1409. https://doi.org/10.2135/cropsci2010.06.0327
Carroll D (2014) Genome engineering with targetable nucleases. Annu Rev Biochem 83:409–439. https://doi.org/10.1146/annurev-biochem-060713-035418
Carson M (1991) Relationship between leaf freckles and wilt severity and yield losses in closely related maize hybrids. Phytopathology:81. https://doi.org/10.1094/Phyto-81-95
Carson M (1995) A new gene in maize conferring the “chlorotic halo” reaction to infection by Exserohilum turcicum. Plant Dis 79. https://doi.org/10.1094/PD-79-0717
Carson ML, Stuber CW, Senior ML (2004) Identification and mapping of quantitative trait loci conditioning resistance to southern leaf blight of maize caused by Cochliobolus heterostrophus Race O. Phytopathology 94(8):862–867. https://doi.org/10.1094/phyto.2004.94.8.862
Chen C, Wang Z, Yang D, Ye C, Zhao Y, Jin D, Weng M, Wang B (2004) Molecular tagging and genetic mapping of the disease resistance gene RppQ to southern corn rust. Theor Appl Genet 108(5):945–950. https://doi.org/10.1007/s00122-003-1506-7
Chen Y, Chao Q, Tan G, Zhao J, Zhang M, Ji Q, Xu M (2008) Identification and fine-mapping of a major QTL conferring resistance against head smut in maize. Theor Appl Genet 117(8):1241–1252. https://doi.org/10.1007/s00122-008-0858-4
Chen W, Zhang M, Li L (2013) The resistance to banded leaf and sheath blight in maize of 282 inbred lines. Afr J Agric Res 8:1547–1552. https://doi.org/10.5897/AJAR2013.6789
Chen G, Wang X, Hao J, Yan J, Ding J (2015) Genome-wide association implicates candidate genes conferring resistance to maize rough dwarf disease in maize. PLoS One 10(11):e0142001. https://doi.org/10.1371/journal.pone.0142001
Chen K, Wang Y, Zhang R, Zhang H, Gao C (2019) CRISPR/Cas genome editing and precision plant breeding in agriculture. Annu Rev Plant Biol 70:667–697. https://doi.org/10.1146/annurev-arplant-050718-100049
Christensen SA, Nemchenko A, Park YS, Borrego E, Huang PC, Schmelz EA, Kunze S, Feussner I, Yalpani N, Meeley R, Kolomiets MV (2014) The novel monocot-specific 9-lipoxygenase ZmLOX12 is required to mount an effective jasmonate-mediated defense against Fusarium verticillioides in maize. Mol Plant-Microbe Interact 27(11):1263–1276. https://doi.org/10.1094/mpmi-06-13-0184-r
Christou P (2013) Plant genetic engineering and agricultural biotechnology 1983-2013. Trends Biotechnol 31(3):125–127. https://doi.org/10.1016/j.tibtech.2013.01.006
Collins N, Drake J, Ayliffe M, Sun Q, Ellis J, Hulbert S, Pryor T (1999) Molecular characterization of the maize Rp1-D rust resistance haplotype and its mutants. Plant Cell 11(7):1365–1376. https://doi.org/10.1105/tpc.11.7.1365
De Groote H, Oloo F, Tongruksawattana S, Das B (2016) Community-survey based assessment of the geographic distribution and impact of maize lethal necrosis (MLN) disease in Kenya. Crop Prot 82:30–35. https://doi.org/10.1016/j.cropro.2015.12.003
Ding J, Wang X, Chander S, Li J (2008) Identification of QTL for maize resistance to common smut by using recombinant inbred lines developed from the Chinese hybrid Yuyu22. J Appl Genet 49(2):147–154. https://doi.org/10.1007/bf03195608
Dovas C, Eythymiou K, Katis N (2004) First report of maize rough dwarf virus (MRDV) on maize crops in Greece. Plant Pathol 53:238–238. https://doi.org/10.1111/j.0032-0862.2004.00973.x
Du L, Yu F, Zhang H, Wang B, Ma K, Yu C, Xin W, Huang X, Liu Y, Liu K (2020) Genetic mapping of quantitative trait loci and a major locus for resistance to grey leaf spot in maize. Theor Appl Genet 133(8):2521–2533. https://doi.org/10.1007/s00122-020-03614-z
Duan C, Wang X, Wu X, Song F, Zhao L, Sun S, Zhu Z (2015) Analysis of maize accessions resistance to Pythium stalk rot and Fusarium ear rot. J Plant Gen Res 16(5):947–954. https://doi.org/10.13430/j.cnki.jpgr.2015.05.004
Duan C, Song F, Sun S, Guo C, Zhu Z, Wang X (2019) Characterization and molecular mapping of two novel genes resistant to pythium stalk rot in maize. Phytopathology 109(5):804–809. https://doi.org/10.1094/phyto-09-18-0329-r
EPPO (2006) Pantoea stewartii subsp. stewartii. EPPO Bulletin 36(1):111–115. https://doi.org/10.1111/j.1365-2338.2006.00920.x
Frey T, Weldekidan T, Colbert T, Wolters PJCC, Hawk JA (2011) Fitness evaluation of Rcg1, a locus that confers resistance to Colletotrichum graminicola (Ces.) G.W. Wils. Using Near-Isogenic Maize Hybrids. Crop Sci 51:1551. https://doi.org/10.2135/cropsci2010.10.0613
Fuchs E, Gruntzig M (1995) Influence of sugarcane mosaic virus (SCMV) and maize dwarf mosaic virus (MDMV) on the growth and yield of two maize varieties. J Plant Dis Prot 102:44–50
Gan D, Ding F, Zhuang D, Jiang H, Jiang T, Zhu S, Cheng B (2014) Application of RNA interference methodology to investigate and develop SCMV resistance in maize. J Genet 93(2):305–311. https://doi.org/10.1007/s12041-014-0364-1
Gao C (2021) Genome engineering for crop improvement and future agriculture. Cell. https://doi.org/10.1016/j.cell.2021.01.005
Gao X, Shim WB, Göbel C, Kunze S, Feussner I, Meeley R, Balint-Kurti P, Kolomiets M (2007) Disruption of a maize 9-lipoxygenase results in increased resistance to fungal pathogens and reduced levels of contamination with mycotoxin fumonisin. Mol Plant-Microbe Interact 20(8):922–933. https://doi.org/10.1094/mpmi-20-8-0922
Gao X, Brodhagen M, Isakeit T, Brown SH, Göbel C, Betran J, Feussner I, Keller NP, Kolomiets MV (2009) Inactivation of the lipoxygenase ZmLOX3 increases susceptibility of maize to Aspergillus spp. Mol Plant-Microbe Interact 22(2):222–231. https://doi.org/10.1094/mpmi-22-2-0222
Gevers HO (1975) A new major gene for resistance to Helminthosporium turcicum leaf blight of maize. Plant Dis Rep 59:296–299
Groth JV, Zeyen R, Davis DW, Christ BJ (1983) Yield and quality losses caused by common rust (Puccinia sorghi Schw.) in sweet corn (Zea mays) hybrids. Crop Prot 2(1):105–111. https://doi.org/10.1016/0261-2194(83)90030-3
Guo R, Dhliwayo T, Mageto EK, Palacios-Rojas N, Lee M, Yu D, Ruan Y, Zhang A, San Vicente F, Olsen M, Crossa J, Prasanna BM, Zhang L, Zhang X (2020) Genomic prediction of kernel zinc concentration in multiple maize populations using genotyping-by-sequencing and repeat amplification sequencing markers. Front Plant Sci 11:534. https://doi.org/10.3389/fpls.2020.00534
Hao Y, Wang H, Yang X, Zhang H, He C, Li D, Li H, Wang G, Wang J, Fu J (2019) Genomic prediction using existing historical data contributing to selection in biparental populations: a study of kernel oil in maize. Plant Genome 12(1). https://doi.org/10.3835/plantgenome2018.05.0025
Helliwell EE, Wang Q, Yang Y (2013) Transgenic rice with inducible ethylene production exhibits broad-spectrum disease resistance to the fungal pathogens Magnaporthe oryzae and Rhizoctonia solani. Plant Biotechnol J 11(1):33–42. https://doi.org/10.1111/pbi.12004
Holland J (2007) Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10(2):156–161. https://doi.org/10.1016/j.pbi.2007.01.003
Holland J, Uhr D, Jeffers D, Goodman M (1998) Inheritance of resistance to southern corn rust in tropical-by-corn-belt maize populations. Theor Appl Genet 96:232–241. https://doi.org/10.1007/s001220050732
Holland JB, Marino TP, Manching HC, Wisser RJ (2020) Genomic prediction for resistance to Fusarium ear rot and fumonisin contamination in maize. Crop Sci 60(4):1863–1875. https://doi.org/10.1002/csc2.20163
Hooker AL (1963) Inheritance of chlorotic-lesion resistance to Helminthosporium turcicum in seedling corn. Phytopathology 53:660–662
Hooker AL (1969) Widely based resistance to rust in corn. Iowa Agric Home Econ Exp Stn Spec Rep 64:28–34
Hooker A (1977) A second major gene locus in corn for chlorotic-lesion resistance to Helminthosporium turicum1. Crop Sci 17. https://doi.org/10.2135/cropsci1977.0011183X001700010035x
Hua K, Zhang J, Botella JR, Ma C, Kong F, Liu B, Zhu JK (2019) Perspectives on the application of genome-editing technologies in crop breeding. Mol Plant 12(8):1047–1059. https://doi.org/10.1016/j.molp.2019.06.009
Hulbert SH (1997) Structure and evolution of the rp1 complex conferring rust resistance in maize. Annu Rev Phytopathol 35:293–310. https://doi.org/10.1146/annurev.phyto.35.1.293
Hurni S, Scheuermann D, Krattinger SG, Kessel B, Wicker T, Herren G, Fitze MN, Breen J, Presterl T, Ouzunova M, Keller B (2015) The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc Natl Acad Sci U S A 112(28):8780–8785. https://doi.org/10.1073/pnas.1502522112
Jamann TM, Luo X, Morales L, Kolkman JM, Chung CL, Nelson RJ (2016) A remorin gene is implicated in quantitative disease resistance in maize. Theor Appl Genet 129(3):591–602. https://doi.org/10.1007/s00122-015-2650-6
Jiang C, Edmeades G, Armstead I, Lafitte H, Hayward M, Hoisington D (1999) Genetic analysis of adaptation differences between highland and lowland tropical maize using molecular markers. Theor Appl Genet 99:1106–1119. https://doi.org/10.1007/s001220051315
Jines MP, Balint-Kurti P, Robertson-Hoyt LA, Molnar T, Holland JB, Goodman MM (2007) Mapping resistance to Southern rust in a tropical by temperate maize recombinant inbred topcross population. Theor Appl Genet 114(4):659–667. https://doi.org/10.1007/s00122-006-0466-0
Johal GS, Briggs SP (1992) Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258(5084):985–987. https://doi.org/10.1126/science.1359642
Jung M, Weldekidan T, Schaff D, Paterson A, Tingey S, Hawk J (1994) Generation-means analysis and quantitative trait locus mapping of anthracnose stalk rot genes in maize. Theor Appl Genet 89(4):413–418. https://doi.org/10.1007/bf00225375
Kaur M, Vikal Y, Kaur H, Pal L, Kaur K, Chawla JS (2019) Mapping quantitative trait loci associated with southern leaf blight resistance in maize (Zea mays L.). J Phytopathol 167(10):591–600. https://doi.org/10.1111/jph.12849
Kelliher T, Starr D, Su X, Tang G, Chen Z, Carter J, Wittich PE, Dong S, Green J, Burch E, McCuiston J, Gu W, Sun Y, Strebe T, Roberts J, Bate NJ, Que Q (2019) One-step genome editing of elite crop germplasm during haploid induction. Nat Biotechnol 37(3):287–292. https://doi.org/10.1038/s41587-019-0038-x
Kibe M, Nair S, Das B, Bright J, Makumbi D, Kinyua J, Suresh LM, Beyene Y, Olsen M, Prasanna B, Gowda M (2020) Genetic dissection of resistance to gray leaf spot by combining genome-wide association, linkage mapping, and genomic prediction in tropical maize germplasm. Front Plant Sci 11:572027. https://doi.org/10.3389/fpls.2020.572027
Kim HC, Kim K-H, Song K, Kim JY, Lee B-M (2020) Identification and validation of candidate genes conferring resistance to downy mildew in maize (Zea mays L.). Genes (Basel) 11(2):191
Konlasuk S, Xing Y, Zhang N, Zuo W, Zhang B, Tan G, Xu M (2015) ZmWAK, a quantitative resistance gene to head smut in maize, improves yield performance by reducing the endophytic pathogen Sporisorium reiliana. Mol Breed 35(8):174. https://doi.org/10.1007/s11032-015-0325-2
Kou Y, Wang S (2010) Broad-spectrum and durability: understanding of quantitative disease resistance. Curr Opin Plant Biol 13(2):181–185. https://doi.org/10.1016/j.pbi.2009.12.010
Krattinger SG, Keller B (2016) Molecular genetics and evolution of disease resistance in cereals. New Phytol 212(2):320–332. https://doi.org/10.1111/nph.14097
Kuki M, Pinto R, Bertagna F, Tessmann D, Amaral A, Scapim C, Holland J (2020) Association mapping and genomic prediction for ear rot disease caused by Fusarium verticillioides in a tropical maize germplasm. Crop Sci:60. https://doi.org/10.1002/csc2.20272
Kumar A, Singh M, Kaur H, Rawal R, Kumar A, Singh P (2017) A review on bacterial stalk rot disease of maize caused by Dickeya zeae. J Appl Nat Sci 9:1214–1225. https://doi.org/10.31018/jans.v9i2.1348
Langner T, Kamoun S, Belhaj K (2018) CRISPR crops: plant genome editing toward disease resistance. Annu Rev Phytopathol 56:479–512. https://doi.org/10.1146/annurev-phyto-080417-050158
Lanubile A, Maschietto V, Borrelli VM, Stagnati L, Logrieco AF, Marocco A (2017) Molecular basis of resistance to Fusarium ear rot in maize. Front Plant Sci 8:1774. https://doi.org/10.3389/fpls.2017.01774
Leng P, Ji Q, Asp T, Frei UK, Ingvardsen CR, Xing Y, Studer B, Redinbaugh M, Jones M, Gajjar P, Liu S, Li F, Pan G, Xu M, Lübberstedt T (2017) Auxin binding protein 1 reinforces resistance to sugarcane mosaic virus in maize. Mol Plant 10(10):1357–1360. https://doi.org/10.1016/j.molp.2017.07.013
Li Y, Wu X, Jaqueth J, Zhang D, Cui D, Li C, Hu G, Dong H, Song Y, Shi Y, Wang T, Li B, Li Y (2015) The identification of two head smut resistance-related QTL in maize by the joint approach of linkage mapping and association analysis. PLoS One 10(12):e0145549. https://doi.org/10.1371/journal.pone.0145549
Li Y, Tong L, Deng L, Liu Q, Xing Y, Wang C, Liu B, Yang X, Xu M (2017) Evaluation of ZmCCT haplotypes for genetic improvement of maize hybrids. Theor Appl Genet 130(12):2587–2600. https://doi.org/10.1007/s00122-017-2978-1
Li C, Zong Y, Wang Y, Jin S, Zhang D, Song Q, Zhang R, Gao C (2018) Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion. Genome Biol 19(1):59. https://doi.org/10.1186/s13059-018-1443-z
Li N, Lin B, Wang H, Li X, Yang F, Ding X, Yan J, Chu Z (2019) Natural variation in ZmFBL41 confers banded leaf and sheath blight resistance in maize. Nat Genet 51(10):1540–1548. https://doi.org/10.1038/s41588-019-0503-y
Liang L, Zhou L, Tang Y, Li N, Song T, Shao W, Zhang Z, Cai P, Feng F, Ma Y, Yao D, Feng Y, Ma Z, Zhao H, Song R (2019) A sequence-indexed mutator insertional library for maize functional genomics study. Plant Physiol 181(4):1404–1414. https://doi.org/10.1104/pp.19.00894
Lin Q, Zong Y, Xue C, Wang S, Jin S, Zhu Z, Wang Y, Anzalone AV, Raguram A, Doman JL, Liu DR, Gao C (2020) Prime genome editing in rice and wheat. Nat Biotechnol 38(5):582–585. https://doi.org/10.1038/s41587-020-0455-x
Liu D, Chen X, Liu J, Ye J, Guo Z (2012) The rice ERF transcription factor OsERF922 negatively regulates resistance to Magnaporthe oryzae and salt tolerance. J Exp Bot 63(10):3899–3911. https://doi.org/10.1093/jxb/ers079
Liu J, Jiang Y, Zeng J (2013) Analysis of northern corn leaf blight epidemic in 2012 and control countermeasures. Plant Prot 39(6):86–90
Liu Q, Liu H, Gong Y, Tao Y, Jiang L, Zuo W, Yang Q, Ye J, Lai J, Wu J, Lübberstedt T, Xu M (2017) An atypical thioredoxin imparts early resistance to sugarcane mosaic virus in maize. Mol Plant 10(3):483–497. https://doi.org/10.1016/j.molp.2017.02.002
Liu M, Shi Z, Zhang X, Wang M, Zhang L, Zheng K, Liu J, Hu X, Di C, Qian Q, He Z, Yang DL (2019) Inducible overexpression of Ideal Plant Architecture1 improves both yield and disease resistance in rice. Nat Plants 5(4):389–400. https://doi.org/10.1038/s41477-019-0383-2
Liu Q, Deng S, Liu B, Tao Y, Ai H, Liu J, Zhang Y, Zhao Y, Xu M (2020a) A helitron-induced RabGDIα variant causes quantitative recessive resistance to maize rough dwarf disease. Nat Commun 11(1):495. https://doi.org/10.1038/s41467-020-14372-3
Liu Y, Hu G, Zhang A, Loladze A, Hu Y, Wang H, Qu J, Zhang X, Olsen M, San Vicente F, Crossa J, Lin F, Prasanna BM (2020b) Genome-wide association study and genomic prediction of Fusarium ear rot resistance in tropical maize germplasm. The Crop J. https://doi.org/10.1016/j.cj.2020.08.008
Lu X, Liu J, Ren W, Yang Q, Chai Z, Chen R, Wang L, Zhao J, Lang Z, Wang H, Fan Y, Zhao J, Zhang C (2018) Gene-indexed mutations in maize. Mol Plant 11(3):496–504. https://doi.org/10.1016/j.molp.2017.11.013
Lübberstedt T, Klein D, Melchinger A (1998) Comparative QTL mapping of resistance to Ustilago maydis across four populations of European flint-maize. Theor Appl Genet 97:1321–1330. https://doi.org/10.1007/s001220051025
Lübberstedt T, Xia XC, Tan G, Liu X, Melchinger AE (1999) QTL mapping of resistance to Sporisorium reiliana in maize. Theor Appl Genet 99(3-4):593–598. https://doi.org/10.1007/s001220051273
Luo M, Gilbert B, Ayliffe M (2016) Applications of CRISPR/Cas9 technology for targeted mutagenesis, gene replacement and stacking of genes in higher plants. Plant Cell Rep 35(7):1439–1450. https://doi.org/10.1007/s00299-016-1989-8
Lv M, Deng C, Li X, Zhao X, Li H, Li Z, Tian Z, Leonard A, Jaqueth J, Li B, Hao J, Chang Y, Ding J (2020) Identification and fine-mapping of RppCML496, a major QTL for resistance to Puccinia polysora in maize. Plant Genome:e20062. https://doi.org/10.1002/tpg2.20062
Ma C, Ma X, Yao L, Liu Y, Du F, Yang X, Xu M (2017) qRfg3, a novel quantitative resistance locus against Gibberella stalk rot in maize. Theor Appl Genet 130(8):1723–1734. https://doi.org/10.1007/s00122-017-2921-5
Ma LS, Wang L, Trippel C, Mendoza-Mendoza A, Ullmann S, Moretti M, Carsten A, Kahnt J, Reissmann S, Zechmann B, Bange G, Kahmann R (2018) The Ustilago maydis repetitive effector Rsp3 blocks the antifungal activity of mannose-binding maize proteins. Nat Commun 9(1):1711. https://doi.org/10.1038/s41467-018-04149-0
Mackay TF, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10(8):565–577. https://doi.org/10.1038/nrg2612
Mageto EK, Crossa J, Pérez-Rodríguez P, Dhliwayo T, Palacios-Rojas N, Lee M, Guo R, San Vicente F, Zhang X, Hindu V (2020) Genomic prediction with genotype by environment interaction analysis for kernel zinc concentration in tropical maize germplasm. G3 (Bethesda) 10(8):2629–2639. https://doi.org/10.1534/g3.120.401172
Mao Y, Botella J, Liu Y, Zhu J (2019) Gene editing in plants: progress and challenges. Natl Sci Rev 6(3):421–437
Martínez-Espinoza AD, García-Pedrajas MD, Gold SE (2002) The Ustilaginales as plant pests and model systems. Fungal Genet Biol 35(1):1–20. https://doi.org/10.1006/fgbi.2001.1301
Maschietto V, Marocco A, Malachova A, Lanubile A (2015) Resistance to Fusarium verticillioides and fumonisin accumulation in maize inbred lines involves an earlier and enhanced expression of lipoxygenase (LOX) genes. J Plant Physiol 188:9–18. https://doi.org/10.1016/j.jplph.2015.09.003
Massman J, Jung H-J, Bernardo R (2013) Genomewide selection versus marker-assisted recurrent selection to improve grain yield and stover-quality traits for cellulosic ethanol in maize. Crop Sci:53. https://doi.org/10.2135/cropsci2012.02.0112
Menkir A, Ayodele M (2005) Genetic analysis of resistance to gray leaf spot of midaltitude maize inbred lines. Crop Sci 45(1):163–170. https://doi.org/10.2135/cropsci2005.0163a
Meuwissen T, Hayes B, Goddard M (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–1829
Mueller D, Wise K, Sisson A, Allen T, Bergstrom G, Bosley D, Bradley C, Broders K, Byamukama E, Chilvers M, Collins A, Faske T, Friskop A, Heiniger R, Hollier C, Hooker D, Isakeit T, Jackson-Ziems T, Jardine D, Warner F (2016) Corn yield loss estimates due to diseases in the United States and Ontario, Canada from 2012 to 2015. Plant Health Prog:17. https://doi.org/10.1094/PHP-RS-16-0030
Nekrasov V, Wang C, Win J, Lanz C, Weigel D, Kamoun S (2017) Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci Rep 7(1):482. https://doi.org/10.1038/s41598-017-00578-x
Nelson R, Wiesner-Hanks T, Wisser R, Balint-Kurti P (2018) Navigating complexity to breed disease-resistant crops. Nat Rev Genet 19(1):21–33. https://doi.org/10.1038/nrg.2017.82
Niks RE, Qi X, Marcel TC (2015) Quantitative resistance to biotrophic filamentous plant pathogens: concepts, misconceptions, and mechanisms. Annu Rev Phytopathol 53:445–470. https://doi.org/10.1146/annurev-phyto-080614-115928
Nyaga C, Gowda M, Beyene Y, Muriithi WT, Makumbi D, Olsen MS, Suresh LM, Bright JM, Das B, Prasanna BM (2019) Genome-wide analyses and prediction of resistance to MLN in large tropical maize germplasm. Genes (Basel) 11(1). https://doi.org/10.3390/genes11010016
Oliva R, Ji C, Atienza-Grande G, Huguet-Tapia JC, Perez-Quintero A, Li T, Eom JS, Li C, Nguyen H, Liu B, Auguy F, Sciallano C, Luu VT, Dossa GS, Cunnac S, Schmidt SM, Slamet-Loedin IH, Vera Cruz C, Szurek B, Frommer WB, White FF, Yang B (2019) Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat Biotechnol 37(11):1344–1350. https://doi.org/10.1038/s41587-019-0267-z
Palloix A, Ayme V, Moury B (2009) Durability of plant major resistance genes to pathogens depends on the genetic background, experimental evidence and consequences for breeding strategies. New Phytol 183(1):190–199. https://doi.org/10.1111/j.1469-8137.2009.02827.x
Pataky J (1995) Evaluation of a silk-inoculation technique to differentiate reactions of sweet corn hybrids to common smut. Phytopathology 85:1323–1328. https://doi.org/10.1094/Phyto-85-1323
Pataky JK, Tracy WF (1999) Widespread occurrence of common rust, caused by Puccinia sorghi, on Rp-resistant sweet corn in the Midwestern United States. Plant Dis 83(12):1177. https://doi.org/10.1094/pdis.1999.83.12.1177d
Pataky JK, Natti TA, Snyder EB, Kurowski CJ (2000) Puccinia sorghi in Sinaloa, Mexico virulent on corn with the Rp1-D gene. Plant Dis 84(7):810. https://doi.org/10.1094/pdis.2000.84.7.810a
Pate MC, Pataky JK, Houghton WC, Teyker RH (2000) First report of Puccinia sorghi virulent on sweet corn with the Rp1-D gene in Florida and Texas. Plant Dis 84(10):1154. https://doi.org/10.1094/pdis.2000.84.10.1154a
Peng A, Chen S, Lei T, Xu L, He Y, Wu L, Yao L, Zou X (2017) Engineering canker-resistant plants through CRISPR/Cas9-targeted editing of the susceptibility gene CsLOB1 promoter in citrus. Plant Biotechnol J 15(12):1509–1519. https://doi.org/10.1111/pbi.12733
Poland JA, Balint-Kurti PJ, Wisser RJ, Pratt RC, Nelson RJ (2009) Shades of gray: the world of quantitative disease resistance. Trends Plant Sci 14(1):21–29. https://doi.org/10.1016/j.tplants.2008.10.006
Pramanik D, Shelake RM, Kim MJ, Kim JY (2020) CRISPR-mediated engineering across the central dogma in plant biology for basic research and crop improvement. Mol Plant. https://doi.org/10.1016/j.molp.2020.11.002
Pu Z (2013) Dynamic analysis on the disease progression of maize leaf blight between Xianyu335 and improved Xianyu335 in Western Region of Heilongjiang Province. J Maize Sci 21(4):119–123
Redinbaugh MG, Stewart LR (2018) Maize lethal necrosis: an emerging, synergistic viral disease. Annu Rev Virol 5(1):301–322. https://doi.org/10.1146/annurev-virology-092917-043413
Ren J, Wu P, Trampe B, Tian X, Lübberstedt T, Chen S (2017) Novel technologies in doubled haploid line development. Plant Biotechnol J 15(11):1361–1370. https://doi.org/10.1111/pbi.12805
Savary S, Willocquet L, Pethybridge SJ, Esker P, McRoberts N, Nelson A (2019) The global burden of pathogens and pests on major food crops. Nat Ecol Evol 3(3):430–439. https://doi.org/10.1038/s41559-018-0793-y
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ, McMahan L, Van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS, Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z, Nagel DH, McCann MC, SanMiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC, Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, McCombie WR, Wing RA, Wilson RK (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326(5956):1112–1115. https://doi.org/10.1126/science.1178534
Shi J, Habben JE, Archibald RL, Drummond BJ, Chamberlin MA, Williams RW, Lafitte HR, Weers BP (2015) Overexpression of ARGOS genes modifies plant sensitivity to ethylene, leading to improved drought tolerance in both arabidopsis and maize. Plant Physiol 169(1):266–282. https://doi.org/10.1104/pp.15.00780
Shi J, Gao H, Wang H, Lafitte HR, Archibald RL, Yang M, Hakimi SM, Mo H, Habben JE (2017) ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol J 15(2):207–216. https://doi.org/10.1111/pbi.12603
Shimatani Z, Kashojiya S, Takayama M, Terada R, Arazoe T, Ishii H, Teramura H, Yamamoto T, Komatsu H, Miura K, Ezura H, Nishida K, Ariizumi T, Kondo A (2017) Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol 35(5):441–443. https://doi.org/10.1038/nbt.3833
Silva JJ, Viaro HP, Ferranti LS, Oliveira ALM, Ferreira JM, Ruas CF, Ono EYS, Fungaro MHP (2017) Genetic structure of Fusarium verticillioides populations and occurrence of fumonisins in maize grown in Southern Brazil. Crop Prot 99:160–167. https://doi.org/10.1016/j.cropro.2017.05.020
Singh A, Andersen A, Jackson-Ziems T, Lorenz A (2016) Mapping quantitative trait loci for resistance to Goss’s bacterial wilt and leaf blight in North American maize by joint linkage analysis. Crop Sci 56. https://doi.org/10.2135/cropsci2015.09.0543
Sitonik C, Suresh LM, Beyene Y, Olsen MS, Makumbi D, Oliver K, Das B, Bright JM, Mugo S, Crossa J, Tarekegne A, Prasanna BM, Gowda M (2019) Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor Appl Genet 132(8):2381–2399. https://doi.org/10.1007/s00122-019-03360-x
Soliman A, Gulden R, Tambong J, Bajracharya P, Adam L, Xu R, Cott M, Daayf F (2018) Developed and validated inoculation and disease assessment methods for Goss’s bacterial wilt and leaf blight disease of corn. Crop Prot 112:159–167. https://doi.org/10.1016/j.cropro.2018.05.022
Song FJ, Xiao MG, Duan CX, Li HJ, Zhu ZD, Liu BT, Sun SL, Wu XF, Wang XM (2015) Two genes conferring resistance to Pythium stalk rot in maize inbred line Qi319. Mol Gen Genomics 290(4):1543–1549. https://doi.org/10.1007/s00438-015-1019-5
Springer NM, Anderson SN, Andorf CM, Ahern KR, Bai F, Barad O, Barbazuk WB, Bass HW, Baruch K, Ben-Zvi G, Buckler ES, Bukowski R, Campbell MS, EKS C, Chomet P, Dawe RK, Davenport R, Dooner HK, Du LH, Du C, Easterling KA, Gault C, Guan JC, Hunter CT, Jander G, Jiao Y, Koch KE, Kol G, Köllner TG, Kudo T, Li Q, Lu F, Mayfield-Jones D, Mei W, DR MC, Noshay JM, Portwood JL 2nd, Ronen G, Settles AM, Shem-Tov D, Shi J, Soifer I, Stein JC, Stitzer MC, Suzuki M, Vera DL, Vollbrecht E, Vrebalov JT, Ware D, Wei S, Wimalanathan K, Woodhouse MR, Xiong W, Brutnell TP (2018) The maize W22 genome provides a foundation for functional genomics and transposon biology. Nat Genet 50(9):1282–1288. https://doi.org/10.1038/s41588-018-0158-0
St Clair DA (2010) Quantitative disease resistance and quantitative resistance Loci in breeding. Annu Rev Phytopathol 48:247–268. https://doi.org/10.1146/annurev-phyto-080508-081904
Storey HH, Howland AK (1957) Resistance in maize to the tropical American rust fungus, puccinia polysora underw. Heredity 11(3):289–301. https://doi.org/10.1038/hdy.1957.26
Sucher J, Boni R, Yang P, Rogowsky P, Büchner H, Kastner C, Kumlehn J, Krattinger SG, Keller B (2017) The durable wheat disease resistance gene Lr34 confers common rust and northern corn leaf blight resistance in maize. Plant Biotechnol J 15(4):489–496. https://doi.org/10.1111/pbi.12647
Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, Song W, Zhang M, Cui Y, Dong X, Liu H, Ma X, Jiao Y, Wang B, Wei X, Stein JC, Glaubitz JC, Lu F, Yu G, Liang C, Fengler K, Li B, Rafalski A, Schnable PS, Ware DH, Buckler ES, Lai J (2018) Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat Genet 50(9):1289–1295. https://doi.org/10.1038/s41588-018-0182-0
Sun X, Qi X, Wang W, Liu X, Zhao H, Wu C, Chang X, Zhang M, Chen H, Gong G (2020) Etiology and symptoms of maize leaf spot caused by Bipolaris spp. in Sichuan, China. Pathogens 9(3). https://doi.org/10.3390/pathogens9030229
Swinnen G, Goossens A, Pauwels L (2016) Lessons from domestication: targeting Cis-regulatory elements for crop improvement. Trends Plant Sci 21(6):506–515. https://doi.org/10.1016/j.tplants.2016.01.014
Tanaka S, Gollin I, Rössel N, Kahmann R (2020) The functionally conserved effector Sta1 is a fungal cell wall protein required for virulence in Ustilago maydis. New Phytol 227(1):185–199. https://doi.org/10.1111/nph.16508
Tao Y, Jiang L, Liu Q, Zhang Y, Zhang R, Ingvardsen CR, Frei UK, Wang B, Lai J, Lübberstedt T, Xu M (2013a) Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol 13:162. https://doi.org/10.1186/1471-2229-13-162
Tao Y, Liu Q, Wang H, Zhang Y, Huang X, Wang B, Lai J, Ye J, Liu B, Xu M (2013b) Identification and fine-mapping of a QTL, qMrdd1, that confers recessive resistance to maize rough dwarf disease. BMC Plant Biol 13:145. https://doi.org/10.1186/1471-2229-13-145
Technow F, Bürger A, Melchinger AE (2013) Genomic prediction of northern corn leaf blight resistance in maize with combined or separated training sets for heterotic groups. G3 (Bethesda) 3(2):197–203. https://doi.org/10.1534/g3.112.004630
Tenaillon MI, U’Ren J, Tenaillon O, Gaut BS (2004) Selection versus demography: a multilocus investigation of the domestication process in maize. Mol Biol Evol 21(7):1214–1225. https://doi.org/10.1093/molbev/msh102
Ullstrup AJ (1965) Inheritance and linkage of a gene determining resistance in Maize to an American race of Fuccinia polysora. Phytopathology 55:425–428
Ullstrup AJ (1972) The impacts of the southern corn leaf blight epidemics of 1970-1971. Annu Rev Phytopathol 10:37–50. https://doi.org/10.1146/annurev.py.10.090172.000345
Vidaver AK, Carlson R (1978) Leaf spot of field corn caused by Pseudomonas andropogonis. Plant Dis Rep 62:213–216
Vivek B, Odongo O, Njuguna J, Imanywoha J, Bigirwa G, Diallo A, Pixley K (2009) Diallel analysis of grain yield and resistance to seven diseases of 12 African maize (Zea mays L.) inbred lines. Euphytica 172:329–340. https://doi.org/10.1007/s10681-009-9993-5
Vivek BS, Krishna GK, Vengadessan V, Babu R, Zaidi PH, Kha LQ, Mandal SS, Grudloyma P, Takalkar S, Krothapalli K, Singh IS, Ocampo ETM, Xingming F, Burgueño J, Azrai M, Singh RP, Crossa J (2017) Use of genomic estimated breeding values results in rapid genetic gains for drought tolerance in maize. Plant Genome 10(1). https://doi.org/10.3835/plantgenome2016.07.0070
Wang X, Duan C (2020) Reorganization of maize disease and causal agent names and disscution on their standardized translation of Chinese names. Sci Agric Sin 53(2):288–316. https://doi.org/10.3864/j.issn.0578-1752.2020.02.006
Wang X, Zhang Y, Xu X, Li H, Wu X, Zhang S, Li X (2014a) Evaluation of maize inbred lines currently used in Chinese breeding programs for resistance to six foliar diseases. The Crop J:2. https://doi.org/10.1016/j.cj.2014.04.004
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu JL (2014b) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32(9):947–951. https://doi.org/10.1038/nbt.2969
Wang F, Wang C, Liu P, Lei C, Hao W, Gao Y, Liu YG, Zhao K (2016) Enhanced rice blast resistance by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor gene OsERF922. PLoS One 11(4):e0154027. https://doi.org/10.1371/journal.pone.0154027
Wang C, Yang Q, Wang W, Li Y, Guo Y, Zhang D, Ma X, Song W, Zhao J, Xu M (2017) A transposon-directed epigenetic change in ZmCCT underlies quantitative resistance to Gibberella stalk rot in maize. New Phytol 215(4):1503–1515. https://doi.org/10.1111/nph.14688
Wang B, Qin J, Shi C, Zheng J, Qin Y, Huang A (2019a) QTL mapping and genetic analysis of a gene with high resistance to southern corn rust. Sci Agric Sin 52(12):2033–2041. https://doi.org/10.3864/j.issn.0578-1752.2019.12.002
Wang B, Zhu L, Zhao B, Zhao Y, Xie Y, Zheng Z, Li Y, Sun J, Wang H (2019b) Development of a haploid-inducer mediated genome editing system for accelerating maize breeding. Mol Plant 12(4):597–602. https://doi.org/10.1016/j.molp.2019.03.006
Wang S, Chen Z, Tian L, Ding Y, Zhang J, Zhou J, Liu P, Chen Y, Wu L (2019c) Comparative proteomics combined with analyses of transgenic plants reveal ZmREM1.3 mediates maize resistance to southern corn rust. Plant Biotechnol J 17(11):2153–2168. https://doi.org/10.1111/pbi.13129
Wanlayaporn K, Authrapun J, Vanavichit A, Tragoonrung S (2013) QTL mapping for partial resistance to southern corn rust using RILs of tropical sweet corn. Am J Plant Sci 04:878–889. https://doi.org/10.4236/ajps.2013.44108
Welz G, Geiger H (2000) Genes for resistance to northern corn leaf blight in diverse maize populations. Plant Breed 119:1–14. https://doi.org/10.1046/j.1439-0523.2000.00462.x
White DG (1999) Compendium of corn diseases, 3rd edition. APS Press,
Wright P, Parker TV, Hedderley (2014) Effect of planting dates and azoxystrobin fungicide application regimes on common rust of maize. N Z J Crop Hortic Sci 42:99–110. https://doi.org/10.1080/01140671.2013.860040
Wu X, Li N, Zhao P, He Y, Wang S (2015) Geographic and genetic identification of RppS, a novel locus conferring broad resistance to southern corn rust disease in China. Euphytica 205. https://doi.org/10.1007/s10681-015-1376-5
Xia X, Melchinger AE, Kuntze L, Lübberstedt T (1999) Quantitative trait Loci mapping of resistance to sugarcane mosaic virus in maize. Phytopathology 89(8):660–667. https://doi.org/10.1094/phyto.1999.89.8.660
Xing Y, Ingvardsen C, Salomon R, Lübberstedt T (2006) Analysis of sugarcane mosaic virus resistance in maize in an isogenic dihybrid crossing scheme and implications for breeding potyvirus-resistant maize hybrids. Genome 49(10):1274–1282. https://doi.org/10.1139/g06-070
Xu M, Melchinger A, Xia X, Lübberstedt T (1999) High-resolution mapping of loci conferring resistance to sugarcane mosaic virus in maize using RFLP, SSR, and AFLP markers. Mol Gen Genet 261(3):574–581. https://doi.org/10.1007/s004380051003
Xu L, Zhang Y, Shao S, Chen W, Tan J, Zhu M, Zhong T, Fan X, Xu M (2014) High-resolution mapping and characterization of qRgls2, a major quantitative trait locus involved in maize resistance to gray leaf spot. BMC Plant Biol 14:230. https://doi.org/10.1186/s12870-014-0230-6
Xu Z, Xu X, Gong Q, Li Z, Li Y, Wang S, Yang Y, Ma W, Liu L, Zhu B, Zou L, Chen G (2019) Engineering broad-spectrum bacterial blight resistance by simultaneously disrupting variable TALE-binding elements of multiple susceptibility genes in rice. Mol Plant 12(11):1434–1446. https://doi.org/10.1016/j.molp.2019.08.006
Xu Z, Hua J, Wang F, Cheng Z, Meng Q, Chen Y, Han X, Tie S, Liu C, Li X, Wang Z, Weng J (2020) Marker-assisted selection of qMrdd8 to improve maize resistance to rough dwarf disease. Breed Sci 70(2):183–192. https://doi.org/10.1270/jsbbs.19110
Yang D, Jin D, Wang B, Zhang D, Nguyen H, Zhang C, Chen S (2005) Characterization and mapping of Rpi1, a gene that confers dominant resistance to stalk rot in maize. Mol Gen Genomics 274(3):229–234. https://doi.org/10.1007/s00438-005-0016-5
Yang Q, Yin G, Guo Y, Zhang D, Chen S, Xu M (2010) A major QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet 121(4):673–687. https://doi.org/10.1007/s00122-010-1339-0
Yang Q, Zhang D, Xu M (2012) A sequential quantitative trait locus fine-mapping strategy using recombinant-derived progeny. J Integr Plant Biol 54(4):228–237. https://doi.org/10.1111/j.1744-7909.2012.01108.x
Yang Q, Li Z, Li W, Ku L, Wang C, Ye J, Li K, Yang N, Li Y, Zhong T, Li J, Chen Y, Yan J, Yang X, Xu M (2013) CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc Natl Acad Sci U S A 110(42):16969–16974. https://doi.org/10.1073/pnas.1310949110
Yang Q, Balint-Kurti P, Xu M (2017a) Quantitative disease resistance: dissection and adoption in maize. Mol Plant 10(3):402–413. https://doi.org/10.1016/j.molp.2017.02.004
Yang Q, He Y, Kabahuma M, Chaya T, Kelly A, Borrego E, Bian Y, El Kasmi F, Yang L, Teixeira P, Kolkman J, Nelson R, Kolomiets M, Dang JL, Wisser R, Caplan J, Li X, Lauter N, Balint-Kurti P (2017b) A gene encoding maize caffeoyl-CoA O-methyltransferase confers quantitative resistance to multiple pathogens. Nat Genet 49(9):1364–1372. https://doi.org/10.1038/ng.3919
Yang N, Liu J, Gao Q, Gui S, Chen L, Yang L, Huang J, Deng T, Luo J, He L, Wang Y, Xu P, Peng Y, Shi Z, Lan L, Ma Z, Yang X, Zhang Q, Bai M, Li S, Li W, Liu L, Jackson D, Yan J (2019a) Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet 51(6):1052–1059. https://doi.org/10.1038/s41588-019-0427-6
Yang P, Praz C, Li B, Singla J, Robert CAM, Kessel B, Scheuermann D, Lüthi L, Ouzunova M, Erb M, Krattinger SG, Keller B (2019b) Fungal resistance mediated by maize wall-associated kinase ZmWAK-RLK1 correlates with reduced benzoxazinoid content. New Phytol 221(2):976–987. https://doi.org/10.1111/nph.15419
Yang P, Scheuermann D, Kessel B, Koller T, Greenwood JR, Hurni S, Herren G, Zhou S, Marande W, Wicker T, Krattinger SG, Ouzunova M, Keller B (2021) Alleles of a wall-associated kinase gene account for three of the major northern corn leaf blight resistance loci in maize. Plant J. https://doi.org/10.1111/tpj.15183
Yao G, Shan J, Cao B, Cui L, Du S, Han Z, Liu T, Li C, Wang L (2013) Mapping the maize southern rust resistance gene in inbred line CML470. J Plant Gen Res 14(3):518–522
Yao L, Li Y, Ma C, Tong L, Du F, Xu M (2020) Combined genome-wide association study and transcriptome analysis reveal candidate genes for resistance to Fusarium ear rot in maize. J Integr Plant Biol 62(10):1535–1551. https://doi.org/10.1111/jipb.12911
Ye J, Zhong T, Zhang D, Ma C, Wang L, Yao L, Zhang Q, Zhu M, Xu M (2019) The Auxin-regulated protein ZmAuxRP1 coordinates the balance between root growth and stalk rot disease resistance in maize. Mol Plant 12(3):360–373. https://doi.org/10.1016/j.molp.2018.10.005
Yin K, Gao C, Qiu JL (2017) Progress and prospects in plant genome editing. Nat Plants 3:17107. https://doi.org/10.1038/nplants.2017.107
Yousef G, Juvik J (2001) Comparison of phenotypic and marker-assisted selection for quantitative traits in sweet corn. Crop Sci:41. https://doi.org/10.2135/cropsci2001.413645x
Zaidi SS, Mukhtar MS, Mansoor S (2018) Genome editing: targeting susceptibility genes for plant disease resistance. Trends Biotechnol 36(9):898–906. https://doi.org/10.1016/j.tibtech.2018.04.005
Zhang H, Chen J, Lei J, Adams M (2001) Sequence analysis shows that a dwarfing disease on rice, wheat and maize in China is caused by rice black-streaked dwarf virus. Eur J Plant Pathol 107:563–567. https://doi.org/10.1023/A:1011204010663
Zhang Y, Xu L, Zhang D, Dai J, Wang S (2010) Mapping of southern corn rust-resistant genes in the W2D inbred line of maize (Zea mays L.). Mol Breed 25(3):433–439. https://doi.org/10.1007/s11032-009-9342-3
Zhang Z, Yang L, Zhou S, Wang H, Li W, Fu F (2011) Improvement of resistance to maize dwarf mosaic virus mediated by transgenic RNA interference. J Biotechnol 153(3-4):181–187. https://doi.org/10.1016/j.jbiotec.2011.03.019
Zhang D, Liu Y, Guo Y, Yang Q, Ye J, Chen S, Xu M (2012a) Fine-mapping of qRfg2, a QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet 124(3):585–596. https://doi.org/10.1007/s00122-011-1731-4
Zhang Y, Xu L, Fan X, Tan J, Chen W, Xu M (2012b) QTL mapping of resistance to gray leaf spot in maize. Theor Appl Genet 125(8):1797–1808. https://doi.org/10.1007/s00122-012-1954-z
Zhang Y, Lubberstedt T, Xu M (2013) The genetic and molecular basis of plant resistance to pathogens. J Genet Genomics 40(1):23–35. https://doi.org/10.1016/j.jgg.2012.11.003
Zhang N, Zhang B, Zuo W, Xing Y, Konlasuk S, Tan G, Zhang Q, Ye J, Xu M (2017a) Cytological and molecular characterization of ZmWAK-mediated head-smut resistance in maize. Mol Plant-Microbe Interact 30(6):455–465. https://doi.org/10.1094/mpmi-11-16-0238-r
Zhang X, Yang Q, Rucker E, Thomason W, Balint-Kurti P (2017b) Fine mapping of a quantitative resistance gene for gray leaf spot of maize (Zea mays L.) derived from teosinte (Z. mays ssp. parviglumis). Theor Appl Genet 130(6):1285–1295. https://doi.org/10.1007/s00122-017-2888-2
Zhao B, Ardales E, Brasset E, Claflin L, Leach J, Hulbert S (2004a) The Rxo1/ Rba1 locus of maize controls resistance reactions to pathogenic and non-host bacteria. Theor Appl Genet 109(1):71–79. https://doi.org/10.1007/s00122-004-1623-y
Zhao B, Ardales EY, Raymundo A, Bai J, Trick HN, Leach JE, Hulbert SH (2004b) The avrRxo1 gene from the rice pathogen Xanthomonas oryzae pv. oryzicola confers a nonhost defense reaction on maize with resistance gene Rxo1. Mol Plant-Microbe Interact 17(7):771–779. https://doi.org/10.1094/mpmi.2004.17.7.771
Zhao M, Gao S, Zhang Z, Rong T, Pan G (2006) Initial identification of quantitative trait loci controlling resistance to banded leaf and sheath blight at elongating and heading date in maize. Fen Zi Xi Bao Sheng Wu Xue Bao 39(2):139–144
Zhao X, Tan G, Xing Y, Wei L, Chao Q, Zuo W, Lübberstedt T, Xu M (2012a) Marker-assisted introgression of qHSR1 to improve maize resistance to head smut. Mol Breed 30(2):1077–1088. https://doi.org/10.1007/s11032-011-9694-3
Zhao Y, Lu X, Liu C, Guan H, Zhang M, Li Z, Cai H, Lai J (2012b) Identification and fine mapping of rhm1 locus for resistance to Southern corn leaf blight in maize. J Integr Plant Biol 54(5):321–329. https://doi.org/10.1111/j.1744-7909.2012.01112.x
Zhao P, Zhang G, Wu X, Li N, Shi D, Zhang D, Ji C, Xu M, Wang S (2013) Fine mapping of RppP25, a southern rust resistance gene in maize. J Integr Plant Biol 55(5):462–472. https://doi.org/10.1111/jipb.12027
Zheng H, Chen J, Mu C, Makumbi D, Xu Y, Mahuku G (2018) Combined linkage and association mapping reveal QTL for host plant resistance to common rust (Puccinia sorghi) in tropical maize. BMC Plant Biol 18(1):310. https://doi.org/10.1186/s12870-018-1520-1
Zhou C, Chen C, Cao P, Wu S, Sun J, Jin D, Wang B (2007) Characterization and fine mapping of RppQ, a resistance gene to southern corn rust in maize. Mol Gen Genomics 278(6):723–728. https://doi.org/10.1007/s00438-007-0288-z
Zhu X, Zhao J, Abbas HMK, Liu Y, Cheng M, Huang J, Cheng W, Wang B, Bai C, Wang G, Dong W (2018) Pyramiding of nine transgenes in maize generates high-level resistance against necrotrophic maize pathogens. Theor Appl Genet 131(10):2145–2156. https://doi.org/10.1007/s00122-018-3143-1
Zhu H, Li C, Gao C (2020) Applications of CRISPR-Cas in agriculture and plant biotechnology. Nat Rev Mol Cell Biol 21(11):661–677. https://doi.org/10.1038/s41580-020-00288-9
Zong Y, Wang Y, Li C, Zhang R, Chen K, Ran Y, Qiu JL, Wang D, Gao C (2017) Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat Biotechnol 35(5):438–440. https://doi.org/10.1038/nbt.3811
Zong Y, Song Q, Li C, Jin S, Zhang D, Wang Y, Qiu JL, Gao C (2018) Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A. Nat Biotechnol. https://doi.org/10.1038/nbt.4261
Zuo W, Chao Q, Zhang N, Ye J, Tan G, Li B, Xing Y, Zhang B, Liu H, Fengler KA, Zhao J, Zhao X, Chen Y, Lai J, Yan J, Xu M (2015) A maize wall-associated kinase confers quantitative resistance to head smut. Nat Genet 47(2):151–157. https://doi.org/10.1038/ng.3170
Zuo W, Ökmen B, Depotter JRL, Ebert MK, Redkar A, Misas Villamil J, Doehlemann G (2019) Molecular interactions between smut fungi and their host plants. Annu Rev Phytopathol 57:411–430. https://doi.org/10.1146/annurev-phyto-082718-100139
Zwonitzer JC, Bubeck DM, Bhattramakki D, Goodman MM, Arellano C, Balint-Kurti PJ (2009) Use of selection with recurrent backcrossing and QTL mapping to identify loci contributing to southern leaf blight resistance in a highly resistant maize line. Theor Appl Genet 118(5):911–925. https://doi.org/10.1007/s00122-008-0949-2
Acknowledgements
We apologize to colleagues whose works were not cited due to space limitations. We thank Prof. Canxing Duan (Chinese Academy of Agricultural Sciences) for providing the photos of NCLB, SCLB, and southern rust. We thank Dr. Gengshen Chen (Huazhong Agriculture University) for sharing the photo of common rust.
Funding
This work was supported by grant from the China Postdoctoral Science Foundation (2019TQ0346) to T.Z and Yunnan Provincial Science and Technology Department (202005AF150026).
Author information
Authors and Affiliations
Contributions
MX and TZ conceived and outlined the review. MX, MZ, and TZ performed the literature search. TZ, MZ, LT, and MX wrote the paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical collection on Maize Genetics, Genomics and Sustainable Improvement
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, M., Tong, L., Xu, M. et al. Genetic dissection of maize disease resistance and its applications in molecular breeding. Mol Breeding 41, 32 (2021). https://doi.org/10.1007/s11032-021-01219-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-021-01219-y