
Abstract
Super-resolution microscopy (SRM) comprises a suite of techniques well-suited to probing the nanoscale landscape of genomic function and dysfunction. Offering the specificity and sensitivity that has made conventional fluorescence microscopy a cornerstone technique of biological research, SRM allows for spatial resolutions as good as 10 nanometers. Moreover, single molecule localization microscopies (SMLMs) enable examination of individual molecular targets and nanofoci allowing for the characterization of subpopulations within a single cell. This review describes how key advances in both SRM techniques and sample preparation have enabled unprecedented insights into DNA structure and function, and highlights many of these new discoveries. Ongoing development and application of these novel, highly interdisciplinary SRM assays will continue to expand the toolbox available for research into the nanoscale genomic landscape.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Abbreviations
| 53BP1 | p53 binding protein 1 |
| ATAC | Assay for transposase-accessible chromatin |
| BaLM | Bleaching/blinking assisted localization microscopy |
| BALM | Binding activated localization microscopy |
| BrdU | 5-Bromo-2'-deoxyuridine |
| CldU | 5-Chloro-2'-deoxyuridine |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CuAAC | Copper catalyzed azide-alkyne cycloaddition |
| dCas9 | Nuclease deficient Cas9 |
| DSB | Double strand break |
| dsDNA | Double stranded DNA |
| dSTORM | Direct stochastic optical reconstruction microscopy |
| EdU | 5-Ethynyl-2'deoxyuridine |
| EM | Electron microscopy |
| FISH | Fluorescence in situ hybridisation |
| fPALM | Fluorescence photoactivated localization microscopy |
| FPs | Fluorescent proteins |
| gRNA | Guide RNA |
| GSDIM | Ground-state depletion with individual molecule return |
| HR | Homologous recombination |
| IdU | 5-Iodo-2'-deoxyuridine |
| MERFISH | Multiplexed error-robust fluorescence in situ hybridization |
| NHEJ | Non-homologous end joining |
| PAINT | Points accumulation for imaging in nanoscale topography |
| PALM | Photoactivated localization microscopy |
| PSF | Point spread function |
| RF | Replication fork |
| SIM | Structured illumination microscopy |
| SMLM | Single molecule localization microscopy |
| SPDM | Spectral precision distance/position determination microscopy |
| sptPALM | Single particle tracking photoactivated localization microscopy |
| SRM | Super-resolution microscopy |
| STED | Stimulated emission depletion microscopy |
| STORM | Stochastic optical reconstruction microscopy |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labelling |
| UVC | Ultraviolet 200–280 nm |
| XdU | Functionalized deoxyuridine nucleotide bases |
Introduction
DNA Organization
The full human genome comprises some two meters of double stranded DNA (dsDNA) containing more than 6 million DNA bases across 46 chromosomes. Remarkably, all of this information is packed into each and every ~10 μm wide cell nucleus where it maintains its ability to comprehensively code for the RNA and protein products necessary for cellular function. Alongside the genomic sequence, the topography of DNA plays important mechanistic roles in DNA biology, including in orchestrating replication and transcription, cell differentiation, and disease [1]. However, our understanding of how DNA is packaged inside the nucleus, and how this organization relates to function, remains limited, predominantly because of the difficulty associated with visualizing this highly compacted and complicated architecture. The proposed models of DNA organization put forward over the last several decades have been based on indirect biochemical assays, simulations, and electron microscopy (EM). Recently, more accurate and direct detection of genomic structures has become possible because of the invention of improved microscopy technologies such as super-resolution microscopy (SRM).
At the most fundamental level, the primary and secondary structures of DNA were discovered 75 years ago using x-ray diffraction patterns of purified DNA [2, 3] (figure 1). Since then, tertiary conformations and many specific protein interactions have also been characterized [4, 5]. Most notably, the fundamental genomic structural component—the nucleosome—was determined using x-ray diffraction and found to comprise eight individual histone proteins assembled as a core encircled by 146 DNA base pairs [6]. Nucleosomes form first order chromatin structures for DNA compaction often referred to as 'beads on a string'. These structures had previously been observed using EM as a 10-nm wide thread. Another DNA structure observed with EM was a 30-nm wide thread which formed the basis for the next order of DNA compaction: quartets of nucleosomes stacked or arranged linearly into a dense fibre [7, 8] (figure 1). This 30-nm fibre quickly became accepted as a text-book model for genomic organization although the internal arrangement of nucleosomes within the fibre was uncertain with multiple possible arrangements of the nucleosomes proposed (Reviewed by Li et al [9]). However in 2008, data generated using cryo-EM demonstrated that the 30-nm fibre is potentially an artefact caused by conventional EM sample preparation and not a legitimate feature of in vivo interphase DNA organization [10, 11] . This demonstrates the importance of choosing an appropriate microscopy strategy that not only provides high spatial resolution but also minimizes perturbation of the sample. In this regard, fluorescence-based microscopies are comparatively less invasive for biological imaging and, with the advent of super-resolution modalities, have become a cornerstone technique for near-molecular resolution studies of genomic structures and function.

Figure 1. A schematic overview of the different levels of DNA compaction. The double helical structure of DNA is wound around eight histone proteins to form nucleosomes. Individual nucleosomes are separated by regions of DNA referred to as linker DNA which together resemble a 'beads on a string' structure. During mitosis, condensed DNA forms characteristic x-shaped chromosomes for segregation during cell division. Intermediary chromatin structures, commonly referred to as higher order chromatin structures boxed in red, remain to be fully characterized.
Download figure:
Standard image High-resolution imageAlthough many higher order structures and compactions of DNA have been proposed and numerous DNA scaffolding proteins discovered, their spatial distributions and behaviours in situ remain unclear [12]. Nonetheless, through conventional microscopy and biochemical research we now understand much about the micron-scale roles and underlying molecular biology of chromatin including 1) the roles of histone modifications in orchestrating compaction and relaxation of chromatin [13], 2) colocalization of transcription and replication machinery within different nuclear regions throughout the cell cycle [14] and 3) the transition between heavily condensed heterochromatin and the more open, less condensed euchromatin [15]. Despite these significant contributions, the use of conventional imaging methods has not allowed for further elucidation of the nanoscale mechanisms and architectures that govern nuclear processes. Recent advancements in SRM and EM provide the necessary spatial resolutions and are now being applied to develop a comprehensive model of the dynamic nuclear environment [16].
DNA damage and repair
Any change to the genetic code of an individual cell runs the risk of affecting the function of an RNA or protein product. Dysfunction of these macromolecules can trigger cell death or impair the cell's ability to propagate and compete with neighbouring cells [17]. Because of this genotoxicity, single-stranded DNA evolved its double-stranded helix structure, engendering a robust in-built redundancy. This allows high fidelity repair of the ~200,000 single-stranded damage events sustained in each replicating human cell every day [18] via templating from the undamaged, complementary strand. However, a small number of DNA lesions—estimated between 10 and 50 per cell, per day [19, 20]—are not so easily repaired because they occur on both DNA strands as a double strand break (DSB). To combat these breaks, several dedicated, coordinated, and highly complicated DSB repair pathways exist within cells. The most prominent repair pathways are high-fidelity, template-based homologous recombination (HR) [21] and the comparatively lower-fidelity ligation-based non-homologous end joining (NHEJ) [19].
These repair pathways are increasingly necessary because, in today's society, the typical level of DSB induction is often exacerbated by exogenous agents, commonly classed as carcinogens (alcohol, cigarettes, etc), which increase either the probability of DSB induction, or the difficulty of repair, and consequently the likelihood of a misrepair event and pathogenesis [22]. Pre-existing mutations (i.e. deficiencies) in DNA replicative, regulatory, and repair proteins can similarly increase the probability of sequence modifications [23]. It is these DNA DSB misrepair events that are the underlying trigger of many mutagenic and apoptotic diseases including cancer and neuro-degeneration [24–27].
In juxtaposition to the often pathogenic role of DNA damage, orchestrated DSB induction in specialist cells is necessary to generate diversity both during gamete production and immune-receptor development for recognition of foreign agents within the body [28]. Furthermore, DSB induction and misrepair is the basis of many chemo- and radio-therapeutics, which preferentially induce high levels of DNA DSBs and forced misrepair in cancerous cells [29]. Harnessing DSBs is also a key aspect of genetic engineering processes such as the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas system [30]. These biological processes have been exploited in order to produce genetically modified organisms with increasing applications in biomedical research, agricultural, and clinical settings.
Extensive research into DSB induction and repair has identified a multitude of specific proteins and mechanisms involved in DNA damage response pathways, building up models of different repair pathways and their crosstalk [22, 31]. However, as with research into DNA organization, in situ studies of DNA DSBs have remained challenging due to the highly dense and complex nuclear environment, as well as the detection limits of conventional techniques. Much of our understanding of DSB induction, mutagenesis, and DSB repair has historically been based on sequencing or other 'omic' data which offers little spatiotemporal information and averages damage events and cells. Additionally in order to generate an amount of DNA damage and repair that can be detected using biochemical techniques, and indeed conventional microscopy approaches, a large number of DSBs typically need to be induced simultaneously [32]. This is often achieved using ionising radiation or micro-irradiation which typically induce widespread cellular damage and clustered DSBs estimated to contain dozens, if not hundreds, of proximate DSBs [33]. Although research utilising these methodologies has been instrumental in describing and understanding many aspects of genomic integrity and DSB repair, this high level of damage is at odds with the sporadic individual DSBs most relevant to disease [34].
Because of the ever-increasing role that DSBs and their (mis)repair play in everyday life, both in causing and treating human disease, there is enormous interest in applying state-of-the-art techniques to further elucidate the spatiotemporal progression of repair and the key underlying molecular mechanisms. Ongoing breakthroughs in the field of genomic instability continue to improve our understanding of cancerous, neuro-degenerative and immune diseases, and by extension, our ability to uncover better preventative and therapeutic approaches, while also building our capacity to specifically and carefully manipulate genomes.
Super-resolution microscopy
The diffraction limit of light dictates that images produced in the far field blur such that spatial resolutions are limited to approximately half the wavelength under observation [35, 36]. This inherent limit of optical microscopy placed a persistent and seemingly insurmountable restriction on what could be seen of the nanoscale landscape within cells. However, the invention of several SRM approaches over the last few decades has enabled circumvention of the diffraction limit, in some cases achieving spatial resolutions of complex cellular features better than 10 nm [37]. Moreover, with the introduction of single molecule localization microscopy in 2006 [38–41], and commercially available SRM setups soon after, SRM techniques are now widely used, powerful research tools.
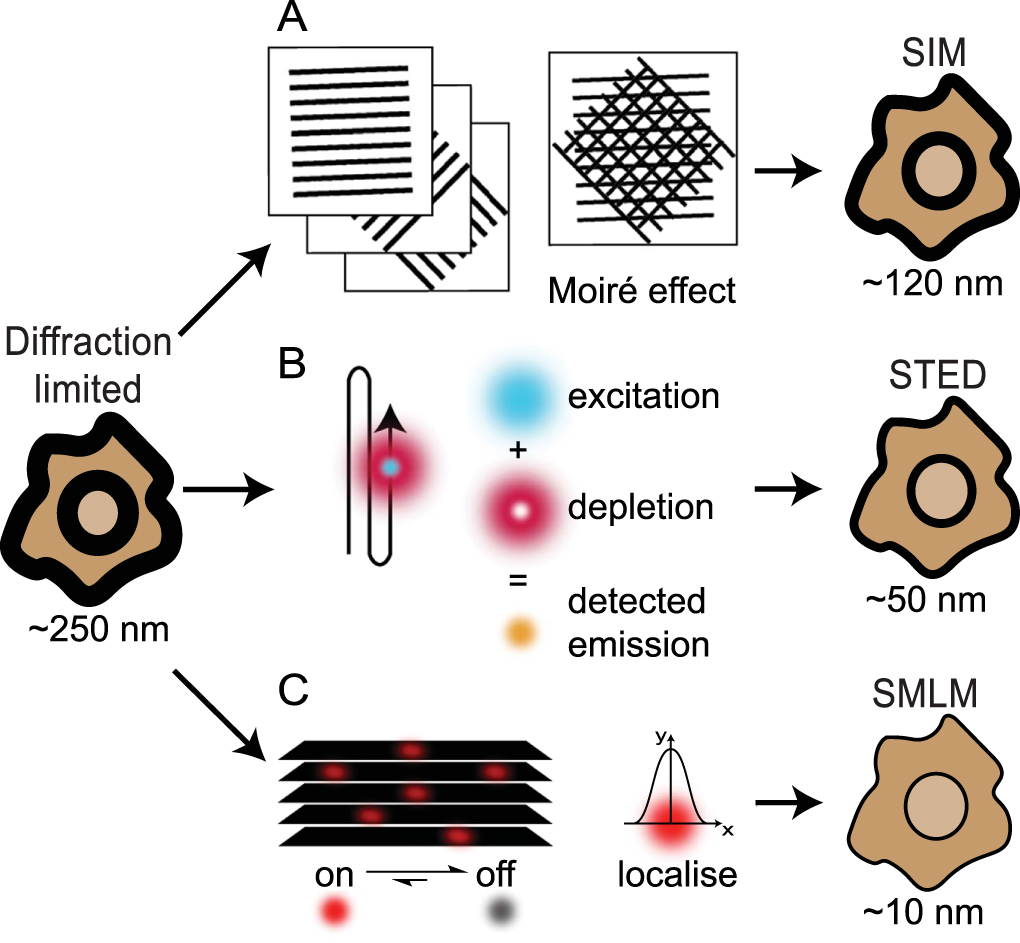
Structured illumination microscopy (SIM) and stimulated emission depletion (STED) are two popular examples of SRMs that rely on sub-diffraction patterning of the excitation beams in order to generate fluorescence emissions constrained to sub-diffraction areas [37]. SIM achieves a ~2–3-fold increase in resolution by imaging samples under a typically striped illumination pattern to create a Moiré effect [42, 43] (figure 2(A)). The illumination is generated using interfering laser beams, and is oriented across the sample of interest in several different configurations in order to generate a final image (Reviewed by Heintzmann & Huser [44]). STED uses a pair of overlapping beams in a confocal microscope configuration (figure 2(B)). The excitation beam excites fluorophores within a diffraction limited focal spot while a second depletion beam is shaped into a torus that overlaps the excitation spot such that only a small diameter of excitation-only illuminated area remains [45, 46]. Although the excitation beam excites fluorophores across the diffraction limited focal area, the depletion beam returns those excited fluorophores under the toroidal beam to the ground state via stimulated emission which can be spectrally separated from the directly emitted fluorescence. Both SIM and STED are particularly useful for live-cell dynamic imaging due to their speed and 3D capabilities, however these techniques can suffer from high levels of phototoxicity and photobleaching and are limited in their practically achievable resolutions (~90 nm for SIM, ~50 nm for STED)[37].
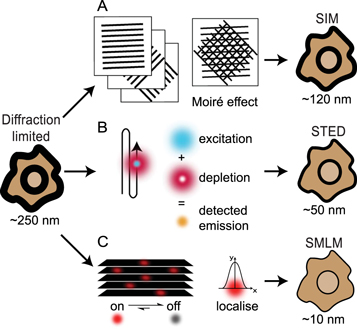
Figure 2. An overview of the main SRM techniques. (A) SIM uses overlapping patterned illumination, in different orientations, to generate Moiré effects. Computational processing of these images generates a ~2–3-fold increase in resolution. (B) The simultaneous scanning of the sample with overlaid excitation and depletion beams used in STED results in detected signals being specific to sub-diffraction areas approaching resolutions of ~40 nm. (C) SMLM relies on the detection, over multiple frames, of sparse single molecule emissions. Raw image stacks of diffraction limited emissions are collected and then analysed to localize each emitter to a few nanometers precision. Following image collection, the coordinates of the single emitters are used to render the final image. Specific methods for achieving single molecule localization that are discussed in this review include bleaching/blinking assisted localisation microscopy (BaLM), binding activated localisation microscopy (BALM), direct stochastic optical reconstruction microscopy (dSTORM), fluorescence photoactivated localisation microscopy (fPALM), ground state depletion with individual molecule return (GSDIM), points accumulation for imaging in nanoscale topography (PAINT), photoactivated localisation microscopy (PALM), spectral precision distance/position determination microscopy (SPDM), and stochastic optical reconstruction microscopy (STORM).
Download figure:
Standard image High-resolution imageInvented almost a decade after SIM and STED, single molecule localization microscopy (SMLM) relies on imaging individual fluorophore emission patterns without any spatial overlap from other fluorophore emissions (figure 2(C)). Single molecule imaging was first pioneered as a biophysics technique in the 1990s [47, 48] and famously used to visualize myosin 'walking' with 1.5 nanometer accuracy [49]. These applications were able to fit single emitter point spread function (PSF) patterns to precisely localize the underlying molecule, however they did not generate an image and only focused on an individual molecule within a diffraction limited area. SMLM extends this work by localizing hundreds or thousands of molecules within a diffraction-limited area. In most variants of SMLM, this is achieved by manipulating the photophysics or photochemistry of the sample such that the vast majority of fluorophores are non-emissive for seconds to minutes at a time before being stochastically returned to an emissive state, although other 'blinking' methods have also been detailed. The speed of this 'blinking' effect is optimally matched with the capture rate of modern scientific cameras and the requirement for enough photons to be detected (~1000 photons per PSF allowing for ~ 10 nm precision) resulting in frame rates ranging 1–100 Hertz [37]. By capturing thousands of images of the sample, each with a different subset of fluorophores emitting, the PSFs of thousands to millions of molecules can be detected. Each PSF is then mathematically fit to determine the underlying coordinates of each molecule, with the final image generated by plotting these localizations with sub-diffraction pixels.
There are currently dozens of SMLM variants, with most adopting a unique acronym to describe the different ways in which the necessary fluorescence blinking is achieved (Reviewed by Li & Vaughan [50]). The four original SMLM variants - photoactivated localization microscopy (PALM) [39], fluorescence PALM (FPALM) [40], stochastic optical reconstruction microscopy (STORM) [38] and PAINT (points accumulation for imaging in nanoscale topography) [41] used photoactivatable fluorescent proteins (FPs), activator-reporter organic dye pairs, or collisional flux of diffusing fluorophores. Direct STORM (dSTORM) was first described two years later in 2008 and is currently one of the most widely used SMLMs because it makes use of conventional organic dyes (typically Alexas and ATTOs) either conjugated to antibodies or orthogonally attached to a target of interest, and easy-to-use photoswitching buffers [51, 52]. Other popular SMLM approaches to emerge include binding activated localisation microscopy (BALM) [53], bleaching/blinking assisted localization microscopy (BaLM) [54], ground-state depletion with individual molecule return (GSDIM) [55] and spectral precision distance/position determination microscopy (SPDM) [56]. Advantageously, SMLM routinely achieves spatial resolutions of 20–30 nm, with localization precisions better than 5 nm, however most SMLM experiments require long acquisition times to capture enough single molecule emissions and are thus restricted to fixed, or slow-moving cellular processes [37].
Collectively, SRM approaches are quickly proving invaluable to researchers working to understand key aspects of genomic organization and instability. The advantageous specificity and sensitivity of conventional fluorescence techniques, combined with the sub-diffraction resolutions of SRM, has already enabled imaging of chromatin distribution and dynamics, alongside individual DNA DSBs and aspects of the mechanisms which underpin their repair. In this review we will detail many of the innovative approaches and findings of this research, with a focus on the particular strengths of SRM of DNA and its future potential.
Novel DNA labelling strategies for SRM
The potential to visualize structures and distributions within the nucleus using SRM has necessitated novel adaptations and improvements to established DNA-labelling strategies alongside approaches for labelling nuclear proteins using antibodies or fluorescent protein co-expression. Furthermore, because fluorescence imaging in the past has been limited in its ability to probe DNA structure, pre-existing fluorescence staining techniques have not always been easily compatible with SRM [57]. Nonetheless, particularly in the last decade, a number of innovative SRM DNA labelling assays have been described.
The first reports of SRM imaging of DNA were achieved using small organic intercalating or groove-binding dyes [58]. Intercalators insert themselves between the planar surfaces of DNA base pairs whereas groove-binding dyes align with the major or minor groove of B-DNA; in many cases DNA labels show a combination of interactions (figure 3(A)). These fluorophores can be used for SRM via the bleaching/blinking assisted localization microscopy (BaLM) approach which involves using very low concentrations so that single fluorophores bind DNA one at a time, are imaged, and then bleach or dissociate before another molecule attaches within the same diffraction limited area [54]. Similarly, binding-activated localization microscopy (BALM) is another SMLM variant that can be used, instead relying on fluorophores such as cyanine dimers which display a marked enhancement of fluorescence upon binding of DNA enabling localization [53, 58, 59]. These techniques were successfully used to image extracted and stretched DNA, chromatin spreads and intact nuclei (Reviewed by Flors [60]). Later photoconversion of common DNA dyes DAPI and Hoechst with 405 nm light was also demonstrated [61]. Recently, BALM has been extended to detect DNA-bound proteins using an inverse BALM (iBALM) strategy [62]. 3D BALM [63] has also been demonstrated on spread chromosomes employing the optical astigmatism approach commonly used for 3D dSTORM [63, 64]. However, imaging of DNA using intercalating and groove-binding dyes remains limited because of the potential for many of these dyes to label RNA and the lack of specificity for targeting DNA sequences or processes of interest [60]. Nonetheless, whole genome imaging of DNA morphology using SRM continues to provide a better benchmark than conventional fluorescence stains for overall nuclear morphology.

Figure 3. Methods for SRM labelling of DNA. (A) Whole genome labelling of DNA using intercalating and groove-binding fluorophores. Intercalators insert between the DNA bases whilst groove-binders bind to the DNA major or minor grooves. (B) Incorporation of nucleotide analogues within DNA provides a method for labelling genomic DNA as it is synthesized. Analogues containing halogens are fluorescently labelled following denaturation using immunofluorescence (right), whereas nucleotides with a terminal alkyne can be detected using the click reaction (left). (C) Visualization of specific genomic loci via FISH involves the hybridization of fluorescent DNA probes complementary to the sequence of interest. (D) Fluorescent proteins fused to nuclease-deactivated Cas9 proteins enable visualization of specific genomic loci by targeting with a guide-RNA complementary to the sequence of interest.
Download figure:
Standard image High-resolution imageAlthough less common than small molecule dye labels for DNA, incorporation of modified nucleotides has been an established methodology, particularly for detecting proliferation and replication, for several decades [65, 66]. This approach uses nucleotides modified with a specific functional group, which are temporarily added to cell media and incorporated into the genomic DNA through replication with minimal toxicity to the cell. After fixation these functional groups can be labelled fluorescently. Originally, halogenated thymine analogues (e.g. 5-bromo-2'-deoxyuridine (BrdU) or chloro/iodo equivalents (CldU/IdU)) that could be detected using antibodies were used (figure 3(B)), however antigen presentation of the halogen group requires extensive DNA denaturation by heat or acid treatment. This denaturation limits the compatibility of XdU labelling of DNA with immunofluorescence of proteins because it often causes protein antigens to lose necessary secondary structure [67]. Moreover, in developing methods for SRM of DNA, it is assumed that such extensive denaturation would significantly alter the nanoscale architecture of the DNA itself.
A novel class of functionalized nucleotides avoids many of these limitations by making use of the copper catalyzed azide-alkyne cycloaddition (CuAAC) reaction, more commonly known as a 'click' reaction [68]. As with halogenated XdU incorporation, nucleotides with an alkyne group (most commonly 5-ethynyl-2'deoxyuridine (EdU)) are incorporated into the genomic DNA during replication, however they do not require denaturation to be detected (figure 3(B)). The direct reaction of small azide-functionalized organic dyes achieves a high yield of one-to-one labelling of incorporated EdUs, does not require perturbing reaction conditions, and is compatible with immunolabelling [67]. Moreover, an improved, copper-free click reaction using a strained ring to promote the cycloaddition has now been demonstrated for low-toxicity labelling in live cells [69]. This labelling strategy has also been applied in conjunction with expansion microscopy to image DNA, RNA and other biomolecules in the cytoplasm and nucleus [70].
EdU DNA labelling for SRM was first reported in 2012 by Zessin et al who highlighted the potential for EdU labelling with dSTORM-compatible fluorophores [71]. They showed that this combination of approaches provides a relatively straightforward and robust method for SRM of DNA in fixed cells. Moreover, they were able to resolve fine chromatin structure, superior to conventional microscopy, and, by using short pulse-durations of EdU incorporation, were able to distinguish individual nascent DNA chromatin structures. The group extended this work to multiplexed DNA, protein and membrane labelling with novel dyes for SRM of bacteria [72] and also went on to demonstrate click labelling of proteins using unnatural amino acids [73].
Interestingly, a comparative SRM, confocal and widefield study of BrdU and EdU labelling assays revealed differences in the staining patterns of the nanoscale architecture of unscheduled DNA synthesis, which occurs following short-wavelength ultraviolet (UVC) induced DNA damage [74]. This DNA synthesis represents the final stage of UVC damage repair whereby new nucleotides are incorporated to fill the lesions enabling detection via XdU stains. In this study the EdU assay showed that repair occurred throughout the chromatin whereas BrdU incorporation was only detected in a few discrete repair foci. Based on these data, the authors concluded that DNA denaturation for BrdU detection is typically incomplete thereby limiting antibody access and detection of bases at many sites throughout the chromatin. Building on this work, we and others have used EdU pulse labelling combined with SMLM to visualize sites of active DNA synthesis at individual DNA replication forks providing a fluorescent marker for replicative processes with good potential for quantification [74–76].
However, as with small intercalating and groove-binding dyes, XdU labelling does not offer specificity for genomic sequences or chromatin structures beyond marking active sites of synthesis. To enable loci-specific SRM imaging of DNA, new approaches have been described that modify conventional fluorescence in situ hybridisation (FISH) assays (figure 3(C)) [77]. FISH has long been used for confocal imaging of genomic loci and relies upon the denaturation of genomic DNA and the hybridization of short complementary strands which carry fluorophores and are designed to be specific to the target sequences of interest. For use with SRM, novel FISH probes have been described which enable visualization of non-repetitive DNA sequences in situ [78]. This approach makes use of short oligos that are complementary to the target sequence and labelled with both an Alexa fluorophore and a quencher. They are specifically designed so that when unbound, they form a hairpin and the Alexa fluorescence is consequently quenched. This approach greatly reduces the signal from non-specifically bound and background oligos.
Recent efforts have further combined SRM and FISH labelling to investigate the nanostructure of telomeres, the important DNA structures that protect the ends of chromosomes. Although the sequences and in vitro structure of telomeres had previously been described, leading to important insights into their functions and potential roles in disease and aging (reviewed by Aubert & Lansdorp [79]), their in situ organization had not been well-visualized. Using SMLM imaging and FISH probes against telomeric repeat sequences, Doksani et al were able to capture SMLM images of characteristic T loops (figure 4) [80]. Removal of the telomere protection protein, TRF2, greatly reduced the number of T loops. Applying similar SRM assays, Chow et al have also identified a novel protein, HP1α, as essential for the protection and regulation of the telomeric structure [81].

Figure 4. Combining FISH labels with SRM imaging enables visualization of telomeric structures. (A) Isolated telomeres from mouse splenocytes following cytocentrifugation reveals the characteristic T loop structures. (B) Pictorial representation of the protective nature of T loop structures. Without the presence of T loops and protector proteins such as TRF2, chromosome ends are susceptible to degradation by DNA damage response pathways, which is hypothesized to lead to human pathologies and aging. Reproduced with permission [80]. Copyright 2013, Elsevier.
Download figure:
Standard image High-resolution imageAnother promising SRM/FISH application that has been described is multiplexed error-robust FISH (MERFISH) which enables high throughput detection of RNA using sequential hybridization schemes involving coded probes [82]. MERFISH reveals transcriptome data at the single cell level with the ability to discern and identify individual RNA molecules. Additionally, correlation analysis between identified RNA species could help predict novel regulatory gene networks. Adaption of the MERFISH protocol enabled the mapping of chromatin interactions of human chromosome 21 [83]. By dividing the chromosome into shorter regions of interest and designing suitably coded probes, Bintu et al found that chromatin formed distinct domains with defined boundaries which varied considerably from cell-to-cell [83]. These boundary positions were lost following depletion of cohesin proteins while the overall domain structure remained. Importantly, their work revealed complex multiple interactions between chromatin loci. Among other revealing applications of MERFISH [84–86], the Zhuang group most recently built on their methodology to enable massively multiplexed and high-throughput imaging which uncovered new insights into the three-dimensional organization of transcription [87].
The targeted genome editing CRISPR/Cas system [88] has also been co-opted for SRM imaging of DNA loci. Widely considered the most promising method for precise editing of genes in the clinical setting, Chen et al exploited the targetable specificity of the combined guide-RNA (gRNA) and the Cas9 protein to label specific DNA sequences [89]. This method utilizes a labelling system that comprises a nuclease deficient Cas9 protein (dCas9) tagged with an FP together with a gRNA that localizes to the target DNA sequence without inducing cleavage (figure 3(D)). The specificity of the dCas9 system was demonstrated by co-labelling mouse telomeric repeats with both dCas9 and FISH probes resulting in colocalization of the two labelling systems [89, 90]. Anton et al further used 3D SIM to visualize telomeres labelled with an FP-dCas9 probe alongside antibody labelled TRF2 showing a telomere morphology consistent with previously reported STORM results [80, 90]. Recent efforts have focused on improving the labelling efficiency of FP-dCas9 constructs; one of which involves recruiting multiple fluorescent constructs to the targeted area using the SunTag peptide array system [91]. Another innovative method recently published utilizes azide functionalized gRNAs [92]. These Cas9-azide gRNA constructs were able to localize to and enrich for the target telomeric regions and although the authors used an FP-tagged Cas9 to visualize the telomeric regions, it will be interesting to see the results from the fluorescently clicked azide gRNAs under SRM conditions.
SRM imaging of sub-diffraction chromatin structure
Over the past decade several groups have focused on using SRM to probe the nanoscale structure of DNA in situ with several landmark papers describing previously unknown chromatin structures and their relationships to cellular function. One important example is the use of STORM imaging of core histone proteins which revealed that nucleosomes are grouped in discrete domains within interphase chromatin and that these domains are separated by nucleosome depleted DNA regions [93]. Some of these regions are associated with active transcription as evidenced by RNA polymerase II co-localization. Ricci et al further demonstrated that formation of these nucleosome domains is dependent upon the cell differentiation state, with stem cells displaying fewer domains overall and generally more open chromatin. They hypothesized that the more open chromatin in these undifferentiated cells allows transcription machinery to access a greater diversity of genes, as gene function has been correlated to chromatin state (figure 5(A)) [94]. As cells become more differentiated, genes that are no longer required are silenced by compaction of the DNA into heterochromatin, as evidenced by larger and denser nucleosome regions [93]. Similar results were later reported in live cells using single particle tracking PALM (sptPALM) [95] to probe chromatin dynamics. This work revealed the persistence of condensed nucleosome domains throughout the cell cycle, including during cell division. Interestingly, Nozaki et al's live cell study [96] (figures 5(B), (C)) determined larger average nucleosome domain sizes than Ricci et al's work in fixed cells [93]. It is not yet clear whether the disparity in domain size is due to a difference in chromatin organization between mammalian cell lines and differentiation states, or a consequence of fixation or imaging artefacts.
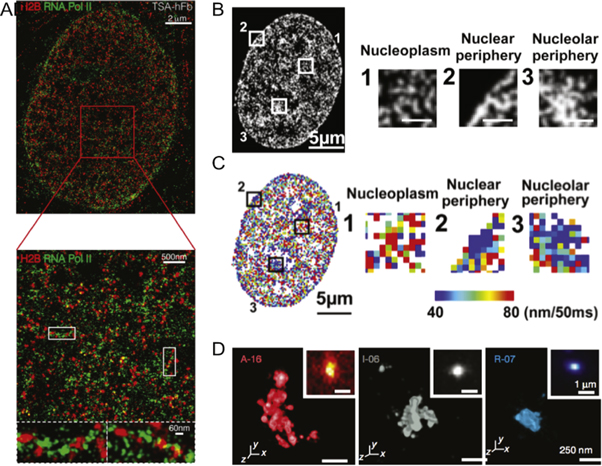
Figure 5. SMLM enables imaging of in vivo nanoscale chromatin structure. (A) STORM colocalization of RNA polymerase II (green) and H2B (red) histones in human fibroblast cells showing strong polymerase signal at weakly compacted chromatin. (B) A representative PALM image of a live HeLa cell expressing a photoactivatable FP-H2B construct with specific nuclear regions magnified (right. Scale bars are 1 μm). (C) Corresponding chromatin heat map following particle tracking for 50 ms. Red regions represent comparatively larger movements whereas blue regions represent comparatively smaller movements. (D) 3D STORM images of active, inactive and repressed chromatin domains with their diffraction limited image inset. (A) reproduced with permission [93], copyright 2015, Elsevier. (B), (C) reproduced with permission [96], copyright 2017, Elsevier. (D) reproduced with permission [97], copyright 2016, SpringerNature.
Download figure:
Standard image High-resolution imageIt is well documented that various epigenetic markers play roles in orchestrating the transcriptional activity at particular genomic loci by manipulating the structure and compaction of chromatin [98]. Recent SRM investigations have successfully described several aspects of the in vivo interplay between the epigenome, chromatin structure, and transcription. Using STORM, Ricci et al demonstrated strong association of RNA polymerase II with weakly compacted histone H2B-labelled euchromatin regions in fibroblast cells (figure 5(B)). On the other hand, they found minimal colocalization between RNA polymerase II and the densest chromatin regions of these cells [93]. These observations were supported by STORM imaging of selected active (H4ac, H3K9ac, H3K4me3 and H3K36me3) and repressive (H3K27me3 and H3K9me) histone modifications that showed active histone marks associating with more open euchromatin domains and more active RNA polymerase II [99]. Similarly, STORM imaging of Drosophila Kc167 cells showed that repressive histone modifications associated with heterochromatin were also spatially excluded from nearby active chromatin [97]. Boettiger et al hypothesized that this effectively created a boundary between active and compacted chromatin to prevent the accidental activation of repressed genes. They also investigated an inactive chromatin state in which histones lacked modifications and exhibited depleted repressive and activator proteins. This inactive chromatin displayed a distinct packing behaviour separate from both the less condensed active, and the fully compact repressed chromatin regions [97]. Fang et al later reported three levels of DNA compaction using EdU labelled chromatin to detect and differentiate dispersed chromatin, clusters of nanodomains and individual nanodomains which could span several kilobases of DNA [100]. It seems likely that these different DNA distributions correspond to the active, repressed and intermediary chromatin states described by Boettiger et al who also determined that the physical three-dimensional size of each chromatin domain correlated to their domain lengths according to power law scaling, with the domain type determining the value of the scaling.
Recently, Otterstrom et al have extended these studies, using SMLM to investigate the effect of histone hyperacetylation on chromatin structure [101]. They found that chromatin regions with high amounts of histone acetylation had less nucleosome-associated DNA, either due to the loss of the nucleosomes themselves or the dismantling of chromatin domains. Furthermore, nucleosome domains that resided within close spatial proximity to each other saw the greatest level of chromatin decompaction upon hyperacetylation, suggesting that unfunctionalized histone tails are required for the maintenance of chromatin domains. From these SRM studies, a new understanding of the multiple levels of DNA compaction is emerging, offering new insights into the specific nucleosome and histone distributions and arrangements associated with actively expressed euchromatin, repressed heterochromatin and intermediary inactive compaction states.
Visualization of the accessible genome has recently been achieved by combining the assay for transposase-accessible chromatin (ATAC) with SMLM, termed 3D ATAC-PALM [102]. ATAC utilizes a Tn5 transposase to digest open (nucleosome depleted) chromatin regions and was tagged with a photoactivatable fluorophore for compatibility with PALM. The accessible chromatin was found to spatially cluster together corroborating earlier studies. However, upon chromatin hyperacetylation the nuclei favoured a more uniform distribution, in agreement with previous findings. By designing DNA-FISH probes for known active and inactive chromatin regions and performing dual colour PALM, Xie et al demonstrated that active accessible chromatin regions colocalized with enriched ATAC and H3K4me3 signals whereas inactive regions displayed little ATAC and H3K4me3 signal [102].
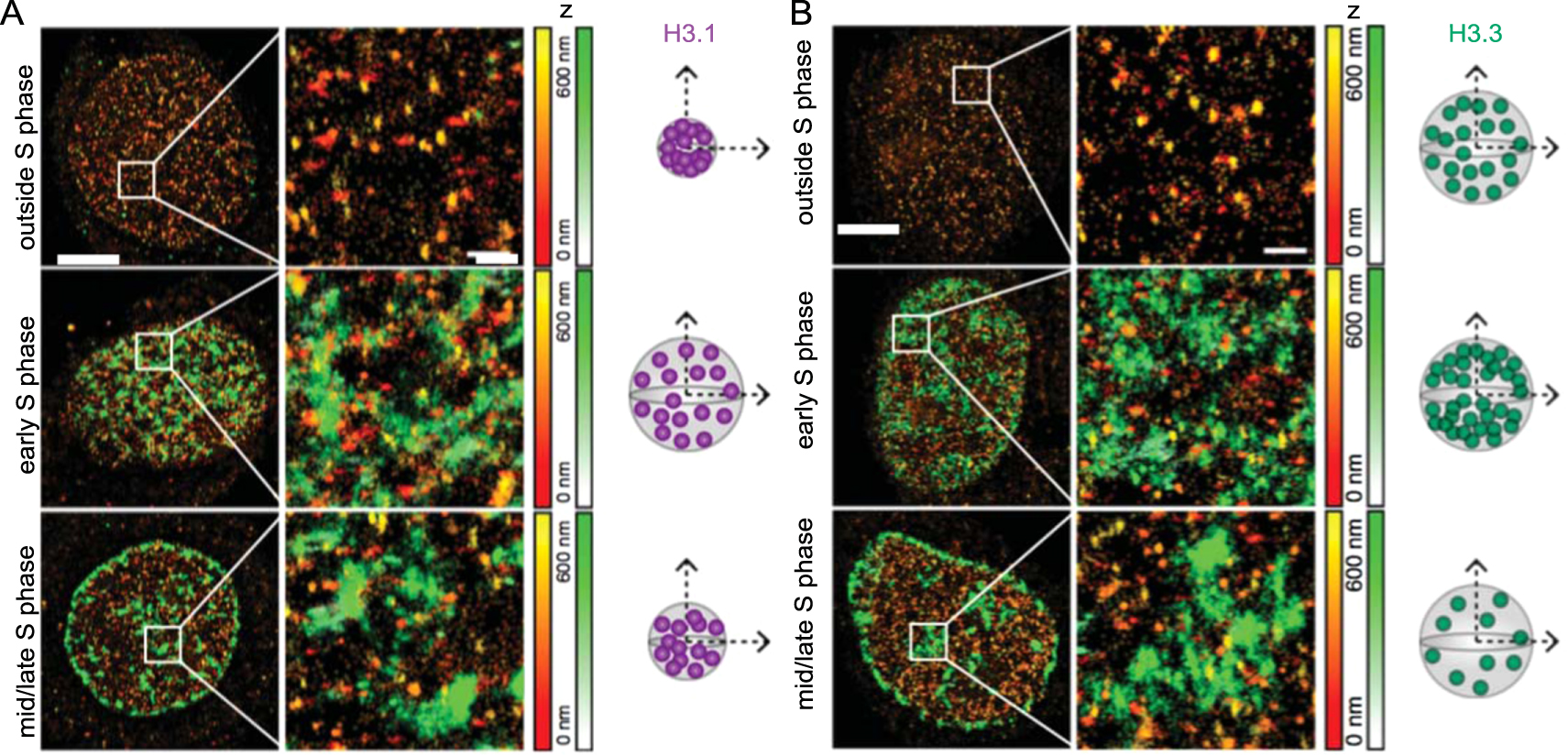
SRM has also been employed to study the movement and recycling of histones during the cell cycle. Clément et al performed 3D STORM of HeLa cells to visualize the histone variants H3.3 and H3.1 which are known to associate with early and late replicating chromatin, respectively [103, 104]. Two distinct types of H3.1 clusters were observed: small dense clusters present throughout the cell cycle indicative of late replicating chromatin, and larger low-density clusters detected only during early S phase corresponding to early H3.3 enriched chromatin (figure 6). They proposed that H3.1 only temporarily marks early replicating chromatin because H3.3 is not incorporated in a DNA synthesis dependent manner [105]. Analysis revealed H3.3 cluster densities decrease from early to late S phase before increasing after S phase. Clément et al conclude that H3.1 histones are deposited throughout the genome during replication but are later replaced in early replicating chromatin regions by H3.3. In addition, replication stress during S phase impaired local histone recycling, revealing altered H3.3 and H3.1 distributions [104]. The spatial context afforded by SRM measurements complemented genome wide analyses of gene expression, particularly in relation to the nanoscale histone dynamics during the cell cycle.

Figure 6. STORM imaging of HeLa cells labelled for replicated DNA (EdU, green) and histone H3 variants (orange) reveals the dynamics of histone recycling. (A) H3.1 and (B) H3.3 display different spatial arrangements at different cell cycle stages. Scale bars represent 5 μm and 600 nm for the zoomed regions on the right. Corresponding z range is given by the colour gradients. Next to each condition is a pictorial representation of the changes in cluster volume and density. Reproduced from [104], 2018, SpingerNature under Creative Commons Attribution 4.0 license.
Download figure:
Standard image High-resolution imageThe effects of DNA damage and repair on chromatin structure
Beyond gene expression and cell cycle effects, changes to chromatin compaction and organization have also been implicated in response to DNA damage, in particular DSBs. However the precise spatiotemporal nature of these rearrangements have proven difficult to study with conventional imaging [106]. Recently, Xu et al used SMLM imaging of intercalating DNA dyes in pathological tissues to quantitate sub-diffraction changes to chromatin structure in early carcinogenesis [107]. Combined with genomic and transcriptomic data, they were able to correlate the initial stages of malignant transformation with subtle, sub-diffraction decompaction and fragmentation of higher order chromatin structures, something which had previously been unobservable with conventional microscopy. Along with the novel characterization of chromatin structure's role in cancer, this research also demonstrates the future potential usefulness of SRMs in a clinical setting.
Other studies have more directly characterized the interplay between DNA damage and chromatin structure, often adapting the long-standing approach for diffraction limited visualization of DNA damage foci by immunofluorescent labelling of the histone variant H2A.X with a phosphorylated serine at the 139 residue (γH2AX) [108]. Bach et al used prolonged low folate levels to induce DSB accumulation which they detected with SMLM imaging of γH2AX and H3K9me3 foci [109]. Using the former as a marker for DSB repair and the latter histone modification as a marker for heterochromatin, they showed a relaxation of DSB clustering over time and a shift of surrounding DNA to less compacted euchromatin. They hypothesized this relaxation allowed for better repair machinery access to the damaged DNA. In a following study, these two histone modifications were used to characterize chromatin structure response to DNA damage by ionizing radiation [110]. Again employing SMLM, Hausmann et al found that the formation of γH2AX foci was most prominent 30 min after damage and that the number and size of foci correlated with damage dose. γH2AX foci co-localized with the H3K9me3 marker which suggested some rearrangement of DSBs to the periphery of condensed chromatin domains. Interestingly, with increased damage, the level of heterochromatin density decreased, with a later report developing a cell independent analysis based on persistent homology further describing the heterochromatin-dependent topology of γH2AX foci [111]. Similar findings have also been reported in yeast, with SIM used to quantify the reduction in total chromatin-associated histones following DNA damage [112]. In this study, the loss of histones was observed to lead to decompaction of chromatin and enhanced chromatin mobility, which is predicted to help facilitate access for homology search and repair. This process was found to be dependent upon activation of the DNA damage checkpoint (a regulatory network of proteins that helps maintain genome integrity), the multi-subunit histone remodeler INO80-C and a functional proteasome for histone degradation. Mutations in the checkpoint kinases Mec1 or Rad53 as well as in INO80-C remodeler specific subunits led to no histone degradation, while inhibition and mutation of 26 s proteasome subunits suppressed histone degradation in response to DNA damage.
Others have made use of SRM of γH2AX to probe more directly the damage foci structure itself leading to a number of contradictory findings, particularly in relation to the existence of single versus clustered DSBs, and the actual, in vivo size of a repair focus. Hagiwara et al dosed cells with high levels of linear energy transfer heavy ion irradiation and characterized 1 μm3 γH2AX foci using SIM [113]. Using the single-stranded bind protein RPA as a more specific mark for a DSB, they concluded these foci comprised multiple forms of damage, and multiple DSBs. While this is expected of such a deleterious damage dose, it remains contentious that this type of damage plays a major role in disease. For this reason, many other studies have focused on lower irradiation doses and damage generated by small molecule drugs. D'Abrantes et al compared SIM, STED and GSDIM (a SMLM variant) alongside high resolution confocal approaches for the characterization of γH2AX foci induced by low linear energy transfer ionizing and laser irradiation [114]. They also visualized the DNA repair proteins 53BP1 and Ku. Although they note that all the SRMs engendered improved resolutions and more accurate detection and counting of foci compared to confocal microscopy, they were unable to detect Ku using SRM and found unexpected differences in the distributions of 53BP1 and γH2AX foci that are inconsistent with current DNA damage response models. Although they were able to report different spatial resolutions, there was no clear determination of the underlying foci size or composition in terms of DSB number.
Also setting out to determine the elementary structural units of damage foci, Natale et al used SIM imaging of γH2AX at both x-ray and Cas-induced DSBs [115]. Building on hypotheses put forward previously ([109, 110]), they found that heterochromatin harbouring DSBs decompacted during repair while retaining compaction-related histone modifications. Moreover, they identified γH2AX nano-foci ~200 nm in diameter that further clustered into approximate groups of four containing only a single DSB (figure 7). These findings somewhat contradicted earlier work by Perez et al which also used SIM and γH2AX, along with heavy ion irradiation, and found elongated sub-foci of ~100 nm which could be further broken down into 40–60 nm bent structures [116]. Other contradicting reports include Reindl et al who employed STED imaging and detected ~540 nm foci in response to high linear energy transfer irradiation and ~390 nm foci in response to low linear energy transfer irradiation [117]. These foci could be further broken down into nanostructures of 135 nm and 119 nm for high and low irradiation, respectively. Finally, using SMLM Sisario et al were able to visualize 45 nm nano-foci comprising only a single nucleosome [118]. The inconsistences in foci size across these several reports likely relate to the different types of damage being investigated, the techniques applied (and the achieved and achievable resolutions) and differences in the terminology of foci, nano-foci, sub-units, etc. A necessary improvement in future applications of SRM to DNA biology will be a consensus on these key aspects, and how to describe and control them. This will be particularly important because many studies rely on γH2AX foci analysis to describe colocalized protein behaviours including Ku [119], MRE11 [120], MDC1 and NBS1 [121] and RAD51 [117, 122].

Figure 7. 3D SIM imaging of chromatin reorganization during DNA DSB repair. (A) Representative HeLa cells immunostained for γH2AX 24 h after ionising radiation damage. Both confocal microscopy (left) and 3D SIM (right) shown. γH2AX foci (red) are shown along with the DAPI channel (grey) overlaid with multi-color coded clusters showing spatially distinct foci. Magnified regions outlined in yellow are provided in the lower panels showing in detail the difference in foci discrimination between confocal and 3D-SIM by the number of different foci (colors) detected. Scale bars = 5 μm and 500 nm for the main and magnified images respectively. (B) 3D SIM images comparing γH2AX formation and detection of DSBs by Ku (left) and TUNEL (right) staining 30 min after ionising radiation exposure. Regions outlined in yellow are magnified below. The nuclear outlines are represented as yellow dashed lines in the halves without the DAPI stain. Scale bars = 5 μm and 500 nm for the main and magnified images respectively. C) Schematic representation of γH2AX reorganization following DSB induction showing the euchromatin to heterochromatin repair trend. Reproduced from [115], 2017, SpingerNature under Creative Commons Attribution 4.0 license.
Download figure:
Standard image High-resolution imageOf particular structural interest, several studies have aimed to characterize the arrangement of p53-binding protein 1 (53BP1) at repair foci. When subjected to ionising radiation and analysed via SIM, this protein, widely considered an HR-antagonist/NHEJ-facilitator, localizes to damage foci to promote repair via NHEJ in early cell cycle stages; however in later cell cycle stages, BRCA1 excludes 53BP1 from DSBs to initiate repair via the HR repair pathway [123]. A similar occurrence of 53BP1 exclusion from DSBs by RAD51 has been observed using STED microscopy: 53BP1 flanks RAD51 foci that, when viewed under diffraction limited conditions, appear colocalized but are spatially distinct at higher resolutions [117, 122]. D'Abrantes et al found that despite the close spatial arrangement of γH2AX and 53BP1, γH2AX foci did not perfectly colocalize with 53BP1 foci, consistent with previously reported observations of diffuse γH2AX but discrete 53BP1 foci [124]. As with varying reports of the structure and size of γH2AX foci, apparent contradictions most likely reflect differences in techniques and damage types. Promisingly, recent in-depth analysis of dSTORM data has enabled empirical counts of γH2AX based on fluorophore photoswitching activity at γH2AX clusters [125]. Such analyses potentially provide a more absolute measure of repair foci.
Ongoing research using SRM promises to uncover the precise mechanisms by which DSB induction results in phosphorylation of surrounding H2AX histones which are yet to be fully described. Similarly, it remains unclear how γH2AX foci coordinate the cascading damage response to prompt and facilitate DSB repair. Previous immunoprecipitation and blotting experiments have indicated proteins capable of binding to γH2AX [126, 127] however these techniques rely on the extraction of proteins and destruction of the nuclear environment therefore losing the spatial configurations within the nucleus. Spatial studies using SRM will be particularly important for understanding the interactions and roles of γH2AX because of its presence in a dense three-dimensional network around single and clustered DSBs.
Single molecule studies of DNA repair pathways
SRM approaches have also been used to elucidate spatiotemporal molecular details of the damage response and repair pathways at DSBs. Rather than focusing on the structure of repair foci, these studies aim to better understand the in vivo associations and progressions of the myriad of repair proteins involved in damage detection, response and repair. Overcoming the previous limitations of signal-to-noise and the dense nuclear environment, several studies have made use of single particle tracking PALM (sptPALM) [95] to visualize the dynamics of DNA repair proteins in live cells. In one such example, Uphoff et al used sptPALM to characterize the behaviours of individual DNA polymerase and ligase proteins in order to reveal their respective reaction rates and provide a molecular model for the repair of gapped DNA in vivo [128]. Utilizing a similar experimental approach, Stracy et al showed that the initiation of a distinct DNA repair pathway in bacterial cells occurs in a two-step process [129]. Specifically, they showed that the UvrA protein has two distinct DNA interactions: firstly in scanning the genome for damaged DNA and secondly in independently localizing to the lesion prior to recruitment of a secondary protein, UvrB [129].
In response to DNA damage, some DNA polymerases are able to synthesize past the site of damage in a process known as translesion synthesis, albeit risking the introduction of errors. To understand how these polymerases gain access to the DNA following damage, Thrall et al tracked individual DNA polymerase IV (Pol IV) proteins in live bacterial cells and found that Pol IV recruitment to damaged DNA is dependent upon the type of damage induced (figures 8(A), (B)) [130]. Pol IV recruitment in cells treated with methyl methanesulfonate, which primarily generates alkyl adducts, required interaction with the β-clamp whereas Pol IV response to guanine adducts generated by nitrofurazone treatment did not. The enrichment of Pol IV at replication sites only occurred following damage induction. Recently, live bacteria sptPALM was further applied to investigate the dynamics of three replication proteins in response to replicative stress (figures 8(C), (D)) [131]. By tracking two polymerases, PolC and DnaE, and another replicative protein DnaX, PolC and DnaX were found to be coupled during normal replication. Upon DNA damage, PolC dissociates from sites of replication suggesting an exchange of polymerases to enable translesion synthesis. SMLM imaging measurements revealed that DnaE contributed a less important, yet still essential, role in bacterial replication than was previously determined via in vitro assays which showed that inactive DnaE inhibits DNA synthesis [132].
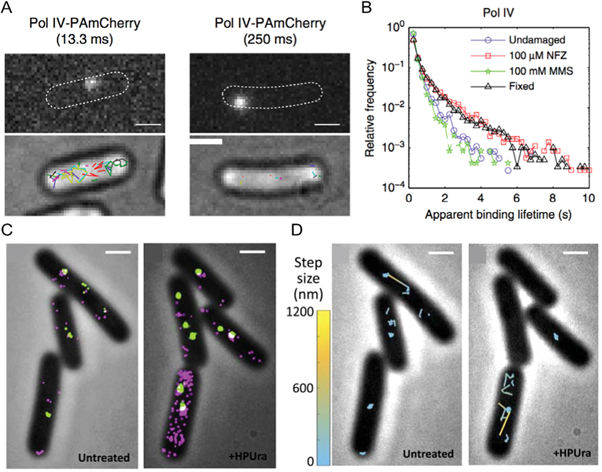
Figure 8. sptPALM enables elucidation of bacterial DNA repair kinetics. (A) Representative images of single FP-Pol IV constructs in Escherichia coli cells (outlined for clarity) imaged with integration times of 13.3 ms (left) and 250 ms (right) with the corresponding brightfield image overlayed with all detected tracks shown in the lower images. Scale bars = 1 μm. (B) Changes in Pol IV construct binding lifetimes in the absence and presence of DNA damage generated using nitrofurazone (NFZ) or methyl methanesulfonate (MMS) treatment, with fixed cells included as a photobleaching control. (C) PALM images of PolC (magenta) and DnaX (green) constructs in control Bacillus subtilis cells (left) and cells treated with 6(p-Hydroxyphenylazo)-Uracil (right) which arrests replication via disruption of PolC. Scale bars = 1 μm. (D) Representative PolC tracks from (C) depicting comparatively fast (yellow) and slow (cyan) movements in untreated (left) and treated (right) cells. (A, B) reproduced from [130], 2017, SpingerNature under Creative Commons Attribution 4.0 license. (C, D) reproduced with permission [131], copyright 2019, Elsevier.
Download figure:
Standard image High-resolution imageOther research groups, including ours, are harnessing the single molecule sensitivity, as well as the superior spatial resolutions, of SMLM to probe subpopulations of DSBs within cells. This has enabled the elucidation of different repair pathways, temporal separation of sequential repair steps, and detection of different damage motifs. Furthermore, because the main cause of endogenous DSBs is DNA replication fork (RF) breakage [34], SRM paired with pulse-labelling of nascent DNA engenders individual RF and DSB imaging which can then be colocalized with various proteins of interest [75, 133].
Using this approach in combination with extensive confocal and biochemical assays, Daddacha et al uncovered a new role for the protein SAMHD1 in HR repair [134]. SAMHD1 was previously reported to restrict viral infections by depleting the nucleotides available for DNA synthesis [135], however, this study demonstrated that SAMHD1 also localizes to DSBs and promotes repair, in part via recruitment of CtIP. SMLM data supported in vitro biochemical assays by enabling visualization of the interaction between SAMHD1 and CtIP at individual damaged RFs. Cells deficient in SAMHD1 were shown to be hypersensitive to DSB inducing agents providing further evidence that SAMHD1 has a key role in HR.
BRCA1 and BRCA2, the protein products of the two breast cancer genes, are extensively researched DNA damage response proteins with recent SMLM studies offering unprecedented insights, particularly in the discovery of previously uncharacterized roles in DNA repair. SMLM studies by D'Alessandro et al showed that BRCA1 associates with DNA:RNA hybrids following DSB induction and the generation of long non-coding RNAs. Furthermore, BRCA2 mediates DNA:RNA hybrid levels through interaction with RNase H2 which together regulates recruitment of other repair proteins [136]. SMLM further revealed the spatial arrangement of γH2AX foci with RNase H2 and detected BRCA1 localizations to DNA:RNA hybrids at DSBs, providing a more detailed view of these foci compared to diffraction limited confocal images. A subsequent study also employing SMLM identified complete RNA polymerase II preinitiation complexes at DSBs and went on to demonstrate that the transcription of long non-coding RNAs at DSBs drives the formation of liquid-liquid phase separation boundaries that support the accumulation of DNA damage response proteins [137]. Other examples of studies using SMLM alongside extensive complementary biochemical assays have also uncovered the role of FBH1 helicase in HR modulation [138] and the formation and role of a TIMELESS-PARP1 complex in DNA damage response [139].
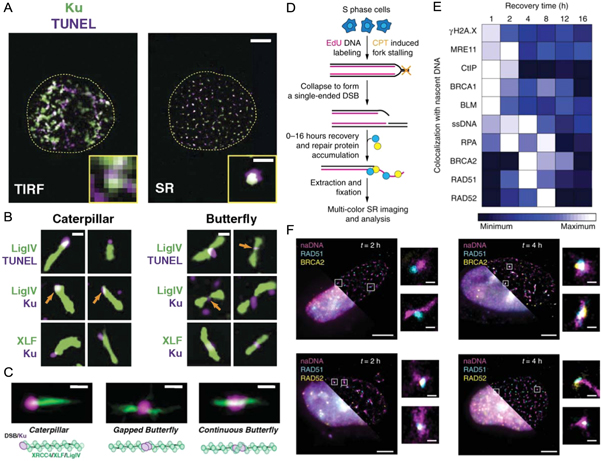
We and others have also developed novel SMLM assays and analyses, including nascent DNA pulse labelling and multi-colour immunolabelling, to map the spatiotemporal progression of different DSB repair pathways [76, 140–144]. Reid et al used both SMLM and single molecule Föster resonance energy transfer experiments to characterize the sequential steps involved in NHEJ, including the generation of XRCC4, XLF and DNA ligase IV filaments (figures 9(A)–(C)) [145]. Similarly, in order to map HR repair, Whelan et al induced single ended DSBs using a small drug, camptothecin, known to mimic endogenous DSBs [34]. By taking temporal snapshots across 16 h of repair and visualizing eight key repair proteins, ssDNA generation, and γH2AX we were able to map the sequences and dependencies of distinct repair steps (figure 9(D)–(F)) [75]. Further spatial analysis revealed key interactions such as MRE11 loading away from the break, and only partial association of RAD51 with BRCA2. This observation agreed with SMLM work by Sánchez et al who found that BRCA2 formed diffuse clusters around RAD51 foci at sites of DNA damage [140]. A separation of BRCA2 and RAD51 was also observed at RPA associated DNA, consistent with BRCA2's role of loading RAD51 onto single stranded DNA to form a filament. In contrast to the study by Sánchez et al, we found that RAD52 mediates RAD51 filament formation on single stranded DNA and in the absence of RAD52, BRCA2 performs this role [75]. However, RAD52 is not able to perform the other roles of BRCA2 necessary for homology search and recombination, providing an explanation as to why little disease phenotype is observed for RAD52 deficient mice whilst a severe phenotype is observed for deficiencies in BRCA2 [146]. Due to the enhanced resolution gained by performing SMLM measurements, the early stages of repair at individual DSB sites could be mapped to reveal the spatiotemporal dynamics between repair proteins. Most recently, we have applied these assays to visualize the different damage motifs that arise in response to low levels of replication stress, successfully differentiating between broken and regressed RFs and identifying several of the key first responders [133].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. SRM visualization of individual DNA damage and repair events. (A) A U2OS human cell nucleus stained for Ku (green) and DSBs using the TUNEL stain (purple) shown in both diffraction limited TIRF (left) and SRM (right). Scale bars = 5 μm and 500 nm for the main and zoomed images, respectively. (B) Individual DSB repair structures showing distinctive caterpillar and butterfly structures with Ku and DSBs displaying as punctate compared to the filaments detected for Ligase IV and XLF. Scale bar = 250 nm. (C) Average particle images showing three distinct structures and a corresponding schematic interpretation. Scale bar = 250 nm. (D) A schematic outline of the pipeline for SRM imaging and analysis for single ended DSBs generated using camptothecin. (E) The resulting heatmap showing the spatiotemporal progression of HR repair of individual DSBs generated as described in (D). (F) Representative three-color whole nucleus images showing diffraction limited (bottom left) and SRM for nascent DNA costained with RAD51 and BRCA2 (top) or RAD51 and RAD52 (bottom) at 2 h (left) and 4 h (right) recovery. Individual foci showing disperse nascent DNA colocalized with proteins also shown. Scale bars = 3 μm for whole nuclei and 250 nm for zoomed foci. (A)–(C) reproduced from [145], copyright, 2015, National Academy of Sciences. (D)–(F) reproduced from [75], 2018, SpingerNature under Creative Commons Attribution 4.0 license.
Download figure:
Standard image High-resolution image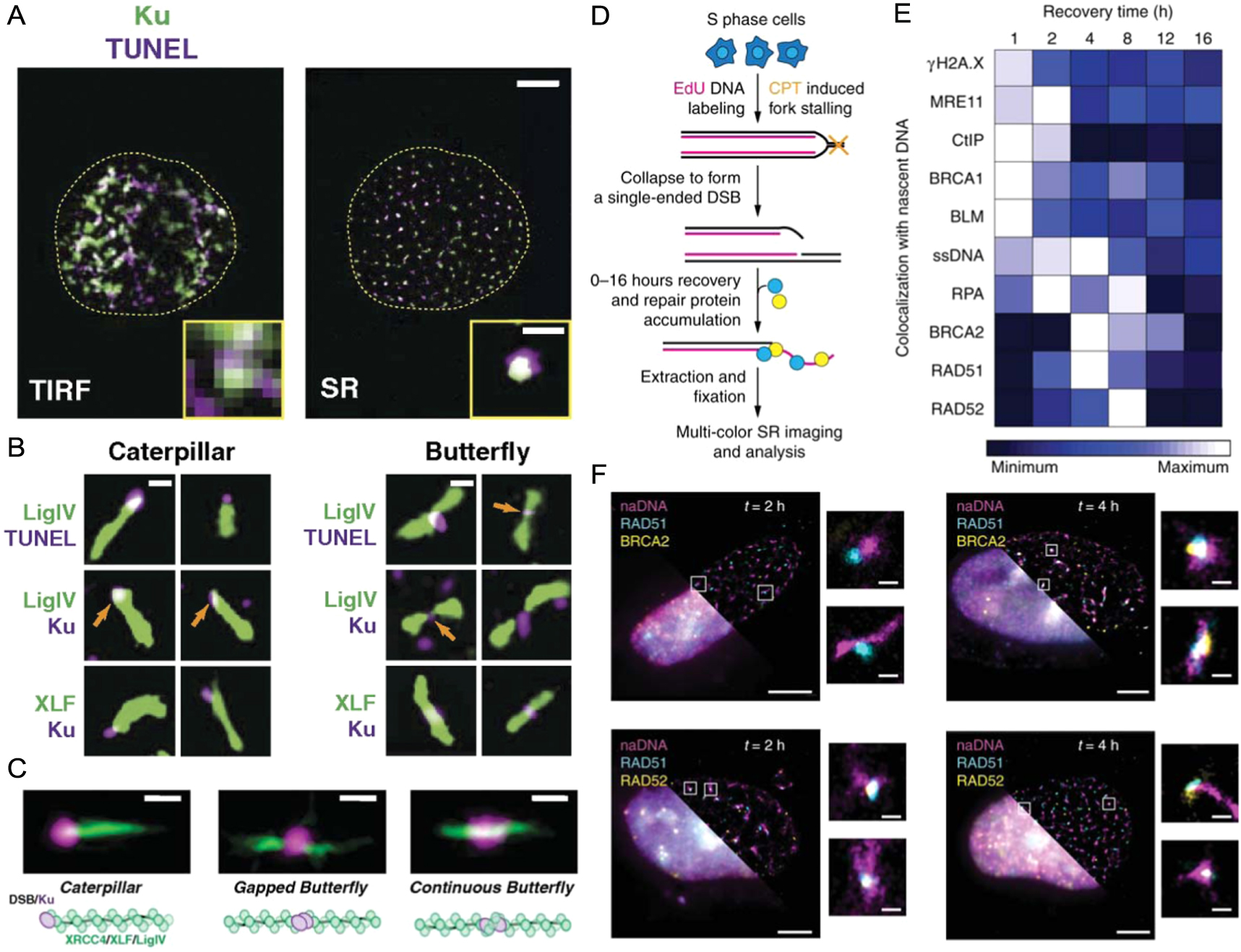{kind=link}
Concluding remarks
The use and popularity of SRM in molecular and cell biology research continues to increase as methodologies become more available and adaptable. Additionally, the requisite interdisciplinary research teams and support facilities have become more established and experienced. In this review, we assessed the recent applications of SRM to image DNA structure with a focus on damage and repair because of its particularly rapid emergence as a technique of choice within this high impact research area. Compared to other important biomolecular architectures, chromatin has long proven one of the most challenging to image due to its high density and relative amorphousness. The improved resolution provided by SRMs has overcome these limitations to an impressive extent as evidenced by the many new insights garnered simply by recapitulating experiments with SRM in place of conventional confocal microscopy.
A significant challenge specific to imaging in vivo DNA that we have discussed extensively is the need for novel labelling assays compatible with SRM modalities. Whereas immunolabelling and FP co-expression are commonplace for protein visualization, and non-specific small molecule dyes have proven sufficient for diffraction limited whole genome imaging, SRM has necessitated the development of several new approaches to target specific nuclear events, chromatin structures and DNA sequences. Combined with improved analyses and imaging modalities, these SRM-suited labelling strategies have underpinned a number of landmark studies over the last decade describing the structure and function of chromatin, particularly as it relates to epigenetic modification and DNA damage responses in different biological contexts.
With the expanding uptake of SRM to study DNA damage and repair, it is increasingly clear that a persistent difficulty within the research area is the size variability of imaged foci which, in the context of DNA damage, can range from tens to several hundreds of nanometers. This stems from the use of different imaging modalities, methods for analysis, and the definitions used for detected foci (nano-foci, nano-structure, etc). As such, a comprehensive assessment and gold standard for SRM determination of damage foci across different damage types and labelling approaches will be invaluable to the field. However, further research into exactly what comprises a focus in the DNA damage field is likely to encounter similar complexities and controversies previously documented in SRM studies of membrane nanoclusters (Reviewed recently by Baumgart et al [147]).
Nonetheless, these challenges also highlight a particular strength of SRM imaging of DNA damage in that the vastly improved sensitivity—down to the single molecule with SMLMs—engenders the ability to study much lower levels of damage [17]. This is an important advantage of SRM studies because it has been a long-standing problem that most human disease related to DNA damage arises from only a handful of DSBs per day, or small increases in this number and/or dysfunctional repair [32]. In contrast, many of the traditional approaches for generating DNA damage, particularly for imaging purposes (e.g. linear energy transfer, laser and x-ray irradiation), generate hundreds of DSBs alongside various other types of damage to the genome and surrounding biomolecules [114]. These techniques generate a level of DNA damage, often clustered, that results in tens if not hundreds of overlapping fluorophores co-labelling a focus that, although less relevant to endogenous damage levels, enables detection with less sensitive imaging methods.
Instead, SRM-based assays allow for much lower, more biologically and disease relevant levels of DNA damage to be generated, detected and characterized. Utilizing the enhanced resolution and sensitivity of SMLM, it is now possible to investigate more subtle levels of DNA damage and repair, such as those sustained daily in a typical cell. Small molecule drugs such as camptothecin and hydroxyurea cause DSBs without destruction of the overall nuclear environment and, at low doses, induce a quantity of DSBs that closely mimics the level of damage endogenously encountered. Similarly, directed DSB induction using modern genome editing systems such as CRISPR/Cas also allows for tight control over the number and nature of DSBs induced. Thus, SMLM in conjunction with controlled DSB induction provides a suitable assay to study responses to native DSB levels through detection and characterization of individual damage events.
Applications of SRM, especially of SMLMs, therefore have the potential to provide an improved understanding of genomic structures and dynamics, yielding new insights into where, when and how DNA-interacting proteins associate with chromatin. Determining whether or not there is competition to commit the damage to one repair pathway over another at the molecular level will provide a better understanding of the dynamic interplay that occurs throughout the cell cycle and what regulations take place concerning repair pathway choice. In addition, elucidating individual protein requirements (e.g. whether another protein is required first before the role can be performed) and redundancies within these pathways (e.g. whether a protein can perform the role of another) would not only strengthen our current models of DSB repair in vivo but also provide insights into the development of more effective therapeutic treatments by targeting vital proteins within these processes.
It should be noted that although SRM can provide molecular insights, a collection of complementary techniques is often required before arriving at conclusions. Increasingly, the combination of SRM with cutting-edge biochemical assays continues to yield promising new insights into nuclear molecular processes. With the growing trend of using SRMs as a quantitative tool, rather than just for visual enhancement, there is a greater demand for image analysis methodologies and standardized workflows. As of yet there are only a few reports comparing SRM image analyses, with many groups electing to develop and implement their own quantitative procedures.
The future prospects of SRM for nuclear and biological studies continue to be developed and optimized in conjunction with novel labelling strategies, and together, these assays will expand our knowledge of intricate nuclear organizations and functions. The shift away from structural to distribution analyses of SRM data will become more prominent as we aim to untangle nuclear regulatory and repair pathways and processes at the molecular level. Undoubtedly the contribution of SRM measurements is deepening our understanding of these complex molecular pathways and will continue to do so for some while yet.
Acknowledgments
DRW is the recipient of an Australian Research Council Discovery Early Career Research Award (DE200100584) funded by the Australian Government. TDMB acknowledges support from the Australian Research Council (DP170104477). ER acknowledges funding support from the National Institute of Health (1R35GM134947-01, 1P01CA247773-01/5491), American Cancer Society (RSG DMC-16-241-01-DMC) and the V foundation for Cancer Research (D2018-020).
Data availability statement
No new data were created or analysed in this study.
Conflict of interest
The authors declare no conflict of interest.