
Abstract
The iridium-catalyzed C-H borylation of diethyl phenylphosphonate results in nonselective mono and bisborylation to afford a near statistical mixture of 3-, 3,5- and 4-boryl substituted aryl phosphonates whereas 3-substituted aryl phosphonates undergo highly regioselective C-H borylation to afford the corresponding meta-phosphonate substituted arylboronic esters as the sole product; the resulting boronic esters were used as nucleophilic reagents in a subsequent palladium-catalyzed Suzuki–Miyaura cross-coupling to generate a range of biarylmonophosphonates. Gratifyingly, the Suzuki–Miyaura cross-coupling can be conducted without purifying the boronic ester which greatly simplifies the synthetic procedure.
Graphical Abstract

Similar content being viewed by others

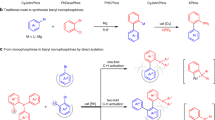
1 Introduction
Arylphosphonates are an important class of molecules as they are key motifs in a host of natural products, bioactive compounds, agrochemicals, and materials as well as intermediates in organic synthesis and precursors to phosphines, which are ubiquitous in organometallic chemistry and catalysis [1,2,3,4,5,6,7,8,9,10,11,12,13]. As such the synthesis and elaboration of aryl phosphonates has attracted considerable attention and several strategies based on C-P bond formation have been reported; these include; nickel, copper and palladium-catalyzed coupling of an aryl halide or pseudo halide with a dialkyl phosphonate [14,15,16,17,18,19,20,21,22,23,24,25,26], nickel, copper and palladium-catalyzed oxidative coupling of arylboronic acids or arylsilanes with dialkyl phosphonates [27,28,29,30,31,32,33], photoinduced transition metal-free coupling of aryl halides with H-phosphonates [34, 35], nickel-catalyzed decarbonylative C-P bond formation [36,37,38], copper-catalyzed P-arylation between diaryliodonium salts and dialkyl phosphonates [39], palladium-catalyzed phosphorylation of aryl C-H bonds [40,41,42,43,44,45,46], dehydrative cross coupling [47], and P-arylation of arynes [48], However, most of these approaches require the use of prefunctionalized substrates and as such are limited in terms of the variety of accessible substrate combinations. Arylphosphonates have also been further elaborated by late-stage modification using either metal catalyzed C-H arylation [49,50,51,52,53,54,55,56,57], where the phosphonate acts as a directing group, or Suzuki Miyaura cross-coupling [58,59,60,61,62,63,64,65,66,67]; the latter is an effective strategy for the synthesis of architecturally important achiral and axially chiral biaryl and heterobiarylphosphines, but it requires the use of a pre-functionalized arylphosphonate electrophile as the coupling partner.
As boronic acids and esters are widely used in synthesis [68], we recently initiated a program to develop ortho-phosphonate aryl and naphthylboronic esters as an entirely new class of nucleophilic coupling partner for the synthesis of substituted aryl and heteroaryl phosphonates (Fig. 1a) [69]. While the use of ortho-phosphonate arylboronic esters as the nucleophilic coupling partner complements existing approaches in which the phosphonate is introduced as the electrophile, the synthesis required multiple steps. Clark and Watson have recently reported a markedly more efficient and general method for the synthesis of o-boryl arylphosphonates via regioselective C-H borylation using a phosphonate as the directing group (Fig. 1b) [70]. This strategy is elegant and highly effective as the starting materials are readily accessible and the catalyst commercially available and it exhibits broad functional group tolerance and is highly flexible in terms of the substitution pattern; moreover, the resulting boronic esters can be converted into a range of products using conventional organic methods. Even though the use of phosphines as directing groups for ortho-selective C-H borylation has previously been reported, the substrate scope is more limited than that accessible using arylphosphonate-directed ortho C-H borylation [71,72,73,74,75,76].

Synthesis of phosphonate-substituted arylboronic esters via a multistep borylation of 2-bromo-substituted aryl phosphonate, b iridium-catalyzed phosphonate-directed ortho C-H borylation and c sterically controlled iridium-catalyzed regioselective C-H borylation of meta-substituted aryl phosphonates
As part of our ongoing program to develop new cross-coupling partners for the synthesis of biaryl-based motifs we have also been exploring the C-H borylation of aryl phosphonates to prepare phosphonate-substituted arylboronic esters as this would avoid the need to prefunctionalise the aryl phosphonate (Fig. 1c). In this regard, the site selectivity of iridium-catalyzed C-H borylation is controlled predominantly by steric factors [77,78,79], which has been exploited to develop the regioselective meta-directed C-H borylation of 1,3-disubstituted arenes to prepare the corresponding 3,5-disubstituted boronic esters; this is now a powerful protocol that has been widely applied in synthesis to access a host of meta-substituted products including phenols [80], arylamine boronate esters [81], arylamines and ethers [82], arylboronic acids, and trifluoroborates [83], disubstituted bromoarenes [84], aryl nitriles [85], alkylarenes [86], pentafluorosulfanyl-substituted potassium aryltrifluoroborates [87], nitrated arenes [88], difluoromethylated arenes [89], trifluoromethylarenes [90, 91] and borylated aryl alkynes [92]. Recently though, alternative approaches have been developed in which meta selective C-H borylation has been directed either by noncovalent secondary interactions between a modified bipyridine and the substate by orientating the iridium towards the metal C-H bond [93, 94], ion pairing between a cationic substrate and an anionic bipyridine [95], a combination of ligand-substate electrostatic interactions and secondary B-N interactions [96] or Lewis acid–base directed borylation with a bifunctional catalyst [97], however, each of these approaches requires the synthesis of rather elaborate ligands. Since the use of steric factors has proven to be a highly reliable strategy for controlling the regioselectivity of C-H borylation, we targeted the iridium-catalyzed C-H borylation of 3-substituted aryl phosphonates to explore whether this protocol would facilitate access to the corresponding phosphonate-substituted arylboronic esters. To this end, the recent report of facile highly selective and divergent acyl and aryl cross-coupling of amides via an iridium-catalyzed C-H borylation/N–C(O) activation sequence to prepare biarylketones and biaryls [98] as well as Clarks aryl phosphonate-directed ortho C-H borylation [70] have prompted us to disclose the results of our preliminary study on the borylation of aryl phosphonates. Herein, we report that the iridium-catalyzed borylation of diethyl phenylphosphonate results in nonselective mono and bisborylation to afford a mixture of 3-, 3,5- and 4-boryl substituted aryl phosphonates while 3-substituted aryl phosphonates undergo highly regioselective C-H borylation to afford the corresponding meta-phosphonate substituted arylboronic esters as the sole product; the resulting boronic esters were also used as the nucleophilic partner in a subsequent palladium-catalyzed Suzuki–Miyaura cross-coupling to generate a diverse range of biaryl monophosphonates.
2 Results and Discussion
This project was initiated to explore whether aryl phosphonates would undergo selective C-H borylation to afford the corresponding aryl phosphonate-based boronate esters directly in a single step as this would avoid the palladium-catalyzed borylation of bromo-substituted aryl phosphonates. The recent development of a highly efficient and versatile preparation of o-phosphonate arylboronic esters via C-H borylation by Clark [70] encouraged us to report details of our preliminary proof of principle studies in this area. The aryl phosphonates required for this investigation were prepared in high yield via palladium-catalyzed C-P cross-coupling between methyl 3-bromobenzoate (1a), 3-bromotoluene (1b), or 3-bromo-1-trifluoromethylbenzene (1c) and diethyl phosphonate, by adapting previously reported procedures (Scheme 1) [26].

Synthesis of metal-substituted arylphosphonates 2a-c via palladium-catalyzed C-P cross-coupling between aryl bromides 1a-c and diethyl phosphonate
2.1 Iridium-Catalyzed Borylation of Diethyl Phenylphosphonate
As C-H borylation is now well-developed and a powerful tool in synthesis [99, 100], preliminary catalytic reactions were conducted using previous literature protocols as a lead [101]. Prior to exploring the C-H borylation of 2a-c, an initial investigation was conducted using diethyl phenylphosphonate 3 as the benchmark substrate to determine the selectivity, undertake optimization studies and establish protocols for analysis. Preliminary reactions were conducted in hexane at room temperature using 5 mol% catalyst generated from (1,5-cyclooctadiene)(methoxy)iridium(I) dimer and 3,4,7,8-tetramethyl-1,10-phenanthroline (tmphen) as the ligand of choice (Table 1, Entry 1), as it has been widely employed for the non-directed iridium-catalyzed C-H borylation of aryl, heteroaryl and alkyl C-H bonds because it is electron rich and its complexes catalyze C-H functionalization much more readily than their less electron-rich counterparts [102,103,104,105]. Under these conditions, a conversion of 97% was obtained after 24 h to afford meta-substituted 4a as the major product (40%) together with its 3,5-diboryl substituted counterpart 4b (22%) as well as para-substituted 4c (35%) (Table 1 entry 1). To this end, [Ir(COD)(OMe)]2 has been reported to generate the most active bipyridine-based catalysts and the room temperature borylation of arenes with these systems is now common place [77, 78]. Gratifyingly, near quantitative conversions could be obtained after only 1 h by increasing the reaction temperature to 50 °C; under these conditions the distribution of regioisomers was similar to that obtained at room temperature (Table 1, Entry 2). A survey of catalyst efficacy as a function of solvent for reactions conducted for 1 h at 50 °C (Table 1, Entries 2–6) revealed that high conversions were obtained in hexane (94%), methylcyclohexane (91%), dioxane (88%) and THF (70%) whereas a markedly lower conversion was obtained in dimethyl formamide (3%). As the highest yields were obtained in hexane, all further studies to identify optimum conditions were conducted in hexane. A reduction in the catalyst loading to 2.5 mol% resulted in a markedly lower conversion of 71% but with a similar distribution of products, under otherwise identical conditions, whereas an increase to 10 mol% reached 96% conversion after only 30 min (Table 1, Entries 7–8).
Although [Ir(COD)(OMe)]2 is the most widely used source of iridium for C-H borylations, reports of efficient catalysis with alternative iridium precursors prompted us to explore the efficacy of systems generated from [Ir(COD)(acac)], [Ir(COD)Cl]2 or [Ir(COD)2]BF4 and tmphen. A screen of each catalyst for the B2pin2 based borylation of diethyl phenylphosphonate 3 in hexane at 50 °C for 1 h revealed that the use of [Ir(COD)(acac)] and [Ir(COD)2]BF4 gave 93% and 77% conversion, respectively, while catalyst generated from [Ir(COD)Cl]2 only reached 7% conversion in the same time, although raising the reaction temperature to 70 °C resulted in a significant improvement in conversion to 51% after 1 h (Table 1, Entries 9–11); for each system examined the selectivity profile was similar to that obtained with [Ir(COD)(OMe)]2. Finally, as high conversions have also been obtained for the borylation of benzene using catalysts generated from [Ir(COD)(OMe)]2 and 4,4'-di-tert-butyl-2,2'-bipyridine (dtbpy), 4,4'-dimethyl-2,2'-bipyridine (4,4'-Me2bpy) or 4,4'-dimethoxy-2,2'-bipyridine [4,4'-(OMe)2bpy] a brief survey of the influence of these ligands on conversion and selectivity was also undertaken, details of which are summarized in Table 1 (Entries 12–17). There was no significant difference between the efficiency of catalysts generated from either [Ir(COD)(OMe)]2, or [Ir(COD)(acac)] with each of the 4,4'-disubstituted 2,2'-bipyridines for a series of reactions conducted at 50 °C for 1 h. There have been reports of dramatic improvements in reactivity for the iridium-catalyzed C-H borylation of phosphines by exchanging the source of boron from bis(pinacolato)diboron (B2pin2) to pinacolborane (HBpin) [75], with the latter proving to be the reagent of choice to obtain high conversions. Under the optimum conditions identified above, the conversions of 95% and 86% obtained in hexane and dioxane, respectively with 2 equivalents of H-Bpin after 1 h at 50 °C and the corresponding selectivity profile for 4a-c matched those obtained with B2pin2 (Table 1, Entries 18–19). Variation of the HBpin:substrate ratio revealed that conversions dropped quite dramatically as the ratio was reduced to 1 (Table 1, Entry 20) while an increase in this ratio to 3 or 5 resulted in complete conversion but with significantly more bisborylation at the two meta positions such that 4b was obtained as the major product from the reaction with 5 equivalents of HBpin (Table 1, Entries 21–22). Moreover, the performance of catalyst generated from [Ir(COD)(OMe)]2 and tmphen was also shown to be affected by the order of addition of the HBpin and ligand, with systems generated by addition of HBpin before the ligand providing the highest conversions (Table entries 2 and 23) whereas those generated by addition of the ligand before the borane gave a slightly lower conversion, consistent with previous reports [101].
Interested in exploring and comparing the progress of the reaction with B2pin2 and HBpin further, the variation of conversion and composition as a function of time at 50 °C in hexane was monitored and products identified and quantified using 31P NMR spectroscopy, as the products and starting material appeared as distinct well-separated resonances (Fig. 2), the results of this study are presented graphically in Figs. 3a and 3b, respectively. The resulting composition-time profile obtained for B2pin2 using 2.5 mol% [Ir(COD)(OMe)]2 and 5 mol% tmphen shows an initial induction period followed by gradual consumption of diethyl phenylphosphonate with concomitant formation of 4a together with 4c while 4b begins to form at longer reaction times (Fig. 3a). This profile is consistent with non-selective C-H borylation as the ratio of 4a + 4b to 4c is approximately two throughout the reaction, as expected for a near statistical mixture resulting from nonselective reaction at the meta and para positions [77]. For comparison, the corresponding composition-time profile with HBpin (Fig. 3b) shows faster consumption of diethyl phenylphosphonate in the early stages of the reaction to afford 4a and 4c and a minor amount of 4b; however, longer reaction times (< 60 min) resulted in a selectivity profile similar to that obtained with B2pin2. In contrast to the induction period for the B2pin2 based borylation shown in Fig. 3a, there is no induction with HBpin; such an induction period with B2pin2 has been attributed to reduction of the 1,5-cyclooctadiene to afford the active trisboryl-tmphen catalyst and this reduction may well be markedly more facile in the presence of HBpin [111]. Indeed, the induction period associated with B2pin2 based borylations catalyzed by [Ir(COD)Cl]2/dtbpy can be eliminated by addition of a catalytic amount of HBpin to the reaction mixture [111]. In addition, a high throughput optimization of iridium-catalyzed C-H borylation demonstrated that borylations with HBpin rival those with B2pin2 provided the HBpin is added before the ligand; as the order of addition of HBpin and ligand have been shown to have a dramatic effect on catalyst performance [101].

31P NMR spectrum of a reaction mixture from the [Ir(COD)(OMe)]2/tmphen catalyzed borylation of diethyl phenylphosphonate (3) after 1 h at 50 °C in hexane showing each of the products together with unreacted 3 as four distinct well-separated resonances

a Reaction profile as a function of time for the iridium-catalyzed borylation of diethyl phenylphosphonate in hexane at 50 °C using 2.5 mol% [Ir(COD)(OMe)]2/5 mol% tmphen and a two mole equivalents of B2(pin)2 showing non-selective formation of 4a, 4b and 4c and b two mole equivalents of HBpin showing rapid consumption of diethyl phenylphosphonate with concomitant formation of 4a, 4b and 4c and ultimately borylation of 4a to afford 4b together with 4c
In contrast to the mild conditions and short reaction times (50 °C, 1 h) required to achieve complete conversion of the diethyl phenylphosphonate using 2.5 mol% [Ir(COD)(OMe)]2/tmphen, albeit to afford a nonselective mixture of meta and para regioisomers, the [Ir(COD)(OMe)]2/[P[3,5-(CF3)2C6H3]3 catalyzed arylphosphonate-directed ortho C-H borylation of diethyl phenylphosphonate was conducted at 120 °C for 24 h and gave a conversion of only 31%; this increased to 42% with the use of [Ir(COD)(acac)] as the source of iridium [70]. Such disparate activity between catalysts based on a bipyridine and a diphosphine has previously been highlighted and attributed to the basicity of the phosphine as catalysts with electron-rich phosphines exhibit low activity whereas those with electron-poor phosphines display moderate activity [106,107,108,109]. Interestingly, the C-H borylation of diethyl o-tolylphosphonate at 100 °C for 24 h using catalyst generated from [Ir(COD)(acac)] and either tmphen or 4,4'-di-tert-butyl-2,2'-bipyridine (dtbpy) resulted in selective borylation meta to the phosphonate to afford the corresponding boronic ester in 38% and 41% yield, respectively; of course, the severe conditions employed for this reaction raises the question of whether the presence of the 2-methyl substituent necessitated this high temperature or might the reaction have occurred under the milder conditions described here? The ability of phosphonate to act as a directing group for iridium-catalyzed C-H borylation clearly depends on the catalyst formulation as this study has shown that [Ir(COD)(acac)]/tmphen is nonselective and affords a statistical mixture of meta- and para-phosphonate phenylboronic esters while [Ir(COD)(acac)]/[P[3,5-(CF3)2-C6H3]3 is selective for ortho-borylation, albeit it under markedly different conditions. We speculate that the active 16e iridium-trisboryl species [Ir(Bpin)3(tmphen)] would not be able to coordinate the phosphonate during the C-H bond activating oxidative addition/σ-bond metathesis step whereas the corresponding 14e monophosphine-coordinated species would have the capacity to expand its coordination sphere to coordinate the phosphonate and thereby direct the C-H activation.
2.2 Iridium-Catalyzed Borylation of 3-Substituted Phosphonates
Having established that the borylation of diethyl phenylphosphonate using iridium catalysts supported by bipyridine ligands was non-selective, 3-substituted phosphonates were identified as the substrates of choice for further studies, reasoning that C-H borylation of these substrates would be under steric control and, as such, would occur with high regioselectivity for meta-substitution [99, 100, 110]. Following the optimum conditions identified above for diethyl phenylphosphonate, a series of borylations were conducted with B2pin2 and aryl phosphonates 2a-c in hexane at 50 °C using 5 mol% catalyst generated from (1,5-cyclooctadiene)(methoxy)iridium(I) dimer and tmphen in order to identify a suitable reaction time; however, only low yields were obtained at this temperature even after extending the reaction time to 20 h. Gratifyingly, when the reaction temperature was raised to 70 °C, high conversions were obtained and the optimum reaction time was determined by monitoring the variation in conversion and composition as a function of time for the borylation of 2a-c using a combination of 1H and 31P NMR spectroscopy; under these conditions 18 h was required to reach complete conversion for each substrate to afford the corresponding meta-phosphonate arylboronic esters 5a-c as the sole regioisomer (Table 2) as shorter reaction times resulted in a mixture of 2a-c and 5a-c. Under these conditions, 5a-c were obtained in high purity after filtration through a short silica plug and did not require further purification prior to modification by Suzuki–Miyaura cross-coupling; this is a particular advantage as it simplifies the procedure and avoids undesired loss of material due to the instability of boronic esters during column chromatography.
While early optimization studies by Ishiyama, Takagi, Hartwig and Miyaura reported that the efficiency of iridium-catalyzed borylations were solvent dependent and that reactions conducted in hexane gave higher conversions than those in either DME or DMF [78], good conversions have recently been reported for phosphine and phosphonate directed C-H borylations conducted in more polar systems including THF and THF/dioxane [72, 74]. As such a comparison of the conversions obtained in THF with those in hexane showed that slightly lower conversions were obtained in THF over the same time (Table 2); this is entirely consistent with a recent high throughput optimization of iridium-catalyzed C-H borylations [101]. A comparison of the performance of in situ generated catalyst with preformed pre-catalyst revealed that polar solvents are suitable for borylations and that the lower conversion obtained with in situ generated catalyst is due the efficiency of catalyst assembly in different solvents rather than the performance of the actual catalyst [101, 102]
As for the borylation of 3, borylation of 2a-c with HBpin under otherwise identical conditions resulted in good conversions over the same time but the yields were marginally lower than those obtained with B2pin2 and the performance of catalyst was also affected by the order of addition of the HBpin and ligand such that the highest conversions were obtained when the HBpin was added before the ligand. Interestingly, while formation of the meta-phosphonate-substituted arylboronic esters 5a-c as the sole product was anticipated, Clark has recently investigated aryl phosphonate-directed ortho C-H borylation and demonstrated that catalysts generated from 3,4,7,8-tetramethyl-1,10-phenanthroline, 4,4'-di-tert-butyl-2–2'-bipyridine or 2-picolylamine and [Ir(COD)(acac)] were also selective for borylation of diethyl o-tolylphosphonate at the meta position whereas phosphine-based catalysts were exclusively selective for ortho C-H borylation [70].
2.3 Palladium Catalyzed Suzuki-Miyaura Cross-Coupling of meta-Phosphonate Substituted Arylboronic Esters
Having already demonstrated that ortho-phosphonate aryl and naphthylboronic esters can be used as nucleophilic partners in the Suzuki–Miyaura cross-coupling [69], the same protocol was applied to diversify 5a-c into a series of substituted biaryl monophosphonates by reaction with a range of aryl bromides. A brief optimization study using 5a and 4-bromoacetophenone as the benchmark combination with a 0.5 mol% loading of XPHOS-based pre-catalyst 6, revealed that the highest conversions were obtained in a 2:1 mixture of THF and water at 70 °C with K3PO4 as base, while reactions conducted in DME, dry THF and toluene under otherwise identical conditions resulted in much lower conversions. A survey of the base revealed that similar conversions were also obtained with K2CO3, Cs2CO3 and NaOH whereas NaOAc, CsF and NBu3 each gave poor conversions. Under these conditions, 6 catalyzed the reaction between electron-rich, electron-poor, and sterically hindered electrophilic partners to afford reasonable yields of the corresponding biaryl monophosphonates as spectroscopically pure oils or solids after purification by column chromatography (Table 3). For example, the yields of biaryl monophosphonate obtained from the reaction between sterically demanding substrates such as 2-bromotoluene or 1-bromonaphthalene and 5a-c matched those obtained with electron deficient substrates. In all case, minor amounts of 2a-c (7–12%) resulting from protodeboration of 5a-c were recovered during purification. At this stage, as the yields are only moderate speculation as to the origin of minor differences in conversions between the various substate combinations is not warranted and further studies will focus on identifying more efficient catalysts and protocols to improve the yields.
3 Conclusions
The iridium-catalyzed C-H borylation of diethyl phenylphosphonate with B2pin2 using catalyst generated from either [Ir(COD)(acac)] or [Ir(COD)(OMe)]2 and tmphen occurs under mild conditions but is nonselective and affords a statistical mixture of meta- and para-borylated products; in contrast, the phosphonate acts as a ortho-directing group with catalyst generated from [Ir(COD)(acac)] and [P[3,5-(CF3)2-C6H3]3 i.e. the ability of phosphonate to act as a directing group depends on the catalyst formulation which is probably associated with the capacity of the active trisboryl species to expand its coordination sphere to accommodate the phosphonate during the C-H activation step. This borylation protocol was also applied to the synthesis of a series of meta-phosphonate-substituted arylboronic esters via a sterically-controlled regioselective C-H borylation of the corresponding 3-substituted aryl phosphonates; the resulting boronate esters were then used as nucleophilic coupling partners in a subsequent palladium-catalyzed Suzuki–Miyaura cross-coupling. This protocol avoids the use of pre-functionalized aryl phosphonates to prepare biaryl monophosphonates and the substitution pattern obtained with this C-H borylation-cross coupling sequence complements that accessible via the phosphonate directed ortho C-H borylation recent developed by Clark; taken together these sequences will provide access to a diverse range of highly substituted biaryl monophosphonates. To this end, studies are currently underway to improve the efficacy of the C-H borylation, undertake kinetic investigations and optimize the Suzuki–Miyaura cross-coupling step, extend the methodology to the synthesis of a much broader range of aryl and heteroaryl monophosphonates and extend the phosphonate-based nucleophilic coupling partners to zinc and magnesium reagents.
4 Experimental
4.1 General Procedures
All chemicals were purchased from commercial suppliers and used without further purification except where indicated. Air-sensitive materials were manipulated using Schlenk line techniques and flame-dried glassware under nitrogen. Where necessary, solvents were dried prior to use. Dichloromethane was distilled from calcium hydride; ethanol from magnesium; 1,4-dioxane and tetrahydrofuran (THF) from sodium/benzophenone; toluene and hexane from sodium. IR spectra were recorded on a Varian 800 FT-IR Scimitar Series infrared spectrometer. 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded on a JEOL ECS 400, Bruker Avance 300, Jeol Lambda 500, or a Bruker Avance 700 MHz instrument. 1H and 13C NMR were referenced against CDCl3. Mass spectrometry analysis was performed in-house using a Micromass LCT premier Mass Spectrometer in Electrospray (ES) mode or by the National Mass Spectrometry Facility in Swansea. Thin layer chromatography was performed on EM reagent 0.25 mm silica gel 60F254 plates. Visualization was accomplished with UV light and aqueous potassium permanganate (VII) solution. Column chromatography of all purified products was carried out using Fluorochem LC3025 (40–63 μm) silica gel. CHN analysis was performed on a Carlo Erba 1108 Elemental Analyser and controlled with Carlo Erba Eager 200 software.
4.2 Experimental Procedures
4.2.1 General Procedure for the Synthesis of Phosphonates 1a-c
A flame-dried nitrogen-filled Schlenk flask was charged with palladium acetate (0.07 g, 0.3 mmol) and triphenylphosphine (0.817 g, 3.10 mmol). Aryl bromide (15.6 mmol), diisopropylethylamine (17.0 mL, 97.3 mmol) and diethyl phosphite (10.0 mL, 77.9 mmol) and dry ethanol (60 mL) were then added, and the reaction mixture was refluxed for 48 h. The reaction mixture was left to cool to room temperature and diluted by the addition of diethyl ether (200 mL). Hydrochloric acid (1 M, 100 mL) was added to wash the reaction mixture. The resulting aqueous layer was extracted with diethyl ether (3 × 200 mL) and the diethyl ether extracts were washed with aqueous sodium hydroxide (2.5 M, 50 mL), brine (50 mL), dried (MgSO4), filtered and the solvent removed under reduced pressure afford a yellow oil. The crude product was purified by column chromatography (ethyl acetate: petrol 2:1).
4.2.2 Methyl 3-(diethoxyphosphoryl)benzoate (2a)
Prepared according to the general procedure described above using palladium acetate (0.07 g, 0.3 mmol), triphenylphosphine (0.817 g, 3.10 mmol), methyl 3-bromobenzoate (3.35 g, 15.6 mmol), diisopropylethylamine (17.0 mL, 97.3 mmol), diethyl phosphite (10.0 mL, 77.9 mmol) and dry ethanol (60 mL). The crude mixture was purified by column chromatography (ethyl acetate: hexanes 2:1) to afford 2a as colourless oil in 76% yield (3.23 g, 11.9 mmol). 1H NMR (300 MHz, CDCl3): δ8.39 (dtd, J = 13.8, 1.7, 0.5 Hz, 1H, Ar–H) 8.13 (ddd, J = 7.9, 3.0, 1.3 Hz, 1H, Ar–H), 7.93 (ddt, J = 12.9, 7.6, 1.4 Hz, 1H, Ar–H), 7.49 (tdd, J = 7.7, 4.0, 0.4 Hz, 1H, Ar–H), 4.14 – 3.98 (m, 4H, -OCH2CH3), 3.85 (s, 3H, C(O)(OCH3), 1.25 (td, J = 7.1, 0.4 Hz, 6H, OCH2CH3); 13C NMR (125 MHz, CDCl3): δ166.0 (d, J = 2.0 Hz), 135.9 (d, J = 10.0 Hz), 133.2 (d, J = 2.8 Hz), 132.7 (d, J = 10.9 Hz), 130.4 (d, J = 15.1 Hz), 130.2 (d, J = 189.8 Hz), 128.7 (d, J = 14.9 Hz), 62.3 (d, J = 5.5 Hz), 52.3 (s), 16.3 (d, J = 6.4 Hz); 31P NMR (162 MHz, CDCl3): δ 17.1 (s); IR (neat) (cm−1): 3043.7, 2983.9, 2955.0, 2906.3, 1724.6, 1579.7, 1560.5, 1156.3, 990.2; Rf (ethyl acetate: hexanes, 2: 1) = 0.31; HRMS (ES+): exact mass calculated for C12H18O5P [M + H]+ requires m/z = 273.0892, found m/z = 273.0891.
4.2.3 Diethyl m-Tolylphosphonate (2b)
Prepared according to the general procedure described above using palladium acetate (0.07 g, 0.3 mmol), triphenylphosphine (0.817 g, 3.10 mmol), 3-bromotoluene (1.90 mL, 15.6 mmol), diisopropylethylamine (17.0 mL, 97.3 mmol), diethyl phosphite (10.0 mL, 77.9 mmol) and dry ethanol (60 mL). The crude mixture was purified by column chromatography (ethyl acetate: hexanes 2:1) to afford 2b as colourless oil in 84% yield (2.99 g, 13.1 mmol). 1H NMR (300 MHz, CDCl3): δ 7.58 – 7.48 (m, 2H, Ar–H), 7.29 – 7.26 (m, 2H, Ar–H), 4.11 – 3.95 (m, 4H, -OCH2CH3), 2.31 (s, 3H, -CCH3), 1.24 (t, J = 7.1 Hz, 6H, -OCH2CH3); 13C NMR (125 MHz, CDCl3): δ 138.2 (d, J = 14.9 Hz), 133.1 (d, J = 3.2 Hz), 132.2 (d, J = 10.0 Hz), 128.7 (d, J = 9.7 Hz), 128.3 (d, J = 15.8 Hz), 128.1 (d, J = 186.8 Hz), 62.0 (d, J = 5.4 Hz), 21.3 (s), 16.3 (d, J = 6.5 Hz); 31P NMR (162 MHz, CDCl3): δ 19.3 (s); IR (neat) (cm−1): 3043.7, 2981.8, 2905.8, 1724.6, 1579.7, 1582.5, 1150.9, 991.5; Rf (ethyl acetate: hexanes, 2: 1) = 0.34; HRMS (ES+): exact mass calculated for C11H17O3P [M + Na]+ requires m/z = 251.0813, found m/z = 251.0814.
4.2.4 Diethyl (3-(Trifluoromethyl)phenyl)phosphonate (2c)
Prepared according to the general procedure described above using palladium acetate (0.07 g, 0.3 mmol), triphenylphosphine (0.817 g, 3.10 mmol), 3-bromobenzotrifluoride (2.18 mL, 15.6 mmol), diisopropylethylamine (17.0 mL, 97.3 mmol), diethyl phosphite (10.0 mL, 77.9 mmol) and dry ethanol (60 mL). The crude mixture was purified by column chromatography (ethyl acetate: hexanes 2:1) to afford 2c as colourless oil in 91% yield (4.01 g, 14.2 mmol). 1H NMR (300 MHz, CDCl3): δ 8.00 (d, J = 13.7 Hz, 1H, Ar–H), 7.93 (dd, J = 13.0, 7.6 Hz, 1H, Ar–H), 7.73 (d, J = 7.8 Hz, 1H, Ar–H), 7.54 (td, J = 7.8, 3.8 Hz, 1H, Ar–H), 4.16—3.99 (m, 4H, -OCH2CH3), 1.26 (t, J = 7.2 Hz, 6H, -OCH2CH3); 13C NMR (125 MHz, CDCl3): δ 134.8 (d, J = 9.4 Hz), 130.8 (dd, J = 32.9 and 15.7 Hz), 130.0 (d, J = 190.4 Hz), 129.1 (s), 129.1 – 128.7 (m), 128.4 (dq, J = 11.2 and 3.6 Hz), 123.5 (qd, J = 272.6 and 2.2 Hz), 62.4 (d, J = 5.5 Hz), 16.1 (d, J = 6.3 Hz); 31P NMR (162 MHz, CDCl3): δ 16.3 (s) IR (neat) (cm−1): 3049.1, 2985.7, 2908.7, 1608.3, 1153.5, 989.5; Rf (ethyl acetate: petrol, 2: 1) = 0.29; HRMS (ES+): exact mass calculated for C11H14O3PF3 [M + H]+ requires m/z = 283.0711, found m/z = 283.0705.
4.2.5 General Procedure for the Iridium-Catalyzed Borylation of Diethyl Phenylphosphonate (3) with B2pin2
A flame-dried nitrogen-filled Schlenk flask was charged with iridium precursor (2.5 mol%), ligand (2.5 mol%) and bis(pinacolato)diboron (0.069 g, 0.28 mmol) or pinacolborane (0.0947 g, 0.74 mmol). Diethyl phenylphosphonate (0.090 g, 0.085 mL, 0.37 mmol) and dry solvent (4 mL) were then added, and the reaction mixture stirred for the allocated time at the specified temperature. After this time, the resulting solution was left to cool to room temperature, diluted by the addition of acetonitrile (2 mL), the resulting solution passed through a silica plug and the solvent removed under reduced pressure. The crude product was dissolved in CDCl3 and analyzed by 31P and 1H NMR spectroscopy to quantify the composition and determine the selectivity profile.
4.2.6 General Procedure for the Iridium-catalyzed Borylation of Diethyl Phenylphosphonate (3) with HBpin
Addition of tmphen before HBpin- A flame-dried nitrogen-filled Schlenk flask was charged with [Ir(COD)(OMe)]2 (0.0061 g, 0.0092 mmol, 2.5 mol%), tmphen (0.0043 g 0.018 mmol, 5 mol%) and dry hexane (4 ml). After stirring for ca. two minute, pinacolborane (0.0947 g, 0.74 mmol) was added followed by diethyl phenylphosphonate (0.090 g, 0.085 mL, 0.37 mmol) and the reaction mixture stirred for the allocated time at the specified temperature. After this time, the resulting solution was left to cool to room temperature, diluted by the addition of acetonitrile (2 mL), the resulting solution passed through a silica plug and the solvent removed under reduced pressure. The crude product was dissolved in CDCl3 and analyzed by 31P and 1H NMR spectroscopy to quantify the composition and determine the selectivity profile.
Addition of HBpin before tmphen- A flame-dried nitrogen-filled Schlenk flask was charged with [Ir(COD)(OMe)]2 (0.0061 g, 0.0092 mmol, 2.5 mol%), pinacolborane (0.0947 g, 0.74 mmol) and dry hexane (4 ml). After stirring for ca. two minute, tmphen (0.0043 g 0.018 mmol, 5 mol%) was added followed immediately by diethyl phenylphosphonate (0.090 g, 0.085 mL, 0.37 mmol) and the reaction mixture stirred for the allocated time at the specified temperature. After this time, the resulting solution was left to cool to room temperature, diluted by the addition of acetonitrile (2 mL), the resulting solution passed through a silica plug and the solvent removed under reduced pressure. The crude product was dissolved in CDCl3 and analyzed by 31P and 1H NMR spectroscopy to quantify the composition and determine the selectivity profile.
4.2.7 General Procedure for the Synthesis of 5a-c via Iridium Catalyzed Borylation of 2a-c
A flame-dried nitrogen-filled Schlenk flask was charged with (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (0.0061 g, 0.00918 mmol), 3,4,7,8-tetramethyl-1,10-phenanthroline (0.00434 g, 0.0184 mmol) and bis(pinacolato)diboron (0.069 g, 0.28 mmol). The 3-substituted diethyl phenylphosphonate (0.37 mmol) and dry hexane (4 mL) were then added, and the reaction mixture stirred for 18 h at 70 °C. After this time, the resulting solution was left to cool to room temperature and diluted by the addition of acetonitrile (2 mL). The resulting solution was passed through a silica plug and the solvent removed under reduced pressure to leave a yellow oil. The crude product was used without further purification.
4.2.8 Synthesis of Methyl 3-(Diethoxyphosphoryl)-5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-yl)Benzoate (5a)
Prepared according to the general procedure described above using (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (0.0061 g, 0.00918 mmol), 3,4,7,8-tetramethyl-1,10-phenanthroline (0.00434 g, 0.0184 mmol), bis(pinacolato)diboron (0.069 g, 0.28 mmol), methyl 3-(diethoxyphosphoryl)benzoate (0.106 g, 0.37 mmol) and dry hexane (4 mL). 1H NMR (300 MHz, CDCl3): δ 8.59 (s, 1H, Ar–H), 8.51 (d, J = 13.2 Hz, 1H, Ar–H), 8.40 (d, J = 12.5 Hz, 1H, Ar–H), 4.22 – 4.03 (m, 4H, -OCH2CH3), 3.91 (s, 3H, -OCH3), 1.32 (s, 12H, Bpin-CH3), 1.30 (t, J = 7.4 Hz, 6H, -OCH2CH3); 31P NMR (162 MHz, CDCl3): δ 17.5 (s); 11B (193 MHz, CDCl3): δ 30.1; HRMS (ES+): exact mass calculated for C18H29BO7P [M + H]+ requires m/z = 399.1744, found m/z = 399.1747.
4.2.9 Synthesis of Diethyl 3-Methyl-[5-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-yl)]Phenyl Phosphonate (5b)
Prepared according to the general procedure described above using (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (0.0061 g, 0.00918 mmol), 3,4,7,8-tetramethyl-1,10-phenanthroline (0.00434 g, 0.0184 mmol), bis(pinacolato)diboron (0.069 g, 0.28 mmol), diethyl m-tolylphosphonate (0.084 g, 0.37 mmol) and dry hexane (4 mL). 1H NMR (300 MHz, CDCl3): δ 7.94 (d, J = 13.9 Hz, 1H, Ar–H), 7.70 (s, 1H, Ar–H), 7.65 (d, J = 14.5 Hz, 1H, Ar–H), 4.14 – 3.94 (m, 4H, -OCH2CH3), 2.31 (s, 3H, -CH3), 1.27 (s, 12H, Bpin-CH3), 1.21 (t, J = 7.5 Hz, 6H, -OCH2CH3); 31P NMR (162 MHz, CDCl3): δ 19.4 (s); 11B (193 MHz, CDCl3): δ 23.4; HRMS (ES+): exact mass calculated for C17H29BO7P [M + H]+ requires m/z = 355.1846, found m/z = 355.1851.
4.2.10 Synthesis of Diethyl (3-(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-yl)-5- (Trifluoromethyl)Phenyl)Phosphonate (5c)
Prepared according to the general procedure described above using iridium ([1,5-cyclooctadiene][methoxy]) dimer (0.0061 g, 0.00918 mmol) 3,4,7,8-tetramethyl-1,10-phenanthroline (0.00434 g, 0.0184 mmol) and bis(pinacolato)diboron (0.069 g, 0.28 mmol), diethyl (3-(trifluoromethyl)phenyl)phosphonate (0.104 g, 0.37 mmol) and dry hexane (4 mL). 1H NMR (300 MHz, CDCl3): δ 7.93 (d, J = 14.1 Hz, 1H, Ar–H), 7.68(s, 1H, Ar–H), 7.64 (d, J = 14.4 Hz, 1H, Ar–H), 4.13 – 3.91(m, 4H, -OCH2CH3), 2.30 (s, 3H, -CH3), 1.26 (s, 12H, Bpin-CH3), 1.19 (t, J = 7.2 Hz, 6H, -OCH2CH3); 31P NMR (162 MHz, CDCl3): δ 18.2 (s); 11B (193 MHz, CDCl3): δ 27.6; HRMS (ES+): exact mass calculated for C17H26BF3O5P [M + H]+ requires m/z = 409.1563, found m/z = 409.1565.
4.2.11 General Procedure for the Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Between m-Phosphonate-Substituted Arylboronic Esters (5a-c) and Aryl Bromides
A flame dried Schlenk flask was cooled to room temperature under vacuum, backfilled with nitrogen and charged with aryl bromide (0.551 mmol), precatalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol) and THF (4 mL). Phosphonate 5a-c (0.37 mmol) and water (2 ml) were then added, and resulting mixture heated for 18 h at 70 °C. After this time, the reaction mixture was cooled to room temperature, diluted with diethyl ether (10 mL) and filtered through a silica plug by flushing with CH2Cl2/MeOH (9/1). The solvent was removed under reduced pressure and the product purified by column chromatography ethyl acetate/hexane (2/1) as eluent.
4.2.12 Methyl 4'-Acetyl-5-(diethoxyphosphoryl)-[1,1'-biphenyl]-3-Carboxylate (7a)
The general procedure described above was followed using 4-bromoacetophenone (0.110 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol) and K3PO4 (0.17 g, 0.734 mmol), 5a (0.147 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford methyl 7a as an off-white solid in 49% yield (0.071 g, 0.181 mmol). 1H NMR (300 MHz, CDCl3): δ 8.47 (dt, J = 6.5 Hz, 1H, Ar–H), 8.27 (dt, J = 13.4 Hz, 1H, Ar–H), 8.07 (dt, J = 8.9 Hz, 4H, Ar–H), 7.75 (dt, J = 8.5 Hz, 2H, Ar–H), 4.34–4.04 (m, 4H, -CH2), 3.98 (s, 3H, -OCH3), 2.65 (s, 3H, -COCH3), 1.36 (t, J = 7.14 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 197.6 (s), 166.07 (s), 143.5, 140.8, 136.8 (s), 134.7 (d, J = 8.0 Hz), 132.0 (s), 131.8 (s), 131.6 (s), 131.4 (s), 130.9 (d, J = 188.2 Hz), 129.2 (s), 127.56(s), 62.7 (d, J = 20.3 Hz), 52.7 (s), 26.9 (s), 16.5 (d, J = 24.8 Hz); 31P NMR (162 MHz, CDCl3): δ 16.7 (s); IR (neat) (cm−1): 2986.4, 1724.4, 1682.3, 1606.6, 1539.1, 1153.1, 991.3; Rf (ethyl acetate: hexanes, 2: 1) = 0.20; Anal. calcd. for C20H23O6P; C, 61.54; H, 5.94. Found: C, 61.98; H 6.36; HRMS (ES+): exact mass calculated for C20H24O6P [M + H]+ requires m/z = 391.1311, found m/z = 391.1311.
4.2.13 Diethyl (4'-Acetyl-5-Methyl-[1,1'-Biphenyl]-3-yl)Phosphonate (7b)
The general procedure described above was followed using 4-bromoacetophenone (0.110 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 6b (0.131 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes, 2/1) to afford 7b as a white solid in 36% yield (0.046 g, 0.133 mmol). 1H NMR (300 MHz, CDCl3): δ 8.02 (dt, J = 7.8 Hz, 1H, Ar–H), 7.85 (d, J = 7.7 Hz, 1H, Ar–H), 7.67 (dt, J = 7.8 Hz, 1H, Ar–H), 7.60 (d, J = 7.1 Hz, 1H, Ar–H), 7.43 – 7.41 (m, 3H, Ar–H), 4.16–3.98 (m, 4H, -CH2), 2.57 (s, 3H, Ar-CH3), 2.41 (s, 3H, -CH3), 1.28 (t, J = 7.5 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 197.9 (s), 144.8 (d, J = 8.7 Hz), 140.4 (s), 140.3 (s), 139.3 (d, J = 8.4 Hz), 136.3 (s), 132.1 (d, J = 8.9 Hz), 129.9 (d, J = 9.1 Hz), 129.1 (s), 128.8 (d, J = 187.6 Hz), 128.8 (d, J = 8.4 Hz) 127.9 (d, J = 9.0 Hz), 127.5 (s), 62.4 (d, J = 20.3 Hz), 26.8 (s), 21.6 (d, J = 8.7 Hz), 16.6 (d, J = 26.0 Hz); 31P NMR (162 MHz, CDCl3): δ 18.7 (s); IR (neat); (cm−1): 2985.7, 1654.6, 1593.2, 1150.8, 989.9; Rf (ethyl acetate: hexanes, 2: 1) = 0.48; Anal. calcd. for C19H23O4P; C, 65.70; H, 6.69. Found: C, 66.13; H 7.07; HRMS (ES+): exact mass calculated for C19H24O4P [M + H]+ requires m/z = 347.1412, found m/z = 347.1413.
4.2.14 Diethyl (4'-Acetyl-5-(Trifluoromethyl)-[1,1'-Biphenyl]-3-yl)Phosphonate (7c)
The general procedure described above was followed using 4-bromoacetophenone (0.110 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5c (0.151 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford 7c as a colourless oil in 39% yield (0.058 g, 0.144 mmol). 1H NMR (300 MHz, CDCl3): δ 8.18 (d, J = 12.0 Hz, 1H, Ar–H), 7.98 (m, 4H, Ar–H), 7.65 (d, J = 9.0 Hz, 2H, Ar–H), 4.13 (m, 4H, -OCH2CH3), 2.59 (s, 3H, C(O)CH3), 1.30 (t, J = 6.0 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 197.4 (s), 142.9 (s), 141.3 (d, J = 15.0 Hz), 136.9 (s), 133.7 (d, J = 10.0 Hz), 131.9 (m), 130.4 (s), 129.2 (s), 127.9 (m), 127.7 (m), 127.5 (s), 123.9 (q, J = 272.0 Hz), 62.7 (d, J = 5.0 Hz), 25.8 (d, J = 6.0 Hz), 16.4 (d, J = 26.0 Hz); 31P NMR (162 MHz, CDCl3): δ 15.8 (s); IR (neat) (cm−1): 2984.5, 1685.1, 1607.1, 1564.2, 1154.1, 991.8; HRMS (ES+): exact mass calculated for C19H20F3O4P [M + H]+ requires m/z = 401.1129, found m/z = 401.1129.
4.2.15 Methyl 4'-Cyano-5-(Diethoxyphosphoryl)-[1,1'-Biphenyl]-3-Carboxylate (8a)
The general procedure described above was followed using 4-bromobenzonitrile (0.100 g, 0.551 mmol), 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5a (0.147 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford methyl 8a as an off-white solid in 41% yield (0.057 g, 0.151 mmol). 1H NMR (300 MHz, CDCl3): δ 8.48 (d, J = 12.2, 1H, Ar–H), 8.44 (s, 1H, Ar–H), 8.24 (d, J = 14.8 Hz, 1H, Ar–H), 7.77 (d, J = 1.9 Hz, 4H, Ar–H), 4.27–4.09 (m, 4H, -CH2), 3.99 (s, 3H, -OCH3), 1.58 (t, J = 7.7 Hz, 6H, -OCH2CH3); 31P NMR (162 MHz, CDCl3): δ 16.4 (s); IR (neat) (cm−1): 2985.8, 2235.7, 1682.3, 1648.9, 1539.1, 1155.8, 993.4; Rf (ethyl acetate: hexanes, 2: 1) = 0.23; Anal. calcd. for C19H20NO5P; C, 61.13; H, 5.40; N, 3.75. Found: C, 61.54; H 5.79, N, 4.12; HRMS (ES+): exact mass calculated for C19H21NO5P [M + H]+ requires m/z = 374.1157, found m/z = 374.1157.
4.2.16 Diethyl (4'-Cyano-5-Methyl-[1,1'-Biphenyl]-3-yl)Phosphonate (8b)
The general procedure described above was followed using 4-bromobenzonitrile (0.100 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5b (0.131 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford 8b as colourless oil in 34% yield (0.041 g, 0.126 mmol). 1H NMR (300 MHz, CDCl3): δ 7.75–7.71 (m, 2H, Ar–H), 7.71 (s, 1H, Ar–H), 7.66 – 7.56 (m, 2H, Ar–H), 7.37–7.35 (m, 2H, Ar–H), 7.67 (dt, J = 7.8 Hz, 1H, Ar–H), 7.60 (d, 1H, Ar–H), 7.43 – 7.41 (m, 3H, Ar–H), 4.19–4.03 (m, 4H, -CH2), 2.41 (s, 3H, Ar-CH3), 2.32 (s, 3H, -CH3), 1.28 (t, J = 7.5 Hz, 6H, -OCH2CH3); 31P NMR (162 MHz, CDCl3): δ 18.4 (s); IR (neat) (cm−1): 2983.1, 2256.6, 1593.2, 1154.7, 990.6; Rf (ethyl acetate: hexanes, 2: 1) = 0.33; HRMS (ES+): exact mass calculated for C18H21NO3P [M + H]+ requires m/z = 330.1259, found m/z = 330.1269.
4.2.17 Diethyl (4'-Cyano-5-(Trifluoromethyl)-[1,1'-Biphenyl]-3-yl)Phosphonate (8c)
The general procedure described above was followed using 4-bromobenzonitrile (0.100 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5c (0.151 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes, 2/1) to afford 8c as a colourless oil in 33% yield (0.047 g, 0.122 mmol). 1H NMR (300 MHz, CDCl3) δ 8.15 (d, 1H, J = 12.0 Hz, Ar–H), 8.02 (d, J = 12.0 Hz, 1H, Ar–H) 7.92 (s, 1H, Ar–H), 7.75–7.72 (m, 2H, Ar–H),7.68–7.65 (m, 2H, Ar–H), 4.21 – 4.01 (m, 4H, -CH2), 1.30 (t, J = 6.0 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3) δ 142.9 (s), 140.5 (d, J = 15.0 Hz), 133.7 (d, J = 10.0 Hz), 136.9 (s), 133.0 (s), 132.1 (s), 132.2–131.9 (m), 129.2 (s), 128.9—128.6 (m), 128.0 (s), 127.8 – 127.5 (m), 122.4 (q, J = 272.0 Hz), 118.4 (s), 62.8 (d, J = 6.0 Hz), 25.8 (d, J = 6.0 Hz), 16.4 (d, J = 26.0 Hz); 31P NMR (162 MHz, CDCl3) δ 15.5 (s); IR (neat) (cm−1): 2984.4, 2229.0, 1608.7, 1509.2, 1152.3, 994.2; HRMS (ES+): exact mass calculated for C18H18NF3O3P [M + H]+ requires m/z = 384.0976, found m/z = 384.0975.
4.2.18 Methyl 5-(Diethoxyphosphoryl)-2'-Methyl-[1,1'-Biphenyl]-3-Carboxylate (9a)
The general procedure described above was followed using 2-bromotoluene (0.094 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5a (0.147 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford 9a as a white solid in 49% yield (0.065 g, 0.181 mmol). 1H NMR (300 MHz, CDCl3): δ 8.37 (d, J = 13.3 Hz, 1H, Ar–H), 8.12 (s, 1H, Ar–H), 7.90 (d, J = 12.2 Hz, 1H, Ar–H), 7.35 – 6.96 (m, 4H, Ar–H), 4.25–3.97 (m, 4H, -CH2CH3), 3.87 (s, 3H, -OCH3), 2.18 (s, 3H, -CH3), 1.27 (t, J = 7.64 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 166.3 (s), 142.8 (s), 139.8 (s), 136.6 (s), 135.3 (s), 134.1 (s), 131.3 (s), 130.6 (s), 129.9 (d, J = 9.2 Hz), 129.2 (d, J = 187.6 Hz), 128.9 (s), 126.2 (s) 125.7 (s), 62.6 (d, J = 20.9 Hz), 52.6 (s), 20.5 (d, J = 8.5 Hz), 16.5 (d, J = 25.6 Hz); 31P NMR (162 MHz, CDCl3): δ 17.1 (s) IR (neat) (cm−1); 2983.1, 1727.6, 1593.2, 1155.9, 988.3; Rf (ethyl acetate: petrol, 2: 1) = 0.30; Anal. calcd. for C19H23O5P; C, 62.98; H, 6.40. Found: C, 63.34; H 6.97; HRMS (ES+): exact mass calculated for C19H24O5P [M + H]+ requires m/z = 363.1361, found m/z = 363.1366.
4.2.19 Diethyl (2',5-Dimethyl-[1,1'-Biphenyl]-3-yl)Phosphonate (9b)
The general procedure described above was followed using 2-bromotoluene (0.094 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5b (0.131 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford 9b as a colourless oil in 37% yield (0.044 g, 0.147 mmol). 1H NMR (300 MHz, CDCl3): δ 7.65 (s, 1H, Ar–H), 7.69 (d, J = 7.8 Hz, 1H, Ar–H), 7.53 (s, 1H, Ar–H), 7.32–7.21 (m, 4H, Ar–H), 4.21–4.06 (m, 4H, -OCH2CH3), 2.44 (s, 3H, -ArCH3), 2.25 (s, 3H, -CH3), 1.34 (t, J = 7.1 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 141.5 (s), 140.0 (d, J = 8.7 Hz), 137.3 (d, J = 8.9 Hz), 137.2 (d, J = 8.5 Hz), 134.2 (s), 133.8 (d, J = 8.4 Hz), 130.0 (s), 129.8 (s), 129.5 (d, J = 186.9 Hz), 128.9 (d, J = 8.6 Hz), 125.9 (s) 124.8 (s), 61.9 (d, J = 20.6 Hz), 20.2 (s), 19.7 (d, J = 8.9 Hz), 16.6 (d, J = 25.1 Hz); 31P NMR (162 MHz, CDCl3): δ 19.1 (s); IR (neat) (cm−1): 2980.6, 1595.7, 1152.3, 992.2; Rf (ethyl acetate: hexanes, 2: 1) = 0.48; HRMS (ES+): exact mass calculated for C18H23O3P [M + H]+ requires m/z = 319.1463, found m/z = 319.1465.
4.2.20 Diethyl (2'-Methyl-5-(Trifluoromethyl)-[1,1'-Biphenyl]-3-yl)Phosphonate (9c)
The general procedure described above was followed using 2-bromotoluene (0.094 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 δ mol), K3PO4 (0.17 g, 0.734 mmol), 5c (0.151 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes, 2/1) to afford 9c as an off-white solid in 35% yield (0.048 g, 0.129 mmol). 1H NMR (300 MHz, CDCl3): δ 7.98 (d, J = 12.0 Hz, 1H, Ar–H), 7.89 (d, J = 12.0 Hz, 1H, Ar–H) 7.71–7.68 (m, 1H, Ar–H), 7.44–7.41 (m, 2H, Ar–H), 7.22–7.17 (m, 5H, Ar–H), 4.23–3.99 (m, 4H, -OCH2CH3), 2.19 (s, 3H, -CH3), 1.29 (t, J = 6.0 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 143.2 (d, J = 14.0 Hz), 139.2 (s), 135.6 (d, J = 10.0 Hz), 135.2 (s), 133.0 (s), 132.3–132.0 (m), 131.3 -131.0 (m), 130.7 (s), 130.6 (s), 129.8–129.6 (m), 129.5 (s), 128.4 (s), 126.9 -126.7 (m), 126.2 (s), 123.6 (q, J = 272.0 Hz), 62.6 (d, J = 6.0 Hz), 20.3 (s), 16.4 (d, J = 6.0 Hz); 31P NMR (162 MHz, CDCl3): δ 16.3 (s); IR (neat) (cm−1): 2963.2, 1602.4, 1149.3, 996.1; HRMS (ES+): exact mass calculated for C18H21F3O3P [M + H]+ requires m/z = 373.1179, found m/z = 373.1180.
4.2.21 Methyl 3-(Diethoxyphosphoryl)-5-(Naphthalen-1-yl)Benzoate (10a)
The general procedure described above was followed using 1-bromonaphthalene (0.114 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5a (0.147 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford 10a as an off-white solid in 49% yield (0.072 g, 0.181 mmol). 1H NMR (300 MHz, CDCl3): δ 8.56 (dt, J = 14.0 Hz, 1H, Ar–H), 8.37 (s, 1H, Ar–H), 8.14 (dt, J = 13.2 Hz, 1H, Ar–H), 7.2 (t, J = 5.4 Hz, 1H, Ar–H), 7.75 (d, 1H, Ar–H), 7.57–7.42 (m, 5H, Ar–H), 4.25–4.04 (m, 4H, -OCH2CH3), 3.96 (s, 3H, -OCH3), 2.41 (s, 3H,—CH3), 1.35 (t, J = 7.8 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 166.3 (s), 141.7 (d, J = 8.7 Hz), 137.9 (s), 137.4 (s), 137.3 (s), 135.0 (s), 133.2 (s), 131.8 (s), 131.3 (d, J = 9.1 Hz), 130.9 (d, J = 187.6 Hz), 130.8 (s), 130.3 (s) 129.2 (s), 128.7 (d, J = 9.2 Hz), 127.5 (s), 126.5 (d, J = 15.2 Hz), 125.4 (d, J = 8.9 Hz), 62.7 (d, J = 20.6 Hz), 52.6 (s), 16.6 (d, J = 26.0 Hz); 31P NMR (162 MHz, CDCl3): δ 17.0 (s); IR (neat) (cm−1): 2982.9, 1725.3, 1592.1, 1152.7, 991.0; Rf (ethyl acetate: hexanes, 2: 1) = 0.27; Anal. calcd. for C22H23O5P; C, 66.33; H, 5.82. Found: C, 66.61; H 6.06; HRMS (ES+): exact mass calculated for C22H24O5P [M + H]+ requires m/z = 399.1361, found m/z = 399.1362.
4.2.22 Diethyl (3-Methyl-5-(Naphthalen-1-yl)Phenyl)Phosphonate (10b)
The general procedure described above was followed using 1-bromonaphthalene (0.114 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5b (0.131 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes, 2/1) to afford 10b as an off-white solid in 39% yield (0.051 g, 0.144 mmol). 1H NMR (300 MHz, CDCl3): δ 7.92 (d, J = 7.9 Hz, 1H, Ar–H), 7.86 (s, 1H, Ar–H), 7.82 (d, J = 8.1 Hz, 1H, Ar–H), 7.72 (m, 2H, Ar–H), 7.55–7.39 (m, 5H, Ar–H), 4.23 – 4.04 (m, 4H, -OCH2CH3), 2.48 (s, 3H, Ar-CH3), 1.35 (t, J = 7.1 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 141.7 (d, J = 8.7 Hz), 137.9 (s), 137.4 (s), 135.0 (s), 134.5 (s), 133.2 (s), 131.8 (s), 131.2 (d, J = 9.1 Hz), 130.8 (d, J = 187.6 Hz), 130.5 (s), 130.3 (s) 129.2 (s), 128.7 (d, J = 9.2 Hz), 127.5 (s), 126.5 (d, J = 15.2 Hz), 125.4 (d, J = 8.9 Hz), 62.7 (d, J = 20.6 Hz), 21.7 (s), 16.6 (d, J = 26.0 Hz); 31P NMR (162 MHz, CDCl3): δ 19.1; (s) IR (neat) (cm−1): 2982.0, 1592.8, 1149.7, 990.7; Rf (ethyl acetate: hexanes, 2: 1) = 0.40; Anal. calcd. for C21H23O3P; C, 71.17; H, 6.54. Found: C, 71.57; H 6.97; HRMS (ES+): exact mass calculated for C21H24O3P [M + H]+ requires m/z = 355.1463, found m/z = 355.1463.
4.2.23 Diethyl (3-Trifluoromethyl-5-(Naphthalen-1-yl)Phenyl)Phosphonate (10c)
The general procedure described above was followed using 1-bromonaphthalene (0.114 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5c (0.151 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes, 2/1) to afford 10c as an off-white solid in 35% yield (0.053 g, 0.130 mmol). 1H NMR (300 MHz, CDCl3): δ 8.05 (d, J = 7.8 Hz, 1H, Ar–H), 7.94 (s, 1H, Ar–H), 7.82 (d, J = 8.1 Hz, 1H, Ar–H), 7.74 (m, 2H, Ar–H), 7.55–7.39 (m, 5H, Ar–H), 4.21 – 4.04 (m, 4H, -OCH2CH3), 1.31 (t, J = 7.1 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 140.9 (d, J = 8.4 Hz), 138.1 (s), 137.5 (s), 135.0 (s), 134.6 (s), 133.7 (s), 131.8 (s), 131.4 (d, J = 9.1 Hz), 130.8 (d, J = 187.6 Hz), 130.6 (s), 130.3 (s) 129.4 (s), 128.7 (d, J = 9.2 Hz), 127.7 (s), 127.1 (d, J = 15.2 Hz), 125.1 (d, J = 8.9 Hz), 122.4 (q, J = 274 Hz), 62.4 (d, J = 20.6 Hz), 16.9 (d, J = 26.0 Hz); 31P NMR (162 MHz, CDCl3): δ 16.1; (s); Rf (ethyl acetate: hexanes, 2: 1) = 0.33; HRMS (ES+): exact mass calculated for C21H21F3O3P [M + H]+ requires m/z = 409.1180, found m/z = 409.1183.
4.2.24 Methyl 5-(Diethoxyphosphoryl)-4'-Methyl-[1,1'-Biphenyl]-3-Carboxylate (11a)
The general procedure described above was followed using 4-bromotoluene (0.100 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5a (0.147 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes 2/1) to afford 11a as a colourless oil in 47% yield (0.063 g, 0.173 mmol). 1H NMR (300 MHz, CDCl3): δ 8.33 (dt, J = 15.8 Hz, 2H, Ar–H), 8.15 (dt, J = 13.6 Hz, 1H, Ar–H), 7.35 (dd, J = 8.0 Hz, 8.7 Hz, 4H, Ar–H), 4.25–4.04 (m, 4H, -OCH2CH3), 3.96 (s, 3H, -OCH3), 2.41 (s, 3H, -CH3), 1.35 (t, J = 7.24 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 166.4 (s), 142.1 (d, J = 8.6 Hz), 138.5 (s), 136.2 (s), 134.4 (d, J = 8.6 Hz), 131.8 (s), 131.7 (s), 131.2 (d, J = 9.1 Hz), 131.1 (s), 130.7 (d, J = 187.6 Hz), 130.5 (s), 130.0 (d, J = 9.0 Hz), 127.2 (s), 62.6 (d, J = 20.6 Hz), 52.6 (s), 21.3 (d, J = 8.7 Hz), 16.5 (d, J = 25.8 Hz); 31P NMR (162 MHz, CDCl3): δ 17.3 (s); IR (neat) (cm−1): 2983.1, 1727.6, 1593.2, 1154.6, 993.4; Rf (ethyl acetate: hexanes, 2: 1) = 0.47; HRMS (ES+): exact mass calculated for C19H23O5P [M + Na]+ requires m/z = 385.1181, found m/z = 385.1182.
4.2.25 Diethyl (4',5-dimethyl-[1,1'-biphenyl]-3-yl)phosphonate (11b)
The general procedure described above was followed using 4-bromotoluene (0.094 g, 0.551 mmol), pre-catalyst 6 (0.5 mol%, 0.0015 g, 1.84 µmol), K3PO4 (0.17 g, 0.734 mmol), 5b (0.131 g, 0.37 mmol), THF (4 mL) and water (2 mL). The crude residue was purified by column chromatography (ethyl acetate: hexanes, 2/1) to afford 11b as a colourless oil in 39% yield (0.046 g, 0.144 mmol). 1H NMR (300 MHz, CDCl3): δ 7.74 (d, J = 7.7 Hz, 1H, Ar–H), 7.54 (s, 1H, Ar–H), 7.49 (s, 1H, Ar–H), 7.30 (m, 4H, Ar–H), 4.15–3.99 (m, 4H, -OCH2CH3), 2.38 (s, 3H, -CH3), 2.33 (s, 3H, -CH3), 1.27 (t, J = 7.5 Hz, 6H, -OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 140.7 (d, J = 8.6 Hz), 138.2 (s), 137.1 (s), 136.8 (s), 131.2 (d, J = 9.0 Hz), 130.8 (d, J = 187.6 Hz), 130.1 (d, J = 8.7 Hz), 128.1 (s), 127.9 (s), 127.2 (s), 126.9 (d, J = 9.1 Hz), 126.3 (s), 62.2 (d, J = 20.7 Hz), 20.6 (s), 20.3 (d, J = 8.6 Hz), 16.6 (d, J = 25.7 Hz); 31P NMR (162 MHz, CDCl3): δ 19.1 (s); IR (neat) (cm−1): 2981.2, 1597.3, 1153.7, 993.4; Rf (ethyl acetate: hexanes, 2: 1) = 0.45; HRMS (ES+): exact mass calculated for C18H24O3P [M + H]+ requires m/z = 319.1463, found m/z = 319.1463.
References
Veale CA, Damewood JR, Steelman GB, Bryant C, Gomes B, Williams J (1995) J Med Chem 38:86
Ortwine DF, Malone TC, Bigge CF, Drummond JT, Humblet C, Johnson G, Pinter GW (1992) J Med Chem 35:1345
Huang WS, Liu S, Zou D, Thomas M, Wang Y, Zhou T, Romero J, Kohlmann A, Li, F, Qi J, Cai L, Dwight TA, Xu Y, Xu R, Dodd R, Toms A, Parillon L, Lu X, Anjum R, Zhang S, Wang F, Keats J, Wardwell SD, Ning Y, Xu Q, Moran LE, Mohemmad QK, Jang HG, Clackson T, Narasimhan NI, Rivera VM, Zhu X, Dalgarno D, Shakespeare WC (2106) J Med Chem 59: 4948
Dang Q, Liu Y, Cashion DK, Kasibhatla J Sr, Jiang T, Taplin F, Jacintho JD, Li H, Sun Z, Fan Y, DaRe J, Tian F, Li W, Gibson T, Lemus R, van Poelje PD, Potter SC, Erion MD (2011) J Med Chem 54:153
Lassaux P, Hamel M, Gulea M, Delbrück HE, Mercuri PS, Horsfall L, Dehareng D, Kupper M, Frère JM, Hoffmann K, Galleni M, Bebrone C (2009) J Med Chem 54:4862
Chen XD, Kopecky DJ, Mihalic J, Jeffries S, Min X, Heath J, Deignan J, Lai S, Fu Z, Guimaraes C, She S, Li S, Johnstone S, Thibault S, Xu H, Cardozo M, Shen W, Walker N, Kayser F, Wang Z (2102) J Med Chem 55: 3837
Wydysh EA, Medghalchi SM, Vadlamudi A, Townsend CA (2009) J Med Chem 53:3317
P.J. Kamer, P.W.N.M. van Leeuwen Phosphorus (III) Ligands in Homogeneous Catalysis, Design and Synthesis (Wiley-VCH: New York, 2012).
Allcock HR, Hofmann MA, Ambler CM, Morford RV (2002) Macromolecules 35:3484
Queffélec C, Petit M, Janvier P, Knight DA, Bujoli B (2012) Chem Rev 112:3777
Troev KD (2006) Chemistry and Applications of H-Phosphonates. Elsevier, Amsterdam
Chou HH, Cheng CH (2010) Adv Mater 22:2468
Hsu FM, Chien CH, Shu CF, Lai CH, Hsieh CC, Wang KW, Chou PT (2009) Adv Funct Mater 19:2834
Gelman D, Jiang L, Buchwald SL (2003) Org Lett 5:2315
Hirao T, Masunaga T, Ohshiro Y, Agawa T (1980) Tetrahedron Lett 21:3595
Hirao T, Masunaga T, Yamada N, Ohshiro Y, Agawa T (1982) Bull Chem Soc Jpn 55:900
Montchamp JL, Dumond YR (2001) J Am Chem Soc 123:510
Peng H, Cai R, Xu C, Chen H, Shi X (2016) Chem Sci 7:6190
Kalek M, Ziadi A, Stawinski J (2008) Org Lett 10:4637
Deal EL, Petit C, Montchamp JL (2011) Org Lett 13:3270
Fu WC, So CM, Kwong FY (2015) Org Lett 17:5906
Xu K, Yang F, Zhang G, Wu Y (2013) Green Chem 15:1055
Bloomfield AJ, Herzon SB (2012) Org Lett 14:4370
Łastawiecka E, Flis A, Stankevič M, Greluk M, Słowik G, Gac W (2018) Org Chem Frontiers 5:2079
Ramírez-López P, Ros A, Estepa B, Fernández R, Fiser B, Gómez-Bengoa E, Lassaletta JM (2016) ACS Catal 6:3955
Bonnaventure I, Charette AB (2008) J Org Chem 73:6330
Zhuang R, Xu J, Cai Z, Tang G, Fang M, Zhao Y (2011) Org Lett 13:2110
Hu G, Chen W, Fu T, Peng Z, Qiao H, Gao Y, Zhao Y (2013) Org Lett 15:5362
Chen TH, Reddy DM, Lee CF (2017) RSC Adv 7:30214
Andaloussi M, Lindh J, Sävmarker J, Sjöberg PJR, Larhed M (2009) Chem Eur J 15:13069
Fu T, Qiao H, Peng Z, Hu G, Wu X, Gao Y, Zhao Y (2014) Org Biomol Chem 12:2895
Luo H, Liu H, Chen X, Wang K, Luo X, Wang K (2017) Chem Commun 53:956
Geng Z, Zhang Y, Zheng L, Li J, Zou D, Wu Y, Wu Y (2016) Tetrahedron Lett 57:3063
Zeng H, Dou Q, Li CJ (2019) Org Lett 21:1301
Yuan J, To WP, Zhang ZY, Yue CD, Meng S, Chen J, Liu Y, Yu GA, Che CM (2018) Org Lett 20:7816
Isshiki R, Muto K, Yamaguchi J (2018) Org Lett 20:1150
Dong J, Liu L, Ji X, Shang Q, Liu L, Su L, Chen B, Kan R, Zhou Y, Yin SF, Han LB (2019) Org Lett 21:3198
Liu C, Szostak M (2017) Angew Chem Int Ed 56:12718
Xu JJ, Zhang P, Gao Y, Chen Y, Tang G, Zhao Y (2013) J Org Chem 78:8176
Feng CG, Ye M, Xiao KJ, Li S, Yu JQ (2013) J Am Chem Soc 135:9322
Hou C, Ren Y, Lang R, Hu X, Xia C, Li F (2012) Chem Commun 48:5181
Khrizanforov M, Strekalova S, Khrizanforova V, Dobrynin A, Kholin K, Gryaznova T, Grinenko V, Gubaidullin A, Kadirov MK, Budnikova Y (2018) Topics in Catal 61:1949
Shaikh RS, Ghosh I, König B (2017) Chem Eur J 50:12120
Hu RB, Wang HL, Zhang HY, Zhang H, Ma YN, Yang SD (2014) Bilstein J Org Chem 10:2071–2076
Wang S, Guo R, Wang G, Chen SY, Yu XQ (2014) Chem Commun 50:12718
Li C, Yano T, Ishida N, Murakami M (2013) Angew Chem Int Ed 52:9801
Xie P, Wang J, Fan J, Liu Y, Wo X, Loh TP (2017) Green Chem 19:1235–2139
Dhokale RA, Mhaske SB (2013) Org Lett 15:2218
Zhang Z, Dixneuf PH, Soule J-F (2018) For a highly insightful review on late-stage modifications of P-containing ligands using transition-metal-catalyzed C-H bond functionalization see. Chem Commun 54:7265
Zhang H, Hu RB, Zhang XY, Li SX, Yang SD (2014) Chem Commun 50:4686
Zhao D, Nimphius C, Lindale M, Glorius F (2013) Org Lett 15:4504
Kuninobu Y, Yoshida T, Takai K (2011) J Org Chem 76:7370
Ma YN, Zhang HY, Yang SD (2015) Org Lett 17:2034
Hu RB, Zhang H, Zhang XY, Yang SD (2014) Chem Commun 50:2193
Zhang HY, Yi HM, Wang GW, Yang B, Yang SD (2013) Org Lett 15:6186
Yuan J, Yue CD, Zhang ZY, Chen J, Yu GA, Che CM (2018) Org Lett 20:1810
Li SX, Ma YN, Yang SD (2017) Org Lett 19:1842
Yin J, Buchwald SL (2000) J Am Chem Soc 122:12051
Shen X, Jones GO, Watson DA, Bhayana B, Buchwald SL (2010) J Am Chem Soc 132:11278
Tang W, Patel ND, Xu G, Xu X, Savoie J, Ma S, Hao MH, Keshipeddy S, Capacci AG, Wei X, Zhang Y, Gao JJ, Li W, Rodriguez S, Lu BZ, Yee NK, Senanayake CH (2012) Org Lett 14:2258
Wu W, Wang S, Zhou Y, Yue YH, Lanning Z, Wan LP, Wang L, Zhou Z, Qiu L (2012) Adv Synth Catal 354:2395
Yamamoto T, Akai Y, Nagata Y, Suginome M (2011) Angew Chem Int Ed 50:8844
Wang S, Li J, Miao T, Wu W, Li Q, Zhuang Y, Zhou Z, Qiu L (2012) Org Lett 14:1966
Zhou Y, Wang S, Wu W, Li Q, He Y, Zhuang Y, Li L, Pang J, Zhou Z, Qiu L (2013) Org Lett 15(15):5508
Zhou Y, Zhang X, Liang H, Cao Z, Zhao X, He Y, Wang S, Pang J, Zhou Z, Ke Z, Qiu L (2014) ACS Catal 4:1390
Zhang Y, Li Y, Pan B, Xu H, Liang H, Jiang X, Liu B, Tse MK, Qiu L (2019) Chemistry Select 4:5122
Ma YN, Yang SD (2016) Chem Record 16:977
D.G. Hall, Boronic Acids (Wiley-VCH; Weinheim, 2011).
Doherty S, Knight JG, Ward NAB, Perry DO, Bittner DM, Probert MR, Westcott SA (2014) Organometallics 33:5209
Xu F, Duke OM, Rojas D, Eichelberger HM, Kim RS, Clark TB, Watson DA (2020) J Am Chem Soc 142:11988
Wen J, Wang D, Qian J, Wang D, Zhu C, Zhao Y, Shi Z (2019) Angew Chem Int Ed 58:2078
Qiu X, Wang M, Zhao Y, Shi Z (2017) Angew Chem Int Ed 56:7233
Qiu X, Wang M, Zhao Y, Shi Z (2019) Angew Chem Int Ed 58:2850
Crawford KM, Ramseyer TR, Daley CJA, Clark TB (2014) Angew Chem Int Ed 53:7589
Wright SE, Richardson-Solorzano S, Stewart TN, Miller CD, Morris KC, Daley CJA, Clark TB (2019) Angew Chem Int Ed 58:2834
Luo X, Yuan J, Yue CD, Zhang ZY, Chen J, Yu GA, Che CM (2018) Org Lett 20:1810
Ishiyama T, Takagi J, Sihida K, Miyaura N, Anastasi NR, Hartwig JF (2002) J Am Chem Soc 124:390
Ishiyama T, Takagi J, Hartwig JF, Miyaura N (2002) Angew Chem Int Ed 41:3056
Cho JY, Tse MK, Holmes D, Maleczka RE, Smith MR (2002) Science 295:305
Maleczka RE, Shi F, Holmes D, Smith MR (2002) J Am Chem Soc 125: 7792.
Holmes D, Chotana GA, Maleczka RE, Smith MR (2006) Org Lett 8:1407
Tzschucke CC, Murphy JM, Hartwig JF (2007) Org Lett 9:761
Murphy JM, Tzschucke CC, Hartwig JF (2007) Org Lett 9:757
Murphy JM, Liao X, Hartwig JF (2007) J Am Chem Soc 129:15434
Liskey CW, Liao X, Hartwig JF (2010) J Am Chem Soc 132:11389
Robbins DW, Hartwig JF (2013) Angew Chem Int Ed 125:967
Joliton A, Carreira EM (2013) Org Lett 15:5147
Li X, Deng X, Coyne AG, Srinivasan R (2019) Chem Eur J 25:8018
Feng Z, Min QQ, Fu XP, An L, Zhang X (2017) Nature Chem 9:918
Litvinas ND, Fier PS, Hartwig JF (2012) Angew Chem Int Ed 51:536
Liu T, Shao X, Wu Y, Shen Q (2012) Angew Chem Int Ed 51:540
Chotana GA, Bastidas JRM, Miller SL, Smith MR, Maleczka RE (2020) Molecules 25: 1754.
Wang J, Torigoe T, Kuninobu Y (2019) Org Lett 21:1342
Kuninobu Y, Ida H, Nishi M, Kanai M (2015) Nature Chem 7:712
Davis HJ, Mihai MT, Phipps RJ (2016) J Am Chem Soc 138:12759
Bisht R, Chattopadhyay B (2016) J Am Chem Soc 138:84
Yang L, Uemura N, Nakao Y (2019) J Am Chem Soc 141:7972
Gao P, Szostak M (2020) Org Lett 22:6010
Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF (2010) Chem Rev 110:890
Hartwig JF (2012) Acc Chem Res 45:864
Preshlock SM, Ghaffari B, Maligres PE, Krska SW, Maleczka RE, Smith MR (2013) J Am Chem Soc 125:7572
Liskey CW, Hartwig JF (2012) J Am Chem Soc 134:12422
Larsen MA, Hartwig JF (2014) J Am Chem Soc 136:4287
Liskey CW, Hartwig JF (2013) J Am Chem Soc 135:3375
Li Q, Liskey CW, Hartwig JF (2014) J Am Chem Soc 136:8755
Liskey CW, Wei CS, Pahls DR, Hartwig JF (2009) Chem Commun 5603.
Ishiyama T, Isou H, Kikuchi T, Miyaura N (2010) Chem Commun 46:159
Ghaffari B, Preshlock SM, Plattner DL, Stapls RJ, Maligres PE, Krska SW, Maleczka Smith MR (2014) J Am Chem Soc 136:14345
Chotana GA, Vanchura BA, Tse MK, Staples RJ, Maleczka RE, Jr, Smith MR, III. (2009) Chem Commun 5731.
Robbins DW, Hartwig JF (2013) Angew Chem Int Ed 52:933
Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF (2005) J Am Chem Soc 127:14263
Acknowledgements
This study was funded by Newcastle University (D.O.P). We also gratefully acknowledge Taibah University, Saudi Arabia for a scholarship to H.Y.A. High resolution mass spectra were obtained at the ESPRC National Mass Spectrometry Service in Swansea. This article is dedicated to the memory of Elwood and Jake, may they rest in peace.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Doherty, S., Knight, J.G., Tran, T.S.T. et al. The Synthesis of Biarylmonophosphonates via Palladium-Catalyzed Phosphonation, Iridium-Catalyzed C-H Borylation, Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling. Catal Lett 152, 398–413 (2022). https://doi.org/10.1007/s10562-021-03643-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03643-3