
Abstract
Complete hierarchy of network amorphization scenarios initiated in AsxS100-x nanoarsenicals within As4S4-As4S3 cut-Sect. (50 ≤ x ≤ 57) is reconstructed employing materials-computational approach based on ab-initio quantum-chemical modeling code (CINCA). Under nanostructurization due to high-energy mechanical milling, the inter-crystalline transformations to nanoscopic β-As4S4 phase accompanied by appearance of covalent-network amorphous matrix are activated. General amorphization trend under nanomilling obeys tending from molecular cage-like structures to optimally-constrained covalent-bonded networks compositionally invariant with parent arsenical. The contribution of amorphization paths in nanoarsenicals is defined by their chemistry with higher molecular-to-network barriers proper to As4S3-rich alloys. The generated amorphous phase is intrinsically decomposed, possessing double-Tg relaxation due to stoichiometric (x = 40) and non-stoichiometric (x > 40) sub-networks, which are built of AsS3/2 pyramids and As-rich arrangement keeping (i) two separated As-As bonds derived from realgar-type molecules, (ii) two neighboring As-As bonds derived from pararealgar-type molecules or (iii) three neighboring As-As bonds in triangle-like geometry derived from dimorphite-type molecules. Compositional invariance of nanoamorphous phase is ensured by growing sequence of network-forming clusters with average coordination numbers Z in the row (As2S4/2, Z = 2.50) – (As3S5/2, Z = 2.55) – (As3S3/2, Z = 2.67). Diversity of main molecular-to-network amorphizing pathways in nanoarsenicals is reflected on the unified potential energy landscape specified for boundary As4S4 and As4S3 components.
Similar content being viewed by others
Introduction
The family of binary arsenic sulphides AsxS100−x [1,2,3,4,5,6], often referred to as arsenicals due to their anticancer therapeutic functionality [7], are accepted as representatives of canonical substances, where diverse structure-conformation tendencies can be revealed in dependence on their chemistry. Thus, at the moderated As content in these thioarsenide alloys close to stoichiometry As2S3 (viz. As40S60), competitive matrix amorphization (vitrification) and layer-type conformation (crystallization) processes occur, while in As-rich AsxS100-x compounds (where x exceeds 40) the latter are replaced by molecular-crystallization tendencies accompanied by stabilization of As4Sn cage-like molecules (n = 5,4,3) [8,9,10].
Under full saturation of covalent chemical bonding in respect to the Mott’s 8-N rule [3], which is characteristic feature of many chalcogenide compounds allowing their consideration in terms of the average coordination number Z (the number of covalent bonds per atom) [5], this As-S system reveals unprecedented glass-forming ability in a vicinity of arsenic trisulphide As2S3 [4]. That is why the glassy g-As2S3 prepared by melt-quenching cannot be crystallized during prolonged thermal treatment even above glass-transition temperature Tg [2, 3], despite the known fact on existing of isocompositional mineral with layered crystalline structure (the orthorhombic orpiment possessing space group P21/n) [8]. Trigonal AsS3/2 pyramids (as main structural motives of this glass-former) interconnected by S atoms in so-called corner- or atom-shared links form basis for under-stoichiometric covalent networks of S-rich AsxS100-x arsenicals (x < 40), realized due to polymerization (bridging) properties of –Sn– chains. Therefore, the compositional domain of stable glass-formers extends in binary As-S system to Z ~ 2.16 dependently on melt-quenching route (it means that g-As16S84 possesses competitive S separation-agglomeration properties) [2,3,4]. Within this range of S-rich AsxS100-x arsenicals up to stoichiometric As2S3 (2.16 < Z ≤ 2.40), this system demonstrates few anomalies like instability onsets in cluster-forming energies [6], which were mistakenly accepted as “signatures” of intermediate optimally-constrained (rigid and stress-free) phases in covalent-bonded network substances [11].
Controversially, under enlarged As content in this AsxS100-x system over As2S3 stoichiometry, the network-forming tendency rapidly disappears at Z ~ 2.44–2.46 on a cost of partially crystallized arrangement of thioarsenide As4Sn molecules (n = 5,4,3) [1,2,3,4]. These cage-like entities (having no dangling bonds or terminated inter-cluster links) serve as main blocks for molecular crystals of some minerals, such as uzonite As4S5 (Z = 2.44), realgar α-As4S4 (Z = 2.50), bonazitte or β-As4S4 (Z = 2.50), pararealgar As4S4 (Z = 2.50), as well as α-/β-dimorphite As4S3 (Z = 2.57) [4, 8]. The former (the uzonite As4S5) along with tetra-arsenic tetrasulphide As4S4 possessing four different crystallographic polymorphs seem to be most plausible candidates for devitrification in the group of melt-quenched AsxS100-x alloys at x > 44.
Notwithstanding, phase equilibria become more complicated with progressive increase in As content in AsxS100-x arsenicals, resulting in the second glass-forming region at 51 ≤ x ≤ 66. Hruby [1] was the first who considered these glasses as metastable plastic phases with Tg below room temperature. In his opinion, thermodynamically stable glasses of these compositions (with Z = 2.51–2.66) do not exist. The second glass-forming region in these thioarsenide alloys was shown to be superimposed with stabilization domain of tetra-arsenic trisulphide polymorphs As4S3 (Z = 2.57) [12, 13], meaning the crucible role of spherically-symmetric As4S3 cage-like molecules [14]. The plastic-crystalline rhombohedral modification of this dimorphite-type As4S3 phase as found by Chattopadhyay et al. [15] and further generalized by Blachnik and Wickel [16] was accepted as responsible for additional rotational disorder in these glass-formers.
This finding was further proved by Aitken for ternary GexAsyS100−x–y glasses with As:Ge ratio approaching ~ 17:1 [17, 18]. It was also shown [19,20,21] that under nanostructurization caused by external influence such as high-energy mechanical milling (nanomilling), the complicated inter-crystalline phase transformations towards nanocrystalline β-As4S4 polymorph (nc-β-As4S4) accompanied by appearance of isocompositional amorphous phase having covalent network were activated in these arsenicals along realgar-dimorphite As4S4–As4S3 cut-Sect. (2.50 ≤ Z ≤ 2.57).
In this work, the amorphization scenarios in AsxS100−x arsenicals along As4S4-As4S3 line (50 ≤ x ≤ 57) will be recognized employing the advanced materials-computational approach [12, 22] developed at the basis of the authorized ab-initio quantum-chemical modeling code (CINCA) [6, 23, 24].
Method: the CINCA Modeling of Molecular-Network Conformations in Covalent Substances
The geometrically-optimized configurations of As4Sn cage-like molecules (n = 4,3) and their network-forming derivatives responsible for amorphization in AsxS100-x arsenicals along As4S4-As4S3 cut-line were simulated with ab-initio quantum-chemical cluster-modeling algorithm CINCA (cation-interlinked network cluster approach) [6, 23, 24]. Network-forming clusters were reconstructed by breaking respective As4S4 or As4S3 molecules on distinct fragments linking them with surrounding matrix by S1/2…S1/2 bridging chains. The HyperChem Release 7.5 program package based on restricted Hartree–Fock self-consistent field method with split-valence double-zeta basis set and single polarization function 6-311G* [25,26,27] was employed for cluster energy calculations. Final geometrical optimization and single-point energy calculations for selected clusters were performed employing the Fletcher-Reeves conjugate gradient method until the root-mean-square gradient of 0.1 kcal/(Å·mol) was reached. The calculated cluster-forming energies Ef were corrected on the energy of terminated H atoms used to transform the network-forming clusters into molecular self-consistent precursors according to the procedure developed elsewhere [27, 28], and finally determined in respect to the energy of single trigonal AsS3/2 pyramid (Ef = − 79.404 kcal/mol) [6, 23].
Thus, the developed cluster-modeling code CINCA allows quantitative comparison between molecular- and network-forming tendencies in covalent substances, parameterizing adequately competitive crystallization-amorphization scenarios.
Results and Discussion
Cluster Modeling of Amophization Paths Derived from Realgar-Type α/β-As4S4 Polymorphs
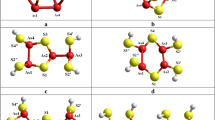
The quantum-chemical model of realgar-type As4S4 molecule (see Fig. 1a) was developed in our preliminary research [29]. This cage-like molecule of D2d symmetry built of 8 heteronuclear As-S bonds and 2 homonuclear As-As bonds in separated mutually orthogonal configuration evolves 8 small rings (4 pentagons and 4 hexagons), thus resulting in under-constrained structural motive possessing average number of topological constraints per atom nc = 2.875 (in respect to the Phillips-Thorpe constraint-counting algorithm [30,31,32] applied to stretching and bending forces ascribed to bonds in this molecule). The same symmetric As4S4 molecule (with intramolecular bond lengths and angles close to those refined from realistic crystalline structures) is character for mineral realgar α-As4S4 [33, 34], synthetic β-As4S4 [35] and its natural analogue (mineral bonazziite As4S4 [36]). The calculated cluster-forming energy Ef for this molecule (in respect to the energy of AsS3/2 pyramid) equals − 0.58 kcal/mol (Table 1) [19, 22], the value dominated over all molecular and network-forming arsenical clusters within compositional range of interest (2.50 ≤ Z ≤ 2.57).

Geometrically-optimized configurations of realgar-type α/β-As4S4 cage-like molecule (a) and its energetically favorable network derivatives formed from As4S5H2 molecule by single × 1-break in S1 position (b), and from As4S7H6 molecule by triple × 3-break in S1-S2-S3 positions (c). The terminated H atoms are grey-colored, S and As atoms are yellow- and red-depicted, and covalent bonds between atoms are denoted by respectively colored sticks [19, 22]
The most plausible amorphization scenarios in arsenic monosulphide AsS (viz. As4S4) are related to high-temperature tetra-arsenic tetrasulphide β-As4S4 polymorph [10, 19, 20]. This specificity is revealed due to lower barrier of α-As4S4-to-β-As4S4 transition [37] as compared with nanostructurization-driven amorphization from each of these phases (low-temperature α-As4S4 or high-temperature β-As4S4), provided direct low-to-high-temperature phase transformation was not inhibited.
Among a group of network-forming derivatives from this realgar-type cage-like molecule (β-As4S4), the smallest cluster-forming energy Ef = −1.29 kcal/mol is achieved for single-broken × 1-β-As4S4 cluster (see Table 1) derived from As4S5H2 molecular precursor as shown on Fig. 1b [19, 22]. This cluster having one hexagon and two pentagons in atomic arrangement is optimally-constrained since nc = 3.00, which is in strict respect to space dimensionality (D = 3). Therefore, covalent-bonded structures bult of such clusters are most favorable for amorphization. Because of low barrier with ground state of As4S4 molecule due to ΔEf = (1.29–0.58) kcal/mol = 0.71 kcal/mol, the expected molecular-to-network transformation occurs even in directly synthesized arsenic monosulphide, being a source of uncontrolled spontaneous amorphization [10, 19, 38,39,40].
An alternative path of induced amorphization in β-As4S4 is related to × 3-β-As4S4 network-forming cluster prototyped by As4S7H6 molecule which appears as triple-broken derivative from realgar-type As4S4 molecule possessing chain structure without small rings (see Fig. 1c) [19, 22]. Despite evidently over-constrained constitution (in view of nc = 3.25), the small Ef value approaching − 1.72 kcal/mol is character for this cluster (see Table 1), resulting in low molecular-to-network barrier ΔEf = 1.14 kcal/mol [19]. This cluster is commensurable (within ± 0.03 kcal/mol error-bar proper to CINCA calculations [23]) with other one, the quadruple-broken × 4-β-As4S4 (or, alternatively, As2S4/2 cluster) obtained by breaking in all S atom positions, thus keeping separated As-As bond in network. These × 3-β-As4S4 clusters surely dominate in nanostructured arsenicals (nanoarsenicals) in view of lower number of covalent bonds destructed in × 0-β-As4S4 molecule.
Thus, full variety of amorphization scenarios derived from β-As4S4 phase can be depicted on potential energy landscape as shown on Fig. 2. The above network-forming derivatives (× 1-β-As4S4 and × 3-β-As4S4) evidently prefer over other variants of network clustering originated in part due to double-breaking of covalent bonds in realgar-type As4S4 molecule (× 2–1-β-As4S4 and × 2–2-β-As4S4) [19].

Potential energy landscape showing amorphization scenarios derived from β-As4S4 phase. The settle-points corresponding to network clusters derived from realgar-type As4S4 molecules by x-fold bond-breaking (keeping small rings, such as pentagons/hexagons nominated in parenthesis) are denoted with respective cluster-forming energies Ef at the right axis. The ground molecular-crystalline state is given in double-well presentation for low-temperature realgar α-As4S4 and high-temperature β-As4S4 polymorph (no amorphization transitions are expected from α-As4S4 state)
Cluster Modeling of Amorphization Scenarios Activated from Pararealgar As4S4 Phase
Pararealgar As4S4 as metastable product of light-induced alteration from both tetra-arsenic tetrasulphide polymorphs (α-As4S4 and β-As4S4) [8, 41, 42], was found to exist (albeit in small amount) in As4S4-As4S3 alloys prepared by melt-quenching and/or nanomilling [20] (following the Kyono’s notation [43], and for sake of convenience, this phase will be marked thereafter as p-As4S4). Notwithstanding, the network-forming tendencies derived from this pararealgar p-As4S4 phase should be accepted in the overall balance of possible amorphization scenarios in the studied arsenicals, provided they were preliminary light-soaked.
The simulated pararealgar-type p-As4S4 cage-like molecule possesses reduced Cs symmetry due to asymmetric adjacent configuration of two neighboring homonuclear As-As bonds (such as As4-As3 and As2–As3 on Fig. 3a) with common As3 atom, the optimized parameters of this configuration in Table 2 being agreed with those determined from the known experimental model [40]. This p-As4S4 molecule includes one tetragon (As2As3As4S1), two pentagons (As1S2As2As3S3 and As1S4As4As3S3) and one hexagon (As1S4As4S1As2S2), thus resulting in under-constrained topological motive with nc = 2.75 (Table 3). In comparison with realgar-type As4S4 molecule (Fig. 1a), the calculated Ef energy for this molecule is higher approaching − 0.94 kcal/mol.

Geometrically-optimized configurations of pararealgar-type p-As4S4 cage-like molecule (a) and its most energetically favorable network derivative separating arsenical matrix on AsS3H3 and As3S5H5 molecules due to quadruple × 4-break in S1-S2-S3-S4 positions. The terminated H atoms are grey colored, S and As atoms are respectively depicted by yellow and red balls, and covalent bonds between atoms are stick-denoted
A great variety of network-forming clusters are expected from this asymmetric configuration of metastable p-As4S4 molecule, their small-ring characteristics (number and type of small rings, hexagons/pentagons/tetragons) and cluster-forming energies Ef being presented in Table 3. The most plausible among them seems to be × 4-p-As4S4 cluster reconstructed from As4S8H8 molecular precursor due to quadruple × 4-break in all S atom positions, which completely destroys small-ring structure of this molecule separating arsenical matrix on AsS3H3 and As3S5H5 molecular precursors (see Fig. 3b). From network-reconstruction viewpoint, this cluster with equivalent bond distances and angles given in Table 4 results in over-constrained topology of overall backbone with nc = 3.25 in average. Realistically, this configuration split into amorphous sub-networks composed of optimally-constrained interlinked AsS3/2 pyramids (Z = 2.40; nc = 3.00) and over-constrained stressed-rigid As3S5/2 chains (Z = 2.55; nc = 3.36). Direct bonded As–S and As–As distances in these clusters fit respectively within ~ 2.26 Å and ~ 2.47 Å, while more essential deviations are observed in bond angles at As atoms. Molecular-to-network barrier for this amorphization pathway is low approaching ΔEf = (1.56–0.94) kcal/mol = 0.62 kcal/mol, this value being comparable with ΔEf for other transitions forming optimally-constrained single-broken × 1–1-p-As4S4 (ΔEf = 0.84 kcal/mol) and triple-broken × 3–1-p-As4S4 derivatives (ΔEf = 1.19 kcal/mol) having nc = 3.00, along with over-constrained double-broken × 2–4-p-As4S4 derivatives with nc = 3.25 (ΔEf = 1.15 kcal/mol). These data testify in favor of rather shallow-level character of respective potential energy landscape on Fig. 4 illustrating amorphization scenarios originated from pararealgar-type p-As4S4 arsenicals. Because of low barriers between these wells, transition towards quadruple-broken × 4-p-As4S4 network-forming clusters possessing Ef = − 1.56 kcal/mol seems to be most favorable.

Potential energy landscape illustrating amorphization scenarios derived from pararealgar-type p-As4S4 arsenical. The shallow-level character of network-forming derivatives reconstructed from pararealgar × 0-p-As4S4 molecule is obvious. The settle-points of network clusters derived by x-fold bond-breaking (keeping small rings, such as tetragons/pentagons/hexagons nominated in parenthesis) are denoted with respective cluster-forming energies Ef at the right axis
Thus, decomposition on two different topological sub-networks occurs first from p-As4S4 arsenical, these being stoichiometric optimally-constrained (Z = 2.40, nc = 3.00) and non-stoichiometric As-rich over-constrained ones (Z = 2.55, nc = 3.36). In the realistic arsenical systems compositionally close to arsenic monosulphide AsS, this process is expected to be merely blocked, provided appearance of pararealgar p-As4S4 phase is inhibited.
Cluster Modeling of Amorphization Paths Derived from Dimorphite α/β-As4S3 Phases
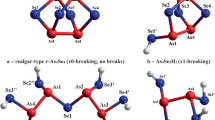
Removing one S atom at the bottom of pararealgar p-As4S4 molecule (within As2-As3-As4-S1 sequence, Fig. 3a), replacing it by one As-As bond, transfers this molecule to other one As4S3-I corresponding to both low- and high-temperature polymorphs of mineral dimorphite α/β-As4S3 [12, 13]. This is most energetically plausible configuration of this As4S3 molecule formed from six heteronuclear As-S and three homonuclear As–As bonds as shown on Fig. 5a. The optimized bond distances and angles in this × 0-As4S3-I molecule obeying triangle-like conformation due to the basal 3-membered As3-ring (triangle) composed of As2–As3–As4 atoms, surmounted by AsS3/2 pyramid composed of As1–(S1–S2–S3) atoms (see Fig. 5a) have been derived from CINCA modeling and gathered in Table 5. It’s seen that direct bonded As-As distances in As3-triangle approach ~ 2.467 Å with all bond angles close to ~ 60°. Direct bonded As-S distances within AsS3/2 pyramid and As-S distances connecting this pyramid with bottom As3-ring approach ~ 2.235 Å, with angle on As1 atom at the apex of AsS3/2 pyramid close to ~ 99.04°. The calculated Ef energy for this molecule (in respect to the energy of AsS3/2 pyramid) was found to be − 1.78 kcal/mol (see Table 6), the value worse than in molecular As4S4-type polymorphs, but comparable and even better than in network-forming derivatives from these cage-like molecules (compare respective energies in Tables 1, 3, 6).

Geometrically-optimized configurations of dimorphite-type α/β-As4S3 cage-like molecule in triangle-like conformation × 0-As4S3-I (a) and its most energetically favorable network derivatives formed from As4S4H2 molecule by single × 1-break in S2 position (b) and separating matrix on AsS3H3 and As3S3H3 molecules due to triple × 3-break in S1-S2-S3 positions (c). The terminated H atoms are grey-colored, S and As atoms are yellow- and red-depicted, and covalent bonds between atoms are stick-denoted
Previously, the crystallographic features of under-constrained As4S3 molecule (nc = 2.71, Table 6) possessing C3v symmetry with four small rings (three pentagons and one triangle) were specified (see, e.g. refs. [8, 14]), and geometrically-optimized parameters of this molecule were derived from ab-initio quantum-chemical model of Kyono [43]. However, the energetic specificity of competitive molecular-network amorphizing tendencies in dimorphite-type As4S3 arsenicals has not been established.
In general, dimorphite α/β-As4S3-type molecule allows three network-forming configurations, which can be reconstructed from × 0-As4S3-I cages shown on Fig. 5a by × 1-, × 2-, × 3-breaking in respective S1, S2, S3 positions, these configurations being parameterized and compared in Table 6.
The optimally-constrained double-broken × 2-As4S3-I clusters having nc = 3.00 in view of one small ring kept in the structure (As3-triangle) cannot be stabilized in realistic amorphous structures because of unfavorable cluster-forming energy Ef = − 11.68 kcal/mol (Table 6). The most plausible amorphization path originated from this α/β-As4S3-type arsenical seems to be related to other possibility of molecular-cage destroying connected with × 3-break in all S atom positions, when arsenical matrix built of × 3-As4S3-I clusters (Z = 2.57; nc = 3.00) are decomposed in two optimally-constrained sub-networks having AsS3H3 and As3S3H3 molecular precursors (Fig. 5c). Like in pararealgar structures, one sub-network is composed of AsS3/2 pyramids (Z = 2.40; nc = 3.00) with identical parameters (compare these parameters for AsS3H3 molecule separated from p-As4S4 in Table 4 and from α/β-As4S3 in Table 7). Other sub-network decomposed from dimorphite-type arsenicals under × 3-break represents optimally-constrained As3S3/2 clusters (Z = 2.67; nc = 3.00) keeping basal As3-ring incorporated by triplicate = As–S– chain links. As it follows from potential energy landscape on Fig. 6a, the calculated molecular-to-network barrier for this amorphization path is low ΔEf = (3.96–1.78) kcal/mol = 2.18 kcal/mol. The only competitive variant for this amorphization scenario in dimorphite-type arsenicals is incomplete destroying of As4S3-I molecule in triangle-like conformation due to × 1-break in S1 position keeping two neighboring small rings (triangle As2As3As4 and pentagon As2As4S3As1S1, see Fig. 5b), thus stabilizing under-constrained amorphous matrix with nc = 2.86 and comparable value of Ef = − 4.24 kcal/mol.

Potential energy landscapes showing diversity of amorphizing network-forming states originated from As4S3 cage-like molecules in different conformations: triangle-like (a), chain-like or zig-zag (b), and star-like (c). The double-well presentation of ground state for triangle-like As4S3-I molecule corresponds to low- and high-temperature modifications of tetra-arsenic trisulphide after Whitfield [12, 13]. The settle-points corresponding to network clusters derived by x-fold bond-breaking (keeping small rings, such as triangles/tetragons/pentagons/hexagons nominated in parenthesis) are denoted with respective forming energies Ef at the left axis
The triangle-like conformation shown in Fig. 5a is not alone arrangement of As4S3 molecular cages, two other under-constrained configurations, the chain-like As4S3-II with nc = 2.71 (Fig. 7a) and star-like As4S3-III with nc = 2.57 (Fig. 8a), being also possible. The optimized geometrical parameters for these molecules were simulated by Kyono [43], however, this author was failed to distinguish energetically realistic configurations among these As4S3-type arsenicals.

Geometrically-optimized configuration of As4S3 cage molecule in chain-like conformation × 0-As4S3-II (a) and its most favorable network derivative × 1–1-As4S3-II formed from prototype As4S4H2 molecule by single × 1-break in S2 position (b). The terminated H atoms are grey colored, S and As atoms are yellow- and red-depicted, and inter-cluster bonds are stick-denoted

Geometrically-optimized configuration of As4S3 cage molecule in star-like conformation × 0-As4S3-III (a) and its most favorable network derivative × 2-As4S3-III formed from prototype As4S5H4 molecule by double × 2-break in S1 and S2 positions (b). The terminated H atoms are grey colored, S and As atoms are yellow- and red-depicted, and inter-cluster bonds are stick-denoted
Removing S atom from one of two equivalent positions S2 or S4 within asymmetric p-As4S4 molecule on Fig. 3a, replacing it by As-As bond, transfers this molecule to other one As4S3-II distinguished by chain-like or zig–zag conformation due to sequent arrangement of all four As atoms (within As1As3As2As4 chain as shown in Fig. 7a). The cluster-forming Ef energies for geometrically-optimized configuration of this molecule and its main network-forming derivatives are gathered in Table 6 and reflected on potential energy landscape (see Fig. 6b). It’s clearly seen that two types of network derivatives (single-broken × 1–1–As4S3-II with nc = 3.00 and triple-broken × 3-As4S3-II with nc = 3.43) are more favorable than parent × 0-As4S3-II molecule, the configuration of optimally-constrained × 1–1-As4S3-II network cluster being shown on Fig. 7b. It means that this chain-like molecular conformation cannot be stabilized realistically, being rather defective one.
The similar finding concerns other conformation possible for molecular As4S3 cages on Fig. 8a. This is so-called star-like conformation of × 0-As4S3-III molecule, which can be reconstructed from pararealgar p-As4S4 molecule (Fig. 3a) removing S atom from S3 position of surmounted pyramid As1(S2S3S4). The character star-like configuration in this molecule is formed by central As4 atom bonded to other three As atoms (As1, As2, and As3) as illustrated on Fig. 8a. As it follows from potential energy landscape on Fig. 6c, this under-constrained × 0-As4S3-III molecule (nc = 2.57) is evidently unfavorable before two over-constrained network derivatives such as double-broken × 2-As4S3-III with nc = 3.14 (Fig. 8b) and triple-broken × 3-As4S3-III with nc = 3.43. Therefore, as in the previous case, the star-like conformation is not expected in realistic As4S3-type arsenical structures.
Competitive Amorphization Scenarios in Nanostructured As4S4-As4S3 Arsenicals
Recent calorimetric heat-capacity studies of nanoamorphization processes in AsxS100−x arsenicals driven by external influences such as nanomilling testify that amorphous phase generated within As4S4–As4S3 domain (2.50 < Z < 2.57) is close to initial melt-quenched arsenical [20, 21, 40]. This fact assuredly serves as main argumentation on the appeared amorphous phase as mixture of network-forming derivatives originated from realgar As4S4-type and dimorphite As4S3-type molecules. Alternatively, this process can be classified as nanomilling-driven amorphization of melt-quenched alloy [19], in contrast to amorphization associated with decomposition of nanostructured phases occurring in these arsenicals enriched on plastic-crystalline β-As4S3 phase [21]. Noteworthy, in respect to temperature-modulated DSC TOPEM® measurements [20, 39], amorphous nanophase generated in these arsenicals possesses double-Tg relaxation, having high-temperature glass-transition mid-point Tg1 close to that of melt-quenched As2S3 alloy and low-temperature glass-transition mid-point Tg2 near solidus of metastable melting in the respective over-stoichiometric arsenical.
Having in mind this specificity of calorimetric events in AsxS100−x alloys, let’s clarify diversity of amorphization scenarios realized in these arsenicals along As4S4–As4S3 cut-line.
In realgar As4S4-type arsenicals with × 0-β-As4S4 molecular cluster-precursor (see Fig. 1a), most energetically favorable network-forming amorphizing cluster × 1-β-As4S4 can be derived from As4S5H2 molecular precursor by single-break in one of S atom positions as illustrated in Fig. 1b. This amorphization path I from molecular under-constrained arrangement (nc = 2.875) to network-forming optimally-constrained arrangement (nc = 3.00) keeping two pentagons is realized with the lowest inter-well barrier of ΔEf = 0.71 kcal/mol (Fig. 9), thus being basic one for amorphizing As4S4-type arsenicals. Competitively, amorphization path II from × 0-β-As4S4 molecular precursor to over-constrained (nc = 3.25) triple-broken derivative (viz. chain-type × 3-β-As4S4 cluster) or, alternatively, but with smaller probability, to quadruple-broken derivative (viz. × 4-β-As4S4 or As2S4/2 cluster) can be realized in this arsenical with barrier of ΔEf = 1.14 kcal/mol. Whichever the case, these network-forming clusters (× 1-β-As4S4, × 3-β-As4S4 or × 4-β-As4S4) are over-stoichiometric ones (Z = 2.50) contributing rather to network modes with low-temperature Tg2 temperature near solidus of metastable melting in As4S4 alloy.

Competitive amorphization scenarios in nanostructured As4S4-As4S3 arsenicals driven from boundary compositions of As-rich molecular precursors As4S4 and As4S3. The molecular precursors of most favorable network-forming clusters are also depicted. The optimally-constrained clusters with nc = 3.00 are inserted in green boxes, while orange and blue colors are used respectively for over-constrained (nc > 3.00) and under-constrained clusters (nc < 3.00). In the cluster marking, terminated H atoms are grey colored, S and As atoms are yellow- and red-colored, and inter-cluster covalent bonds are denoted by respectively-colored sticks
The only variant on phase-decomposed amorphous substance which can be derived from As4S4-type alloys appears due to adjacent (neighboring) arrangement of both As–As bonds in pararealgar p-As4S4 structure (Fig. 3). Network decomposition occurs from p-As4S4 cage-like molecule (Fig. 3a) owing to × 4-break in all S atom positions resulting in × 4-p-As4S4 cluster, separating matrix on two sub-networks composed of optimally-constrained AsS3/2 pyramids (nc = 3.00, Z = 2.40) and over-constrained As3S5/2 chains (nc = 3.36, Z = 2.55) with low barrier of molecular-to-network transition approaching 0.62 kcal/mol, as depicted by amorphization pathway III on Fig. 9. Thus, the amorphous phase generated in this case possesses double-Tg relaxation with high-temperature Tg1 close to glass-transition mid-point of g-As2S3 (~ 208 °C [39]) and low-temperature Tg2 near glass-transition mid-point corresponding to solidus of metastable melting of over-stoichiometric As55S45 (~ 130 °C as determined by Hruby [1]). Realistically, such transformations in As4S4-type arsenicals are possible just from so-called χ-phase As4S4 considered as intermediate precursor of pararealgar p-As4S4 phase in light-induced alteration from α/β-As4S4 phases [42, 44,45,46]. This χ-phase As4S4 was indeed identified in arsenic monosulphide employing the Raman scattering spectroscopy [20].
From the other side, in dimorphite α/β-As4S3-type arsenicals, amorphization can be realized only from As4S3 molecules possessing triangle-like conformation of neighboring As-As bonds (see Fig. 5a). The most favorable amorphization path IV for this × 0-As4S3-I molecule with ΔEf = 2.18 kcal/mol is connected with matrix decomposition in two optimally-constrained sub-networks (nc = 3.00) depicted on Fig. 5c, these being composed of AsS3/2 pyramids (Z = 2.40) and As3S3/2 clusters (Z = 2.67) keeping three neighboring homonuclear As-As bonds in triangle-like geometry. The latter are responsible for sharp 275 cm−1 band ascribed to symmetric-stretch breathing mode of the basal As3-rings in the Raman spectra of As4S3-type alloys [14, 17, 18, 20, 21, 28]. Thereby, in the current case, the amorphizing substance also demonstrates strong glass-forming ability due to optimally-constrained topology of covalent backbone (nc = 3.00), possessing double-Tg relaxation with distinguishable glass-transition mid-points Tg1 and Tg2 as for × 4-p-As4S4 clusters derived from pararealgar p-As4S4 molecule.
This amorphization path IV dominates in dimorphite As4S3-type arsenicals (see Fig. 9), the only competitive amorphization path V being related to incomplete destroying of As4S3-I molecule due to single × 1-break stabilizing under-constrained matrix (nc = 2.86) with neighboring small rings in triangle and pentagon configurations (as shown in Fig. 5b). This variant is to be considered rather as defective in view of higher molecular-to-network barrier ΔEf = 2.46 kcal/mol.
Thus, nanoamorphization in As4S4-As4S3 arsenicals dominates by clear tendency towards optimally-constrained structural network with nc = 3.00 stabilized via pathways I, III and IV (see Fig. 9), respective molecular-to-network transitions being accompanied by arsenical decomposition on stoichiometric (Z = 2.40) and over-stoichiometric (Z > 2.40) sub-networks via path III and IV. Competitive contribution of these amorphization scenarios is defined preferentially by arsenical composition, where higher barriers of molecular-to-network transition in dimorphite-type As4S3 arsenicals (ΔEf > 2 kcal/mol) allow As4S4-As4S3 alloys nominated as higher-crystalline ones, in full respect to terminology of Hruby [1].
The unified potential energy landscape showing diversity of molecular-to-network amorphization scenarios in the studied nanostructured arsenicals realized for boundary As4S4 and As4S3 components is shown on Fig. 10.

Unified potential energy landscape showing diversity of main molecular-to-network amorphization transitions in nanostructured As4S4-As4S3 arsenicals, restricted for boundary As4S4 (left) and As4S3 (right) compositions. The calculated cluster-forming energies (Ef) are right-side denoted near energy wells corresponding to molecular clusters and their most favorable network derivatives pointed out by arrows (the atom-averaged topological constraints nc are indicated)
Under nanostructurization due to high-efficient external influence such as nanomilling, the role of As4S4-type arsenicals is mainly revealed through amorphization transitions from under-constrained realgar-type × 0-β-As4S4 molecule (nc = 2.875) supplemented by transitions from under-constrained pararealgar-type × 0-p-As4S4 ones (nc = 2.75). The former dominates by appearance of network-forming optimally-constrained × 1-β-As4S4 clusters (having nc = 3.00 and molecular-to-network barrier approaching ΔEf = 0.71 kcal/mol) along with over-constrained × 3-β-As4S4 clusters (having nc = 3.25 and ΔEf = 1.14 kcal/mol). The latter dominates mainly by transformation towards × 4-p-As4S4 clusters separating the arsenical matrix on two sub-networks composed of stoichiometric optimally-constrained AsS3/2 trigonal pyramids (Z = 2.40, nc = 3.00) and As-rich non-stoichiometric over-constrained As3S5/2 chains (Z = 2.55, nc = 3.36). In this case, the degree of network decomposition in nanoarsenical (and, respectively, the parameters of double-Tg relaxation) is defined by completeness of preliminary alteration from α/β-As4S4 phase to metastable pararealgar p-As4S3 phase through its intermediate precursor (such as χ-phase As4S4).
In As4S3-type arsenicals, main amorphization scenario is connected with transition from under-constrained dimorphite-type × 0-As4S3-I molecules (Z = 2.57, nc = 2.71) possessing triangle-like configuration in the nearest arrangement of three neighboring homonuclear As-As bonds (as depicted in Fig. 5a) to their triple-broken × 3-As4S3-I derivatives. The respective structural transformations separate arsenical matrix on two optimally-constrained sub-networks (both having nc = 3.00) composed of AsS3/2 pyramids (Z = 2.40) and over-stoichiometric As-rich As3S3/2 clusters (Z = 2.67) keeping the basal As3-rings. This amorphization scenario can be only slightly disturbed (if any) by incomplete destroying of As4S3-I molecules owing to single × 1-breaking in one of S atom positions, thus stabilizing intrinsically uniform but rather under-constrained (in view of nc = 2.86) network with cluster-forming energy approacging Ef = − 4.24 kcal/mol (see Fig. 9).
Conclusions
Complete hierarchy of network-forming amorphization scenarios in AsxS100-x nanoarsenicals within As4S4-As4S3 cut-Sect. (50 ≤ x ≤ 57) is reconstructed employing materials-computational approach based on ab-initio quantum-chemical modeling code (CINCA).
Under nanostructurization caused by external influence such as high-energy mechanical milling, complicated inter-crystalline phase transformations towards nanoscopic high-temperature β-As4S4 polymorph accompanied by appearance of compositionally-invariant covalent-network amorphous phase are activated in these alloys. General amorphization trend under nanomilling obeys tending from molecular cage-like structures to optimally-constrained covalent networks which are isocompositional to the parent alloy. Optimal topological constitution of amorphous phase generated in nanoarsenicals is ensured by diversity of respective small-ring configurations incorporated in their covalent networks. Contribution of network-forming amorphization pathways in As4S4-As4S3 nanoarsenicals is defined by their chemistry with higher molecular-to-network transition barriers proper for As4S3-rich alloys (more than ~ 2 kcal/mol). The generated amorphous phase is intrinsically decomposed, possessing double glass-transition temperature Tg relaxation due to stoichiometric (x = 40) and non-stoichiometric (x > 40) sub-networks, which are respectively built of AsS3/2 pyramids and As-rich entities keeping two separated As-As bonds derived from realgar-type molecules, two neighboring As-As bonds derived from pararealgar-type molecules and/or three neighboring As-As bonds in triangle-like geometry derived from dimorphite-type molecules. Compositional invariance of nanomilling-derived amorphous phase in AsxS100-x arsenicals within As4S4-As4S3 cut-section is ensured by network-forming clusters generated in a growing sequence with average coordination number Z: (As2S4/2, Z = 2.50) – (As3S5/2, Z = 2.55) – (As3S3/2, Z = 2.67). Full diversity of molecular-to-network amorphization pathways in the studied As4S4-As4S3 nanoarsenicals is specified on unified potential energy landscape for boundary As4S4 and As4S3 components.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
References
A. Hruby and J. Non-Cryst (1978). Solids 28, 139.
Z. U. Borisova, Glassy Semiconductors. (Plenum Press, New York, 1981), pp. 1–505.
A. Feltz, Amorphous Inorganic Materials and Glasses. (VCH Publ, New York, 1993), pp. 1–446.
A. L. Emelina, A. S. Alikhanian, A. V. Steblevskii, and E. N. Kolosov (2007). Inorg. Mater. 43, 95.
J.-L. Adam, X. Zhang (Eds.), Chalcogenide glasses: Preparation, properties and applications. (Woodhead Publ. Ser. in Electronics and Optical Mater., Philadelphia-New Delhi, 2013), pp. 1–716.
O. Shpotyuk and M. Hyla (2017). J. Optoelectron. Adv. Mater. 19, 48.
P. J. Dilda and P. J. Hogg (2007). Cancer Treat. Rev. 33, 542.
P. Bonazzi and L. Bindi (2008). Z. Ktistallogr. 223, 132.
O. Shpotyuk, P. Demchenko, Y. Shpotyuk, Z. Bujňáková, P. Baláž, and J. Non-Cryst (2019). Solids 505, 347.
O. Shpotyuk, P. Baláž, Z. Bujňáková, A. Ingram, P. Demchenko, and Y. Shpotyuk (2018). J. Mater. Sci. 53, 13464.
P. Chen, C. Holbrook, P. Boolchand, D. G. Georgiev, K. A. Jackson, and M. Micoulaut (2008). Phys. Rev. B 78, 224208.
H.J. Whitfield (1970). J. Chem. Soc. A, 1800.
H.J. Whitfield (1973). J. Chem. Soc. Dalton, 1737.
A. C. Wright, B. G. Aitken, G. Cuello, R. Haworth, R. N. Sinclar, J. R. Stewart, J. W. Taylor, and J. Non-Cryst (2011). Solids 357, 2502.
T. N. Chattopadhyay, E. Gmelin, and H. G. von Schnering (1983). Phys. Stat. Sol. A 76, 543.
R. Blachnik and U. Wickel (1984). Thermochim. Acta 81, 185.
B. G. Aitken and J. Non-Cryst (2004). Solids 345–346, 1.
S. Soyer-Uzun, S. Sen, and B.G. Aitken (2009). J. Phys. Chem. C 113, 6231.
O. Shpotyuk, P. Demchenko, Y. Shpotyuk, Z. Bujňáková, P. Baláž, M. Hyla, and V. Boyko (2019). Mater Today Commun 21, 100679.
O. Shpotyuk, P. Demchenko, Y. Shpotyuk, S. Kozyukhin, A. Kovalskiy, A. Kozdras, Z. Lukáčová Bujňáková, and P. Baláž (2020). J. Non-Cryst. Solids 539, 120086.
O. Shpotyuk, S. Kozyukhin, P. Demchenko, Y. Shpotyuk, A. Kozdras, M. Vlcek, A. Kovalskiy, Z. Lukáčová Bujňáková, P. Baláž, V. Mitsa, and M. Veres (2020). J. Non-Cryst. Solids 549, 120339.
O. Shpotyuk, M. Hyla, V. Boyko, Y. Shpotyuk, and V. Balitska (2020). Appl. Nanosci. 10, 4689.
O. Shpotyuk, M. Hyla, and V. Boyko (2013). J. Optoelectron. Adv. Mater. 15, 1429.
O. Shpotyuk, M. Hyla, and V. Boyko (2015). Comput. Mater. Sci. 110, 144.
W. J. Hehre, R. F. Stewart, and J. A. Pople (1969). J. Chem. Phys. 51, 2657.
A. D. McLean and G. S. Chandler (1980). J. Chem. Phys. 72, 5639.
K. Jackson (2000). Phys Stat Solidi B 217, 293.
R. Holomb, M. Veres, and V. Mitsa (2009). J. Optoelectron. Adv. Mater. 11, 917.
O. Shpotyuk, A. Ingram, and P. Demchenko (2015). J. Phys. Chem. Solids 79, 49.
J. C. Phillips and J. Non-Cryst (1979). Solids 34, 153.
M. F. Thorpe and J. Non-Cryst (1983). Solids 57, 355.
M. F. Thorpe and J. Non-Cryst (1995). Solids 182, 135.
T. Ito, N. Morimoto, and R. Sadanaga (1952). Acta Cryst. 5, 775.
D. J. E. Mullen and W. Nowacki (1972). Zeit. Krist. 136, 48.
E. J. Porter and G. M. Sheldrick (1972). J. Chem. Soc. Dalton Trans. 13, 1347.
L. Bindi, G. Pratesi, M. Muniz-Miranda, M. Zoppi, L. Chelazzi, G. O. Lepore, and S. Menchetti (2015). Mineral. Mag. 79, 121.
P. Baláž, W. S. Choi, and E. Dutková (2007). J. Phys. Chem. Solids 68, 1178.
P. Baláž, M. Baláž, O. Shpotyuk, P. Demchenko, M. Vlček, M. Shopska, J. Briančin, Z. Bujňáková, Ya. . Shpotyuk, B. Selepová, and L. Balážová (2017). J. Mater. Sci. 52, 1747.
O. Shpotyuk, Z. Bujňáková, M. J. Sayagués, P. Baláž, A. Ingram, Ya. . Shpotyuk, and P. Demchenko (2017). Mater. Charact. 132, 303.
O. Shpotyuk, A. Kozdras, P. Baláž, Z. Bujňáková, and Ya. . Shpotyuk (2019). J. Therm. Anal. Calorim. 135, 2945.
P. Bonazzi, S. Menchetii, and G. Pratesi (1995). Am. Mineral. 80, 400.
P. Bonazzi, S. Manchetti, G. Pratesi, M. Muniz-Miranda, and G. Sbrana (1996). Am. Mineralogist 81, 874.
A. Kyono (2013). Phys. Chem. Minerals 40, 717.
D. L. Douglass, C. Shing, and G. Wang (1992). Am. Mineralogist 77, 1266.
K. Trentelman and L. Stodulski (1996). Anal. Chem. 68, 1755.
M. Muniz-Miranda, G. Sbrana, P. Bonazzi, S. Menchetti, and G. Pratesi (1996). Spectrochim. Acta A 52, 1391.
Acknowledgements
The paper is part of scientific research performed within the project No 0119U100357, subject of Scientific Program funded by Ministry of Education and Science of Ukraine (2019-2022).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shpotyuk, O., Hyla, M., Shpotyuk, Y. et al. Cluster Modeling of Network-Forming Amorphization Pathways in AsxS100−x Arsenicals (50 ≤ x ≤ 57) Diven by Nanomilling. J Clust Sci 33, 1525–1541 (2022). https://doi.org/10.1007/s10876-021-02077-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-021-02077-6