
Occurrence and Release of Trace Elements in Pyrite-Rich Waste Rock


1
Applied Geochemistry, Luleå University of Technology, 971 87 Luleå, Sweden
2
Ore Geology, Luleå University of Technology, 971 87 Luleå, Sweden
*
Author to whom correspondence should be addressed.
Minerals 2021, 11(5), 495; https://doi.org/10.3390/min11050495
Submission received: 28 March 2021
/
Revised: 2 May 2021
/
Accepted: 4 May 2021
/
Published: 7 May 2021
(This article belongs to the Special Issue Environmental Geochemistry in the Mining Environment)
Abstract
:Waste rock can contain high concentrations of deleterious trace elements, which upon oxidation can be released, having a significant impact on water quality. Therefore, knowledge about their occurrence and overall mobility is crucial to ensure suitable environmental protection measures. Sulfide-rich waste rock was characterized and quantified using automated mineralogy (QEMSCAN). Selected pyrite grains were analyzed for trace element occurrence using LA-ICP-MS before, during, and after leaching the waste rock in 10 L small-scale test cells for two years to assess trace element occurrence and mobility. Sequential extraction was used to estimate elemental sequestration during the experiment. The high abundance of pyrite (66%) and scarcity of buffering minerals resulted in low pH (<1.3) leachate with high concentrations of trace elements such as As (21 mg/L), Cu (20 mg/L), Hg (13 µg/L, Pb (856 µg/L), Sb (967 µg/L), Tl (317 µg/L ), and Zn (23 mg/L) in solution with limited retention in secondary minerals, primarily due to these elements’ association with pyrite either as inclusions or impurities showing an average abundance of 193 ppm As, 15 ppm Cu, 13 ppm Hg, 20 ppm Pb, 24 ppm Sb, 26 ppm Tl, and 74 ppm Zn in the waste rock. The occurrence of Cu and Zn as inclusions associated with the pyrite led to their extensive mobilization of 79% and 72%, respectively, despite their low abundance in the waste rock. Provided the overall leachability of S (11%) and limited formation of secondary minerals, the average oxidation rate suggests depletion of the pyrite within approximately 18 years. In conclusion, this study shows the importance of detailed mineralogical investigations and early preventive measures of waste rock to ensure sustainable mine waste and water management.
Keywords:
LA-ICP-MS; QEMSCAN; sequential extraction; pyrite; trace elements; trace metals; sulfide oxidation; waste rock1. Introduction
The oxidation of sulfide minerals with the subsequent generation of acid rock drainage (ARD), characterized by a low pH and high concentrations of metal(loids), is considered one of the most challenging environmental consequences of mining [1,2,3]. ARD develops in sulfide-bearing mine waste where the neutralization capacity (mainly in the form of carbonate minerals) is absent, depleted due to ongoing sulfide oxidation, or when carbonate dissolution is inefficient due to limited contact between the neutralizing and acid-producing minerals.
Although mining is focused on ore mineral extraction, it is common minerals such as pyrite, ending up as mine waste, which are mainly responsible for ARD generation. As the demand for metals and minerals intensifies, ore with increasing complexity is mined, resulting in increasingly complex mine waste, which risks resulting in problematic leachate. Storage of reactive mine waste under ambient conditions until remediation results in the impending risk of accelerated sulfide oxidation and the likelihood of potentially detrimental elements being released into the environment. Hence, traditional remediation methods such as dry covers or subaqueous disposal are not suitable for highly reactive or complex mine waste because the barrier is usually applied at the end of the mine’s life. Sulfide passivation or microencapsulation by forming coatings on the sulfide surfaces capable of protecting the core from O2 and Fe3+ attacks [4] could be a solution during operation to control ARD formation. Several additives for enhancing coatings have been explored, both organic and inorganic, in solid or liquid form, sprinkled, mixed, or layers on top [1]. However, despite promising results, these methods are still only under the development stage and studies are dominated by batch-type laboratory tests focusing on single mineral systems such as pure pyrite. However, a single mineral system does not consider interactions between minerals in more complex mineral systems, such as waste rock.
A study by Nyström et al. [5] showed that small additions of lime kiln dust (LKD) (5 wt.%) could limit sulfide oxidation for more than two years in pyritic waste rock, likely through the passivation of reactive surfaces by the formation of hydrous ferric oxides (HFO). The treatment resulted in a general improvement of the leachate quality and decreased concentrations of most elements. However, to understand the effect of passivation through HFO formation and its potential long-term stability, one must first understand the geochemical behavior of the waste rock itself.
Pyrite, the most abundant mineral in waste rock [5], can host trace elements that can be either valuable or toxic to humans and the ecosystem [6], and their release can have a tremendous impact on the environment. Hence, it is important to study pyrite to show these elements’ occurrence and potential release from waste rock to determine the passivation’s potential to trap these elements, or if there is a continuous risk that these elements are released into the environment. A complete mineralogical and geochemical characterization is therefore needed to evaluate potential environmental impact. This study aimed to identify trace elements associated with pyrite and their overall mobility in waste rock to display the occurrence and mobility of trace elements from pyrite and the subsequent ARD evolution.
2. Materials and Methods
2.1. Waste Rock
The pyrite-rich waste rock originated from a Boliden Mineral AB-owned Zn–Cu–Au–Ag open-pit mine in northern Sweden. The ore deposit is a volcanic-associated massive sulfide deposit hosted in the Skellefte group submarine volcanic rocks with an age of approximately 1.89 Ga. Ore is hosted in a quartz–feldspar porphyritic rhyolite, which occurs as discrete occurrences in the mining area. Since mining began in 2000, 9.9 million tons of waste rock have been generated, with the expectation that 10 million tons of waste rock will be present at the end of the mine’s life; of which 9.3 million tons is predicted to be potentially acid-producing.
Alakangas et al. [7] and Nyström et al. [8] have previously described the sampling of waste rock, which included a screening of waste rock piles with a handheld X-ray Fluorescence (XRF) device from Olympus Innov-x systems, USA, to select waste rock with high sulfur content. Alakangas et al. [7] characterized the waste rock for its major and minor elements, showing an average sulfur content of 30%. Nyström et al. [8] characterized waste rock by its major mineral content, showing that it was dominated by pyrite and quartz with lesser amounts of muscovite (sericite), chlorite, and calcite. The waste rock showed limited neutralizing ability correlating to the abundance of pyrite and scarcity of buffering minerals such as calcite.
ALS Vancouver, Canada, determined sulfidic sulfur for three samples using 25% hydrochloric leach followed by leco furnace melt followed by analysis of the samples by inductively coupled plasma optical emission spectroscopy (ICP-OES).
Polished uncovered thin sections (26 mm × 46 mm) of crushed waste rock (5–30 mm sieve size) were prepared by Vancouver Petrographics Ltd., Vancouver, BC, Canada, with special care taken not to dissolve water-soluble phases in the sample. Optical examination of polished thin sections was performed in reflected and transmitted light using a standard Nikon Eclipse E600POL petrographic microscope (Nikon Instruments INC., Tokyo, Japan) to characterize minerals present in the waste rock optically. Automated quantitative mineralogical characterization by QEMSCAN® 650 with two Bruker EDX detectors was performed at Boliden Mineral AB, using their mine site database as a reference. A total of four carbon-coated thin sections (each holding at least seven waste rock pieces) were analyzed. QEMSCAN step size was set to 6 µm as a compromise between the number of analysis points yielded and the time for analysis (approximately 7 h/thin section). The scan area comprised 29,585 µm × 19,175 µm with 117 field measures/thin section.
2.2. Leaching of Waste Rock
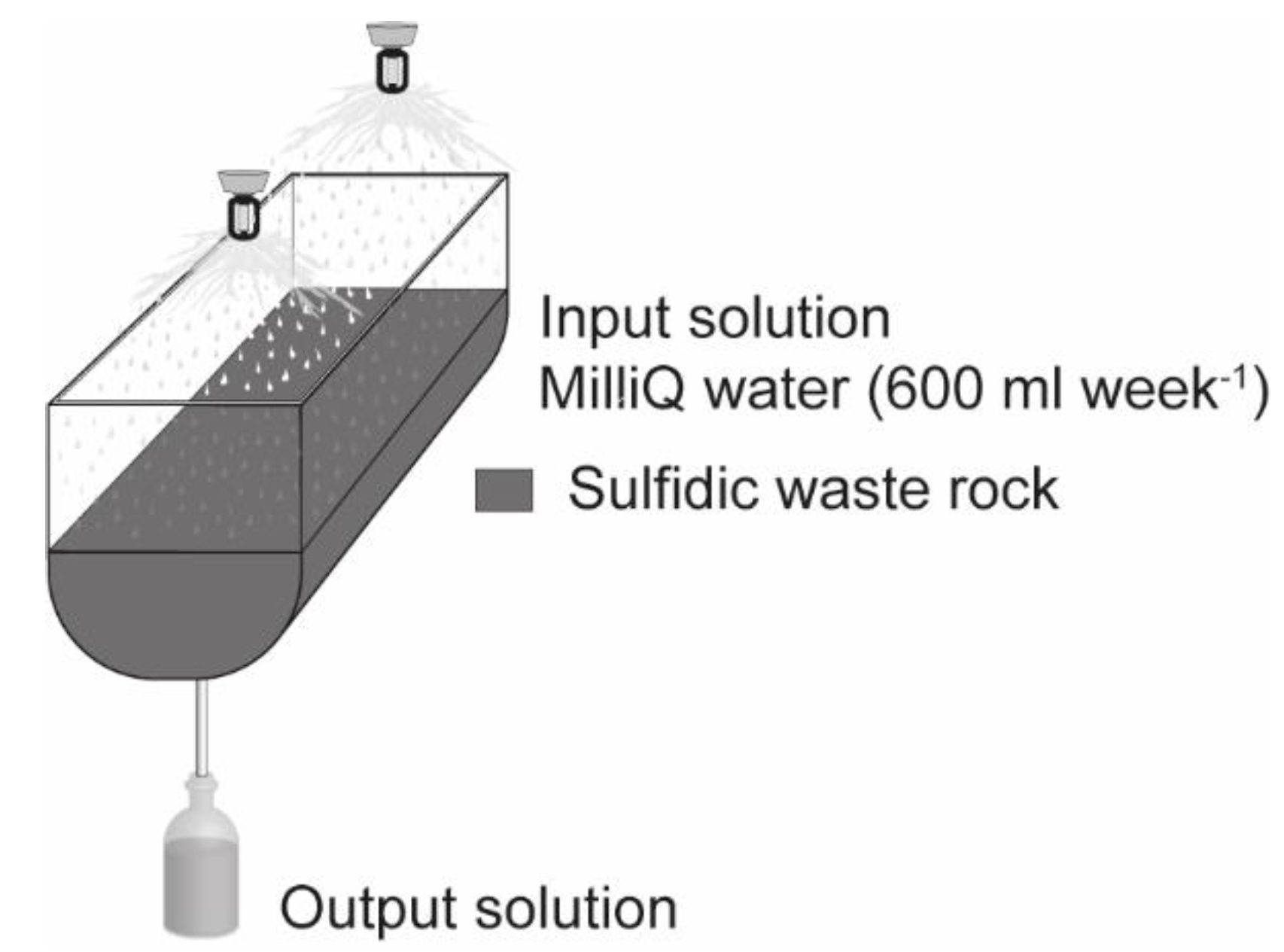
Kinetic testing was conducted in non-saturated small-scale test cells to study pyrite-rich waste rock oxidation. The experimental setup deviated from standard column testing and was chosen explicitly to study secondary mineral formation for sulfide oxidation preventive purposes [5,8]. It has been shown that precipitation of, e.g., hydrous ferric oxides (HFO) is predominant at the interface between mine waste and alkaline source [9,10]; therefore, it was desirable to have a large cell surface area. The experimental design consisted of high-density polyethylene cells with surface areas of 513 cm2 and a total volume of 10 L (Figure 1). The cells were irrigated weekly with 600 mL of MilliQ water (0.05 µS/cm), corresponding to average annual precipitation in the area and equivalent to a yearly L/S weight ratio of approximately 4. The bottom of each cell was lined with geotextile to avoid clogging of the tap. A total of four cells were constructed and filled with 7.55 kg of crushed waste rock (5–30 mm sieve size). Results from the reference cell’s long-term leaching (solely waste rock) are presented here to illustrate the extent of trace elements’ mobility related to their presence and exposure in pyrite. Three of the cells were used to prevent sulfide oxidation and have been published previously [5,8].
Solid samples (grab) were taken from the cell after leaching the waste rock for one and two years (weeks 52 and 103). Samples were taken at approximately 2 cm depth of the waste rock profile (8 cm in total) and dried at <30 °C for a total of five hours before being subjected to small amounts of pressured air. The treatment was adopted to differentiate between secondary mineral formation on the pyrite and bulk for inhibition studies [5,8]. For comparison, grab samples from the reference cell were treated similarly.
Leachates were collected from the tap located at the bottom front of the cell weekly the day after irrigation. The collected leachates’ pH, electrical conductivity (EC), and temperature were measured in closed containers to avoid air exposure. The pH and EC were measured using a WTW Multi 3420 multimeter equipped with Sentix® 940 (pH) and TetraCon® 925 (EC) electrodes. Leachate samples were filtrated through a 0.22 µm nitrocellulose membrane into high-density polyethylene bottles using vacuum filtration. The filter equipment was washed with 5% nitric acid and the filters in 5% acetic acid. Filtrated samples were stored cold (4 °C) and in darkness until analysis. Before analysis, the samples were acidified with 1 mL nitric acid (suprapur) per 100 mL sample. Selected samples were analyzed for major and trace element composition using inductively coupled plasma atomic emission spectroscopy (ICP-AES, Agilent ICP-OES 725) and inductively coupled plasma sector field mass spectrometry (ICP-SFMS, ELEMENT 1, Finnigan MAT) at the SVEDAC-accredited laboratory ALS Scandinavia in Luleå. The analysis was performed according to US EPA Method 200.7 (modified) and 200.8 (modified) or quantitative screening analysis for over 70 elements.
2.3. Trace Element Association
Trace element occurrence and distribution in pyrite grains were performed on thin sections of the initial waste rock and grab samples after one and two years of leaching (week 52 and 103). An ESI NWR 193 nm excimer laser microprobe coupled to a Thermo Scientific iCAP Q quadrupole ICPMS at Luleå University of Technology, Sweden, was used. Analyses on pyrite were performed by laser-ablating spots of 35 μm in diameter, repetition rate of 5 Hz, and laser beam energy maintained between 3 and 4 Jcm−2. Analysis time was restricted to 56 s, comprising 20 s background (laser shutter closed) and 100 laser bursts/36 s analysis (laser shutter open). Imaging of pyrite was performed by ablating a set of parallel lines arranged in a grid over the sample where line space corresponded to beam size. Beam size was 10 μm2 to investigate zoning in pyrites of less than 500 μm2. The beam was rastered over the lines at half speed of the beam size (i.e., spot = 10 μm2, speed = 5 μm/s−1), at 5 Hz repetition rate. Acquisition times were as follows: major silicate elements (i.e., Mg, Al, Si, Ca) and S = 0.005 s; major elements (i.e., Fe, Ni) = 0.005 s; Ag and Au = 0.5 s; other elements = 0.01 s; with a total sweep time of approximately 0.5 s. Data reduction was carried out using Iolite4 software [11]. Iron was used as the internal standard (assuming stoichiometric Fe of pyrite). The primary calibration standard used was NIST 612 [12], with UQAC FeSb [13], MASS1, USGS [14], and NIST 610 [12] used as secondary standards. Standards were analyzed every one-and-a-half hours to correct for instrument drift.
2.4. Secondary Mineral Formation
2.4.1. Solid-Phase Sequential Extraction
Quantitative information on trace element distribution in secondary minerals was gained by subjecting samples of the initial waste rock and oxidized waste rock (after weeks 52 and 103) to a seven-step sequential extraction at the SGS laboratory (Pittsburgh, PA, USA). The sequential extraction by Dold [15] was adapted to the specific primary and secondary mineralogy associated with copper-sulfide ores but has also been applied to other types of mine waste. Here, only the first four steps were considered (Table 1). A residual fraction was defined as the difference between the whole rock element content (NaOH fusion) and the sum of steps I–IV. Such residual fractions include mainly primary sulfide and silicate minerals [15].
2.4.2. Geochemical and Mass Calculations
Geochemical calculations, including aqueous species distribution and mineral saturation state, were carried out using PHREEQC version 3.4.0 [16] using the wateq4f.dat thermodynamic database [17]. Ion imbalance for considered samples was within ±15%. Redox potential (Eh) results were used to estimate the speciation of Fe(II) and Fe(III) reported suitable at a pH below 4 [18]. Calculations involving Fe(III) minerals, thermodynamic data for Fe(III) mineral solubility, were calculated as the following: precipitations of gypsum, ferrihydrite, goethite, and jarosite were considered as reported in the wateq4f database [17]. The solubility constant of more ordered ferrihydrites, namely 2- and 6-line, were used as reported by [19], while for schwertmannite, the solubility constant from Majzlan et al. [20], originating from Bigham et al. [21], was used.
3. Results and Discussion
3.1. Waste Rock Characterization
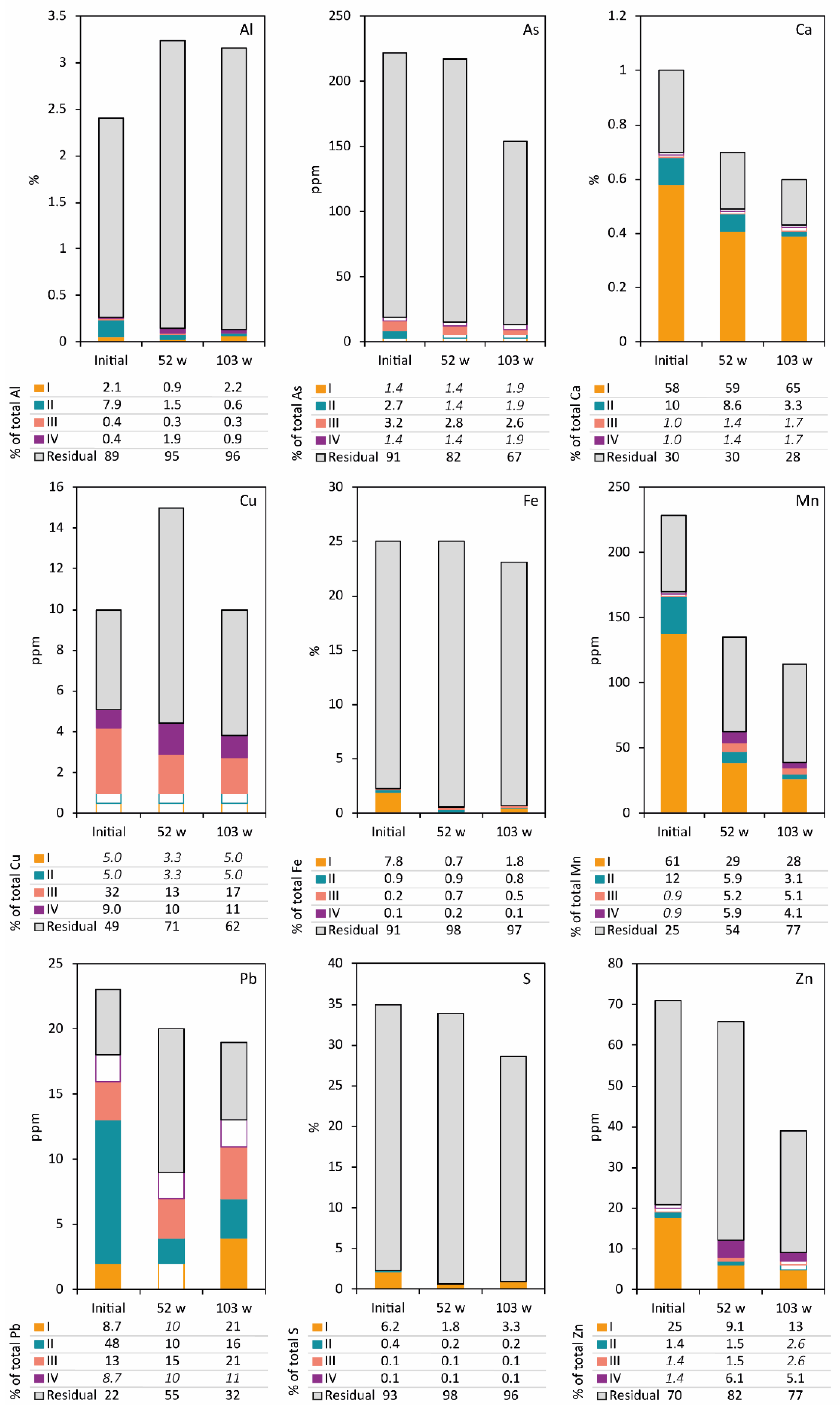
Optical microscopy followed by quantitative automated mineralogy (QEMSCAN®) revealed that the waste rock was dominated by pyrite (66%) with smaller amounts of quartz (17%), muscovite (6%), chlorite (4%), and calcite (1%). Other sulfides found at trace amounts in the waste rock included arsenopyrite (FeAsS, 1.8 × 10−4%), bournonite (PbCuSbS3, 4.1 × 10−4%), chalcopyrite (CuFeS2, 12 × 10−4%), pyrrhotite (Fe1-xS, 2.4 × 10−4%), and sphalerite (ZnS, 2.6 × 10−4%) resulting in a low overall content of As-, Cu-, Pb-, and Zn-bearing minerals in the waste rock (Table 2). Under preliminary observation in the optical microscope, no direct weathering signs were shown, despite storage under ambient conditions for approximately one year before sampling (Figure 2). However, sequential extraction tests performed on the material before leaching showed the presence of secondary minerals with approximately 9% Fe and 7% S associated with steps I–IV (Figure 3). Assuming that no primary Fe and S minerals occur in steps I–IV, the results show partial sulfide weathering of the initial waste rock. The occurrence of Al in step II displays the waste rock’s kaolinite content (Figure 3, Table 2). Primary minerals such as calcite (step II) can also be found in steps that otherwise would be assumed to display element association to secondary minerals. The comparably high presence of Ca in steps I and II and S in step I suggests the formation of gypsum (step I) derived from the weathering of calcite (step II) and sulfides. Manganese was also found in step II, suggesting its association with carbonate minerals; in this case, calcite. Figure 3 shows that most elements are associated with step I compared to steps II–IV, suggesting that the secondary minerals mainly consist of readily soluble salts such as gypsum, melanterite, and other metal sulfates. A small amount of Al, Fe, and S were found in steps III–IV, indicating the limited presence of secondary minerals such as HFO, Al hydroxides, and Al hydroxysulfates.
3.2. Sulfide Oxidation and Subsequent Element Mobilization
3.2.1. First Year
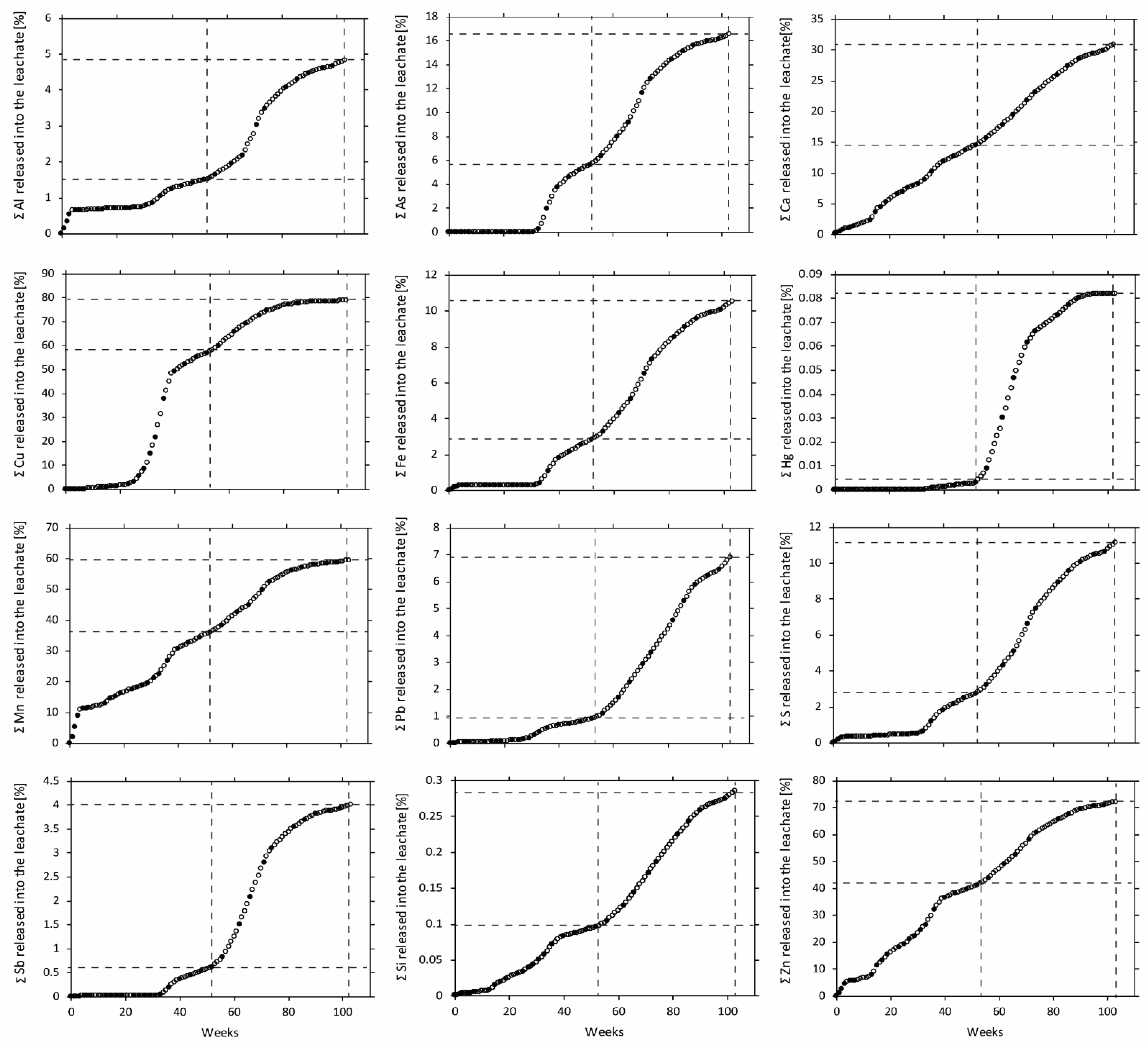
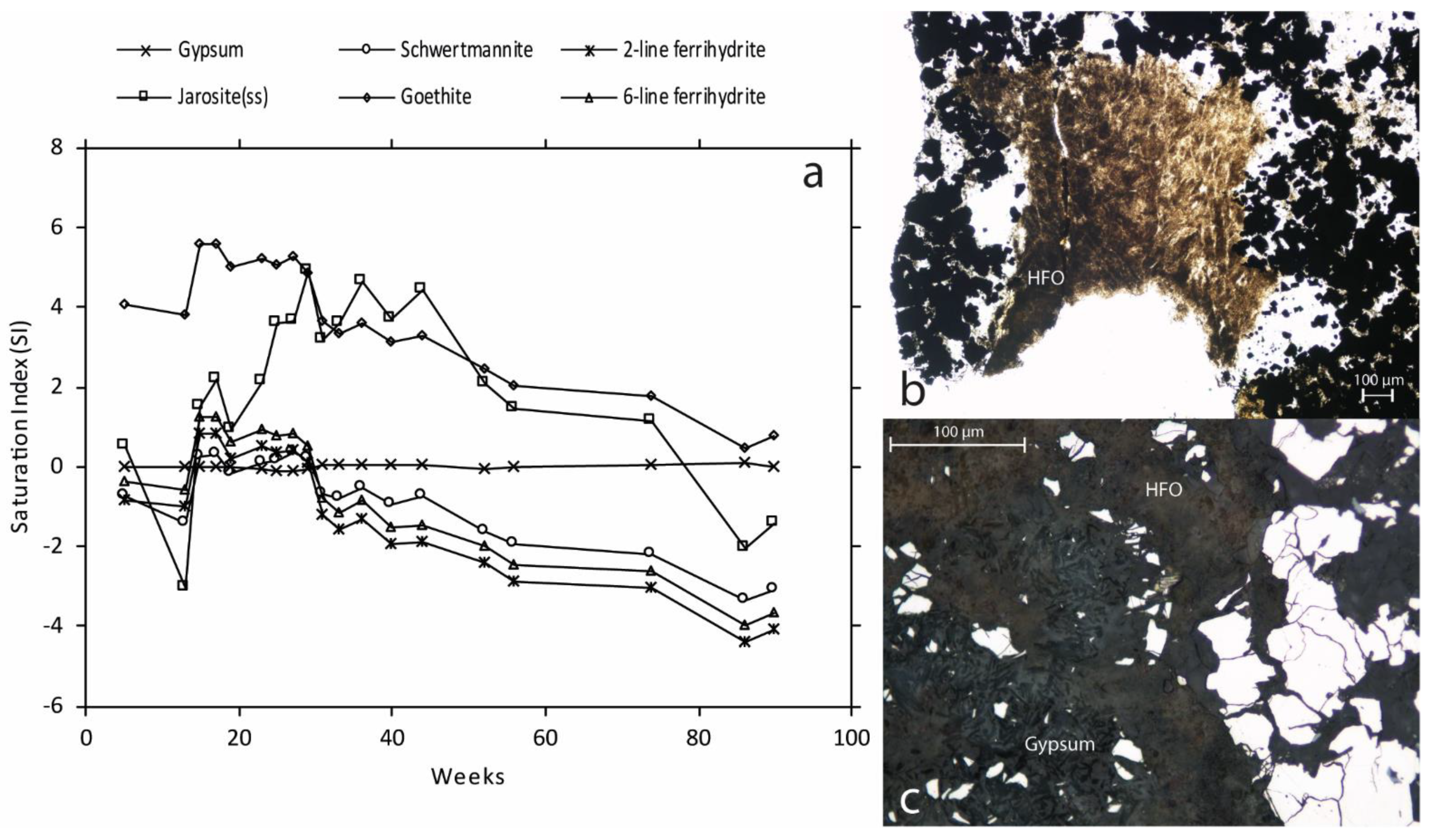
Initially, the leachate was characterized by a pH around 3 and high electrical conductivity (EC) around 25 mS/cm [5], and concentrations of Fe, Mn, and S spiked but slowly decreased, suggesting a wash-out of easily soluble minerals (Figure 4). The pH increased until week 15 to its highest value at 4.6. During this time, increasing pH and declining Fe concentrations in the leachate compared to S suggests the precipitation of HFO such as schwertmannite, 2- and 6-line ferrihydrite, supported by geochemical calculations (Figure 5a). Despite the high pyrite content and paucity of carbonate minerals in the waste rock, it took approximately 29 weeks of leaching before signs of accelerated sulfide oxidation occurred, shown by a decreasing pH (<3.5) and increasing EC along with increasing element content released into the leachate (Figure 4). After 29 weeks, Fe and S’s molar ratios changed from below 0.05 to around 0.5, indicating that the system was dominated by pyrite oxidation. The acceleration of sulfide oxidation occurred when the pH was below 3.5, when Fe3+ and acidophilic bacteria usually dominate [22,23]. At this time, Fe and S concentrations increased to concentrations between 0.24 and 18 g/L and 0.75 and 17 g/L, respectively [5]. The accelerated sulfide oxidation reached a plateau where the pH, EC, and most element concentrations in the leachate leveled out (Figure 4), likely due to temperature fluctuation in the laboratory. A decrease in temperature has been shown to slow down the sulfide oxidation rate [24,25,26], explaining the changes in element concentrations at weeks 35–48. During the first 52 weeks of leaching, it is suggested that a non-negligible fraction of the mobilized Fe and S originated from the dissolution of secondary phases present in the initial waste rock, such as melanterite (step I). Moreover, the accelerating sulfide oxidation with declining pH (<1.5) suggests a dissolution of HFO phases from week 29 and onward [27], resulting in the mobilization of Fe and limited increase in steps III–IV in sequential extraction (Figure 3 and Figure 5). During the first 52 weeks of leaching, Fe and S were mobilized, corresponding to approximately 3% of their initial content.
When studying waste rock samples in an optical microscope after 52 weeks of leaching, inferred HFO formation was preferentially occurring, associated with silicate minerals such as muscovite (Figure 5b), limiting the silicate weathering. The dissolution of silicates and aluminosilicates provides some neutralization potential. However, even in acid water, the dissolution of such minerals is usually slow [28] which, together with the inferred HFO formation, suggests a limited contribution to the buffering capacity. The previous statement is supported by the behavior of silicate-associated elements such as Al and Si, which during the first year of leaching were mobilized at 84–253 mg/L and 21–84 mg/L, respectively [5].
Overall, silicate-associated elements such as Al, Ca, and Mn show variations in content amongst solid samples (Table 2, Figure 4), suggesting a heterogeneous distribution of these minerals in the waste rock, which may interfere in interpreting the results. Analysis of Si is lacking in sequential extraction, but approximately 0.1% was mobilized during the first years of leaching (Figure 4). The amount of Al mobilized during the first year of leaching corresponded to approximately 1.5%. Only a small amount of Al was retained in what can be assumed to be secondary phases. Figure 3 shows an increase in step IV, likely due to the formation of more crystalline Al-(oxy)hydroxides such as diaspore. During the first 52 weeks of leaching, it is suggested that silicates are only mildly affected by the sulfide oxidation, resulting in limited ability to buffer the pH.
3.2.2. Second Year
During the second year of leaching, both Fe and S concentrations in the leachate increased by a magnitude of four, resulting in 11% of their initial content being mobilized (Figure 4). The formation of easily soluble salts such as gypsum and melanterite increased between the first and second year of leaching due to the low pH and high elemental concentrations in the leachate [29]. The test-cell was only irrigated once per week; therefore, precipitation and dissolution of easily soluble minerals such as melanterite or other metal sulfates are believed to occur weekly. The continuous dissolution of HFO phases and Al-(oxy)hydroxides (Figure 5a) due to the low pH can be seen as decreasing contents of Al and Fe in steps III–IV (Figure 3), suggesting that noteworthy decrease of the sulfide oxidation rate through HFO formation is unlikely.
Overall, silicate weathering was continuously limited during the second year of leaching, resulting in approximately 5% Al and 3% Si being mobilized (Figure 4). Other silicate-associated elements such as Ca (31%) and Mn (60%) showed extensive weathering and mobilization. At this time, Ca and Mn concentrations varied between 399 and 566 mg/L and 6.7 and 36 mg/L, respectively [5]. Throughout leaching, geochemical calculations showed that gypsum formation and dissolution largely controlled the concentration of Ca and SO42− in the leachate (Figure 5a). Before week 29, calcite dissolution buffered the system, leading to SO42− concentration controlling the gypsum formation, whereas the opposite was indicated after the accelerated sulfide oxidation with Ca controlling the formation. There is a distinct association of Mn with Ca; therefore, the mobilization variations are likely due to gypsum precipitation acting as a scavenger for Ca (Figure 3, Figure 5 and Figure 6). Despite the increased mobilization of silicate-associated elements during the second year, the effect on the pH and overall mobilization of elements are minimal. Hence, suggesting that processes that could substantially affect the sulfide oxidation rate and overall quality of the leachate are limited. Provided the overall release of S and limited formation of secondary minerals, an extrapolation of the average oxidation rate suggests depletion of the pyrite within approximately 18 years.
3.3. Occurrence and Mobility of Trace Elements
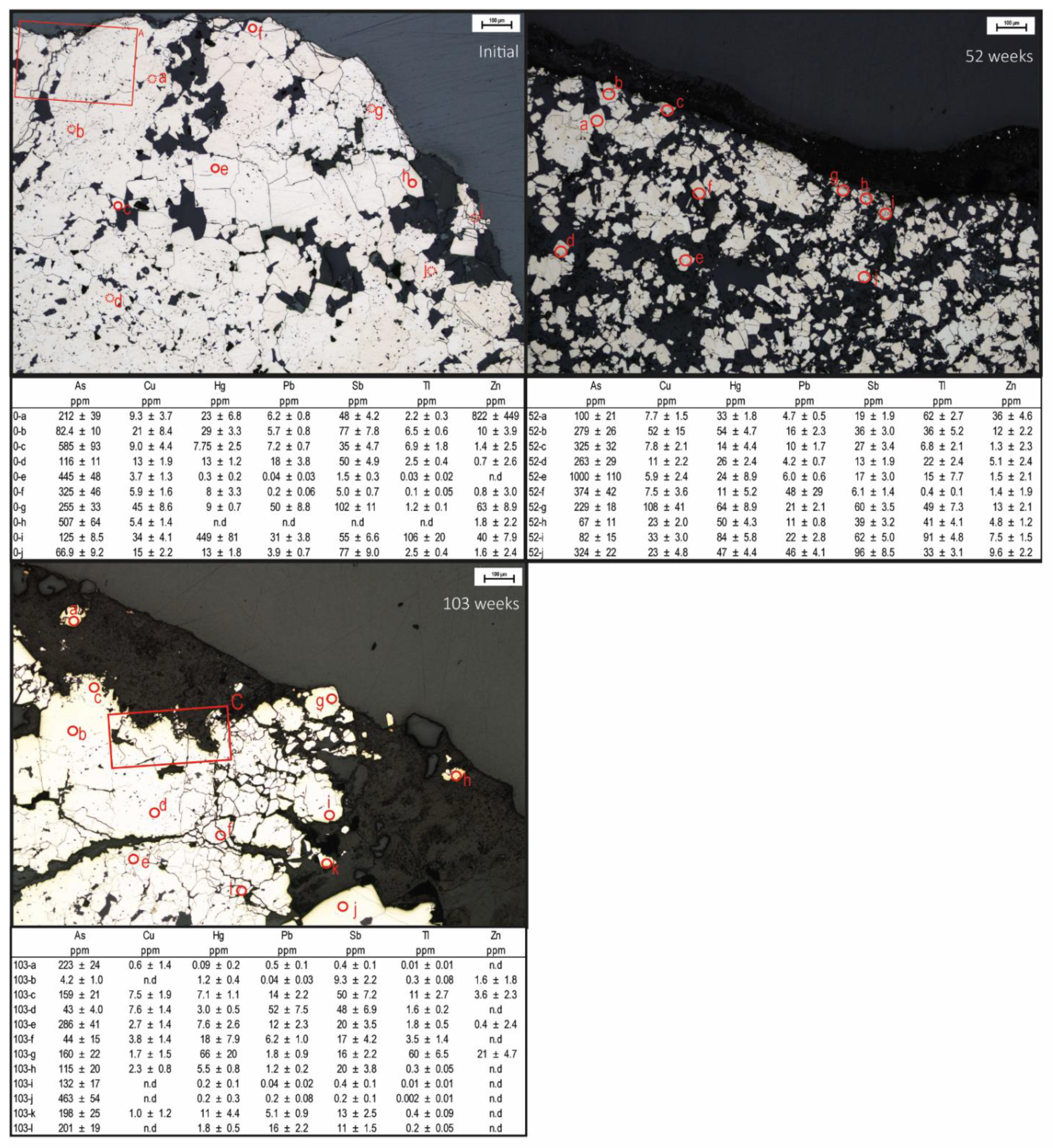
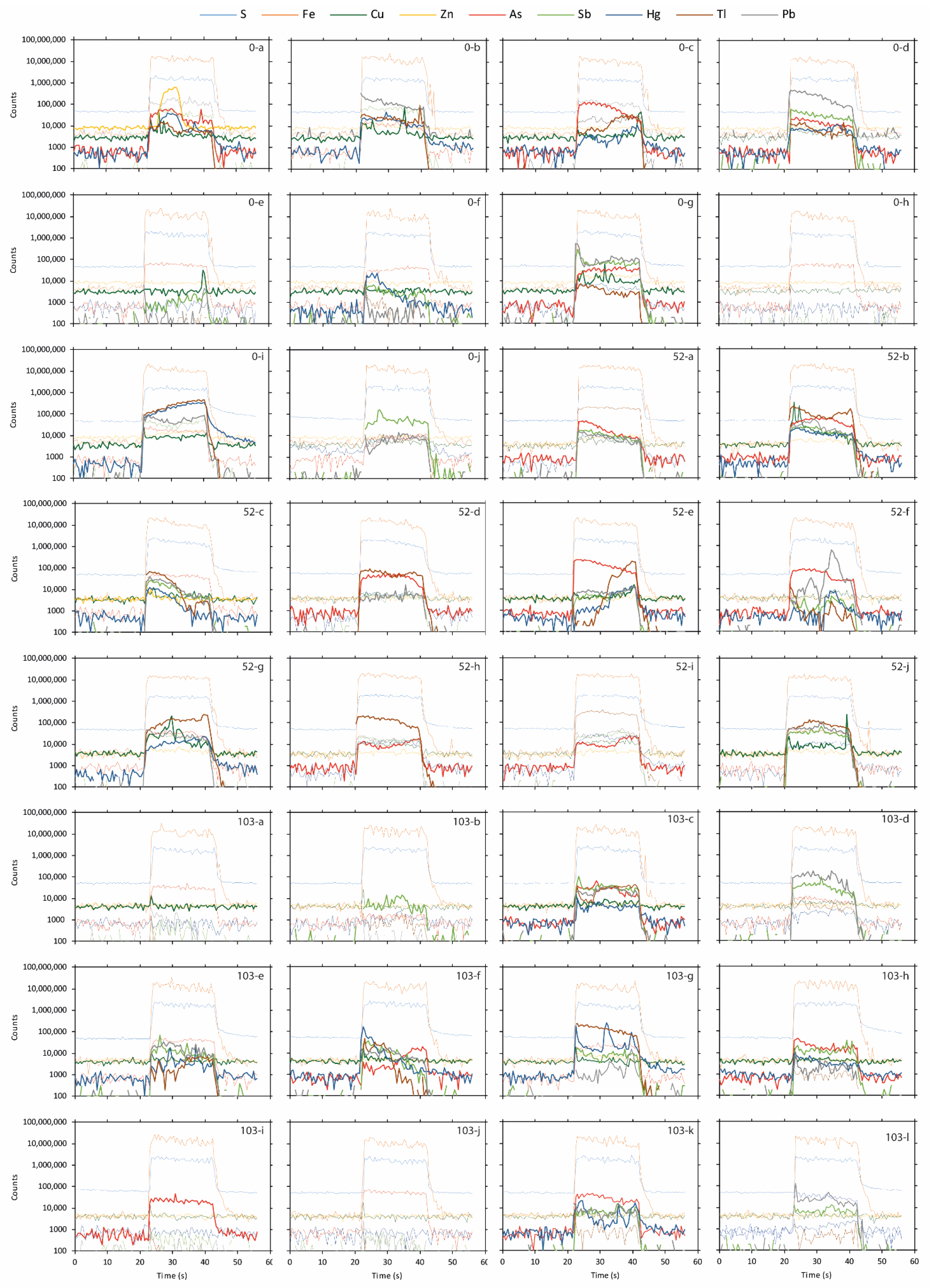
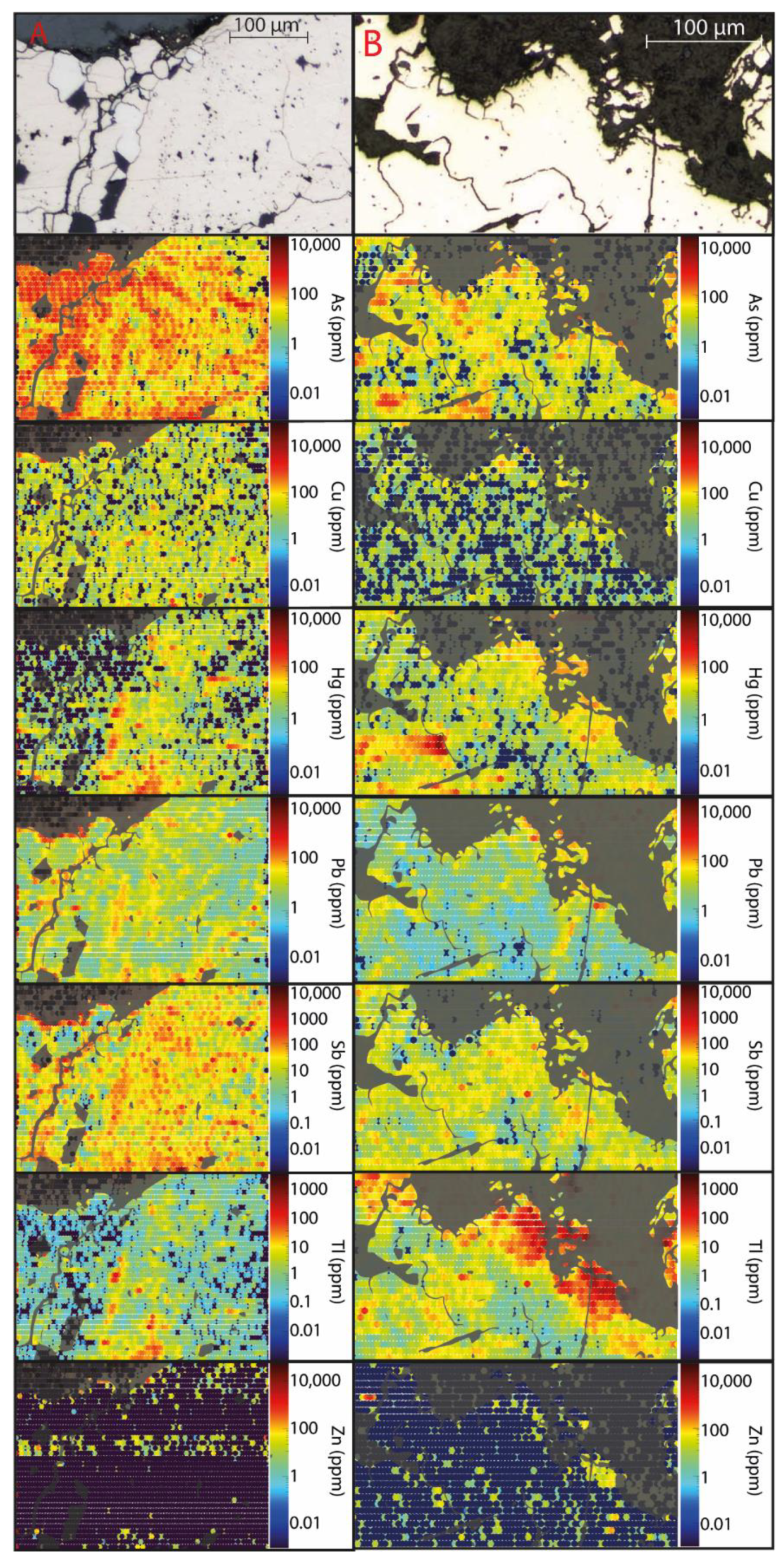
The whole-rock chemical composition showed an elevated abundance of As (217 ppm), Hg (17 ppm), Sb (38 ppm), and Tl (26 ppm) with smaller Cu (19 ppm), Pb (22 ppm), and Zn (68 ppm) content (Table 3). However, neither of the minerals identified occurred in sufficiently high quantities to explain the element concentrations observed. Pyrite was the most abundant mineral in the waste rock; therefore, knowledge about trace element composition and deportment within the pyrite grains are important. Impurities and mineral inclusions can lead to physical stress in the crystal structure, making the mineral more susceptible to oxidation [30], profoundly affecting the leachate quality. Charting count data of spot analyses tracks how trace element content changes during an analysis as deeper portions of the grains are sampled by the laser (Figure 6). Observations using charting count data show that the pyrite has several types of zonations containing trace elements. Changes in pyrite composition from As-rich to Hg- and Tl-rich and an overall weak negative covariance could be seen for As and Tl. A quantitative analysis of pyrite using LA-ICP-MS (Figure 2) showed that although As was present in all spot analyses, there were large variations in concentrations, with the highest observed in crystalline pyrite. As seen in Figure 2, concentrations of As in the initial pyrite varied between 67 and 585 ppm, resulting in an average concentration of 272 ppm. Additionally, pyrite oxidized for 52 weeks showed higher concentrations of As than the initial pyrite (Figure 2). Variations in concentrations during the first year are likely the result of internal variations combined with the previously proposed limited oxidation. Similar observations can be made for other trace elements such as Cu, Pb, and Tl (Figure 2). Mapping of the pyrite showed that As was the most evenly distributed element of those analyzed (Figure 7A), suggesting the occurrence of arsenic-bearing pyrite, which can increase the pyrite’s oxidation rate extensively [31]. Leaching mobilized As, resulting in concentrations between 0.2 µg/L and 21 mg/L [5].
During the second year, the leachate’s overall elemental concentrations increased, following a similar pattern as Fe, and S, with approximately 16% of the initial As content being released after 103 weeks of leaching (Figure 4). In general, the occurrence of As in pyrite makes it more susceptible to oxidation [28,30]. The increased susceptibility for the pyrite to leach could explain why a slightly higher fraction of As was mobilized compared to Fe or S. Comparing petrographic images of the initial pyrite and pyrite oxidized for 103 weeks revealed that weathering causes fracturing which likely accelerates the weathering [32] (Figure 2). The occurrence and overall mobility of As suggest that it was mainly associated with pyrite, which overrode any impact, e.g., arsenopyrite, had on the leachate quality. The decrease in As associated with secondary phases follows Fe, suggesting its adsorption to or co-precipitation with HFO. The correlation implies that As’s association with secondary phases has a limited effect on its mobilization (Figure 3). In other words, As in the pyrite affects the pyrite’s overall oxidation and stability. Conversely, it is shown that the pyrite has a profound effect on the mobilization of As.
As seen in Figure 7, Cu is more sporadically distributed in the pyrite than As. Spot analysis showed large spatial variations of Cu concentrations in the pyrite, varying between 4 and 45 ppm (average 16 ppm). Moreover, chart counting data show that although Cu was found as impurities in the pyrite, higher concentrations were associated with mineral inclusions, likely as chalcopyrite (Figure 6). Copper displayed a high mobilization rate throughout the leaching period, with concentrations between 0.1 and 20 mg/L [5]. The release of Cu was extensive throughout leaching (79%), with the majority being mobilized during the first year (57%). Despite Cu’s low occurrence in pyrite compared to As, their release rates were similar. As seen in Figure 4, Cu concentrations increased in the leachate before the sulfide oxidation rate accelerated (around week 29), suggesting one or more critical sources of Cu in the waste rock. A large fraction of Cu is assumed to originate from the pyrite. However, approximately 32% of the Cu content was associated with minerals such as chalcopyrite and bournonite (Table 2 and Table 3), which is expected to affect the weathering and consequently the build-up of Cu in secondary minerals. Chalcopyrite is more weathering-resistant than pyrite and arsenopyrite at neutral and low pH and when Fe3+ is the primary oxidant [29,34]. However, when sulfides are in contact with each other, electrons move between the sulfides, creating a galvanic cell. As a result, the mineral with the highest rest potential is protected from oxidation, and the mineral with the lowest rest potential is weathered more strongly [6,35,36,37,38]. In systems combining pyrite with chalcopyrite, the latter is more prone to oxidize, resulting in Cu being mobilized [39,40,41]. In other words, it is likely that the combination of pyrite and chalcopyrite results in the high mobilization of Cu (Figure 4). Additionally, Cu’s preferential occurrence in and around fractures and the pyrite’s more porous parts are likely to further contribute to the mobilization (Figure 7). Figure 3 shows that the accumulation of Cu associated with secondary phases decreases with time, following the same pattern as Fe, suggesting capturing in HFO due to its affinity to adsorb to or co-precipitate with the minerals schwertmannite and goethite [42]. Similar observations can be made for Zn. The proposed dissolution of HFO with time dissolves Cu, leading to an increase in the leachate. However, the dissolution of secondary minerals is comparably small compared to the overall mobilization of Cu. Therefore, it can be concluded that even though pyrite is not the direct source of Cu in the waste rock, Cu-rich inclusions in the pyrite are the main reason for Cu being mobilized.
Similar to Cu, both Pb and Zn concentrations in the pyrite varied substantially between 0and 50 ppm (average 14 ppm) and 1 and 822 ppm (average 105 ppm), respectively (Figure 2), with impurities matching low concentrations. In contrast, high concentrations are likely due to the presence of inclusions associated with the pyrite’s more porous parts. A weak covariance could be seen for Pb and Sb, suggesting that some impurities could consist of bournonite (Figure 6). Additionally, Pb could occur as inclusions of galena, despite not showing up during automated quantitative mineralogical analysis. The step size was slightly smaller than the size for spot analysis, and therefore, surficial inclusions of galena could have been missed during automated quantitative mineralogy. Correspondingly, impurities of Zn likely consisted of sphalerite.
Similar to Cu, Pb was mainly found in and around fractures in the more porous pyrite. However, Pb was mobilized at much lower concentrations, varying between 16 and 856 µg/L [5], corresponding to an approximate mobilization of 7% where the majority was released during the second year (Figure 4). The overall mobilization pattern of Pb resembled those of As, Fe, and S, but at a lower degree (Figure 4). The lower amount of Pb in the leachate could derive from its precipitation into easily soluble salts (step I, Figure 3) due to elevated concentrations in the leachate [29]. However, it should be noted that mobilized Pb increased around week 25, four weeks before the proposed sulfide oxidation acceleration, suggesting at least one other influential Pb source (Figure 4). Lead and Zn’s weathering behaviors differed notably from each other, with Zn being released at concentrations varying between 2 and 42 mg/L with a total mobilization resembling Cu (70%) (Figure 4). Approximately 3% of the Zn content was associated with sphalerite, which is expected to have impacted Zn’s mobility. Sphalerite is considered more easily weathered than pyrite, arsenopyrite, and chalcopyrite [43]. Due to problems when mapping using LA-ICP-MS, the quantification of Zn was inconclusive. However, it is likely that similar reasons as for Cu, with electrons moving between sulfides, are behind the high mobilization of Zn.
Antimony, Hg, and Tl were, similarly to Cu, Pb, and Zn, also found in and around fractures in the more porous pyrite (Figure 2). However, whereas Cu and Zn were mainly found as inclusions in the pyrite, Hg, Sb, and Tl were predominantly found as impurities within the pyrite. Antimony content varied between 2 and 102 ppm, and Hg and Tl varied between 0 and 449 ppm and 0 and 106 ppm, respectively, mainly as outliers with a weak covariance present (Figure 2 and Figure 7A). Quantification of Hg by LA-ICP-MS is difficult, and the quantified ppm values here may be subject to higher error than other elements presented [44]. Leaching of the waste rock resulted in Sb concentrations varying between 0.9 and 967 µg/L [8], with an overall mobilization of 4% (Figure 4). Although Sb followed the same mobilization pattern as As, Fe, Pb, and S, the overall release was lower. Unlike Pb, where a lower degree of mobilization could be explained by the element being retained in secondary minerals, sequential extraction for Sb was missing due to the high detection limit (<5 ppm), leading to any possible correlations being missed. Considering the amount of Sb found in the waste rock, a relatively large amount of secondary minerals could have precipitated despite the results being below the detection limit (13%). Similar observations were made for Hg (6%). However, the overall high content of Hg in the waste rock correlated with a seemingly limited leachability (0.08%) (Figure 4), which is somewhat misleading because measured concentrations of Hg released into the leachate (13 µg/L) far exceeded the MCL (maximal contamination level) from USEPA for drinking water of 2 µg/L [45]. Additionally, extremely high concentrations of Tl were observed in the leachate (317 µg/L), which can be compared to the MCL of 2 µg/L or the MCLG (maximum contamination level goals) of 0.5 µg/L [45]. Thallium is often undetected by standard analytical methods and rarely studied, even though it is considered more toxic to humans than both Cd and Hg [46]. Under extremely acidic conditions, such as those operating here, Tl’s adsorption onto surfaces is restricted, suggesting limited retention in secondary minerals [47]. Although the correlation between Hg and Tl was observed in pyrite, the leachability of Hg was substantially lower than for Tl, suggesting an additional source of Hg not related to and more resilient to weathering than pyrite. Due to the high detection limit of sequential extraction, it is also possible that the weathered Hg precipitated into secondary mineral without showing in the results.
There do not seem to be any other correlations between element releases other than that attributed to sulfide oxidation, suggesting that there are most likely several different minerals being leached, but these are overridden by the pyrite oxidizing to a large extent. The low pH, the abundance of Fe3+, and mineral exposure are the main mechanisms responsible.
3.4. Implications
The coupling of mineralogical and geochemical interactions is essential in understanding parameters controlling ARD formations and developing effective and sustainable preventive methods [48]. As previously discussed, most techniques for microencapsulation study the ability to prevent oxidation in pyritic tailings. When a buffering material is added to pyrite, oxidation can occur in circumneutral pH, favoring the precipitation of HFO on the pyrite’s surface, resulting in a limitation of the sulfide oxidation [49]. When pyrite is the sole mineral, trace element mobilization and attenuation reactions reflect reactions within the pyrite. However, preventing oxidation in a more complex material, such as waste rock consisting of numerous minerals, is more complicated because electrochemical processes most often protect the pyrite, favoring the oxidation of other sulfides [6,35,36,37,38]. In other words, trace element allocation is a vital aspect of the prevention’s success in limiting their release. It is evident that in the waste rock studied herein, it is not the pyrite itself but rather inclusions within the pyrite that are most vulnerable to oxidation due to the previously discussed aspects of electrochemical processes. Therefore, in the event of passivation of sulfidic surfaces using HFO, there is an imminent risk that the release of trace elements will not be prevented by remaining in the mineral. Instead, there is a probability of these elements being preferentially bound in or to secondary minerals. For example, the waste rock studied herein treated with LKD, Cu, and Zn, mainly present as inclusions, had the highest attenuation in secondary minerals of the trace elements studied [5]. Therefore, there is a risk of them being released in changing geochemical environments, such as when dry-covered. Altogether, this shows a need to understand interactions between minerals when implementing preventing methods such as microencapsulation to ascertain the method’s success.
4. Conclusions
- Trace elements found at elevated levels in the waste rock coincide with those found in pyrite, either as impurities or as inclusions. Hence, trace element content in a single mineral can have a noticeable impact on the chemistry of the waste rock as a whole, especially if that mineral has a high abundance in the whole rock;
- Waste rock leachate showed high element concentrations despite relatively low abundance in the solid phase. An estimation showed that approximately 78% Cu, 72% Zn, and 16% As had been depleted after only two years of leaching;
- The high leachability and low retention in secondary minerals after only two years of leaching suggest that metals such as Cu and Zn are highly interesting elements to be recovered by accelerating the sulfide oxidation, despite their overall low abundance in the waste rock. However, the viability of such measures needs to be further studied;
- Leaching of this waste rock, although selectively sampled, clearly shows that applying preventive measures to inhibit the sulfide oxidation during the delay time from mining to remediation is crucial for limiting the overall environmental impact.
Author Contributions
Conceptualization, E.N. and L.A.; methodology, E.N., C.W. and H.T.; software, E.N. and H.T.; writing—original draft preparation, E.N.; writing—review and editing, E.N., H.T., C.W. and L.A.; visualization, E.N.; supervision, L.A.; project administration, L.A.; funding acquisition, E.N. and L.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was carried out as a part of the Strategic Innovation Programme for the Swedish Mining and Metal Producing Industry (SIP-STRIM) of Vinnova, Formas, and the Swedish Energy Agency, with additional financial support from Boliden Mineral AB, Centre of Advanced Mining and Metallurgy (CAMM2) at Luleå University of Technology and J. Gust. Richert foundation.
Data Availability Statement
Data for this study are presented in the paper and cited references.
Acknowledgments
The authors would like to thank Susanne Bauer for her invaluable help in the laboratory.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Park, I.; Tabelin, C.B.; Jeon, S.; Li, X.; Seno, K.; Ito, M.; Hiroyoshi, N. A review of recent strategies for acid mine drainage prevention and mine tailings recycling. Chemosphere 2019, 219, 588–606. [Google Scholar] [CrossRef]
- Kefeni, K.K.; Msagati, T.A.M.; Mamba, B.B. Acid mine drainage: Prevention, treatment options, and resource recovery: A review. J. Clean. Prod. 2017, 151, 475–493. [Google Scholar] [CrossRef]
- INAP (The International Network for Acid Prevention) Global Acid Rock Drainage (GARD) Guide. Available online: http://www.gardguide.com/images/5/5f/TheGlobalAcidRockDrainageGuide.pdf (accessed on 18 January 2021).
- Zhang, Y.L.; Evangelou, V.P. Formation of ferric hydroxide-silica coatings on pyrite and its oxidation behavior. Soil Sci. 1998, 163, 53–62. [Google Scholar] [CrossRef]
- Nyström, E.; Kaasalainen, H.; Alakangas, L. Prevention of sulfide oxidation in waste rock by the addition of lime kiln dust. Environ. Sci. Pollut. Res. 2019, 26, 25945–25957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraitis, P.K.; Pattrick, R.A.D.; Vaughan, D.J. Variations in the compositional, textural and electrical properties of natural pyrite: A review. Int. J. Miner. Process. 2004, 74, 41–59. [Google Scholar] [CrossRef]
- Alakangas, L.; Andersson, E.; Mueller, S. Neutralization/prevention of acid rock drainage using mixtures of alkaline by-products and sulfidic mine wastes. Environ. Sci. Pollut. Res. 2013, 20, 7907–7916. [Google Scholar] [CrossRef]
- Nyström, E.; Kaasalainen, H.; Alakangas, L. Suitability study of secondary raw materials for prevention of acid rock drainage generation from waste rock. J. Clean. Prod. 2019. [Google Scholar] [CrossRef]
- Sahoo, P.K.; Tripathy, S.; Panigrahi, M.K.; Equeenuddin, S.M. Inhibition of acid mine drainage from a pyrite-rich mining waste using industrial by-products: Role of neo-formed phases. Water Air Soil Pollut. 2013, 224, 1–11. [Google Scholar] [CrossRef]
- Pérez-López, R.; Nieto, J.M.; Álvarez-Valero, A.M.; de Almodóvar, G.R. Mineralogy of the hardpan formation processes in the interface between sulfide-rich sludge and fly ash: Applications for acid mine drainage mitigation. Am. Mineral. 2007, 92, 1966–1977. [Google Scholar] [CrossRef]
- Paton, C.; Hellstrom, J.; Paul, B.; Woodhead, J.; Hergt, J. Iolite: Freeware for the visualisation and processing of mass spectrometric data. J. Anal. At. Spectrom. 2011, 26, 2508–2518. [Google Scholar] [CrossRef]
- Pearce, N.J.G.; Perkins, W.T.; Westgate, J.A.; Gorton, M.P.; Jackson, S.E.; Neal, C.R.; Chenery, S.P. A compilation of new and published major and trace element data for NIST SRM 610 and NIST SRM 612 glass reference materials. Geostand. Newsl. 1997, 21, 115–144. [Google Scholar] [CrossRef]
- Savard, D.; Bouchard-Boivin, B.; Barnes, S.J.; Garbe-Schönberg, D. UQAC-FeS: A new series of base metal sulfide quality control reference material for LA-ICP-MS analysis. In Proceedings of the 10th International Conference on the Analysis of Geological and Environmental Materials, Sydney, Australia, 8–13 July 2018. [Google Scholar]
- Wilson, S.A.; Ridley, W.I.; Koenig, A.E. Development of sulfide calibration standards for the laser ablation inductively-coupled plasma mass spectrometry technique. J. Anal. At. Spectrom. 2002, 17, 406–409. [Google Scholar] [CrossRef]
- Dold, B. Speciation of the most soluble phases in a sequential extraction procedure adapted for geochemical studies of copper sulfide mine waste. J. Geochem. Explor. 2003, 80, 55–68. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s guide to PHREEQC—A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. US Geol. Surv. Water-Resour. Investig. Rep. 1999, 99, 4259. [Google Scholar]
- Ball, J.W.; Nordstrom, D.K. User’s manual for WATEQ4F with revised thermodynamic database and test cases for calculating speciation of major, trace and redox elements in natural waters. US Geol. Surv. 1991, 189, 91–183. [Google Scholar]
- Maest, A.S.; Nordstrom, D.K. A geochemical examination of humidity cell tests. Appl. Geochem. 2017, 81, 109–131. [Google Scholar] [CrossRef] [Green Version]
- Stefánsson, A. Iron(III) hydrolysis and solubility at 25 °C. Environ. Sci. Technol. 2007, 41, 6117–6123. [Google Scholar] [CrossRef] [PubMed]
- Majzlan, J.; Navrotsky, A.; Schwertmann, U. Thermodynamics of iron oxides: Part III. Enthalpies of formation and stability of ferrihydrite (~Fe(OH)3), schwertmannite (~FeO(OH)3/4(SO4)1/8), and e-Fe2O3. Geochim. Cosmochim. Acta 2004, 68, 1049–1059. [Google Scholar] [CrossRef]
- Bigham, J.M.; Schwertmann, U.; Traina, S.J.; Winland, R.L.; Wolf, M. Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochim. Cosmochim. Acta 1996, 60, 2111–2121. [Google Scholar] [CrossRef]
- Nordstrom, D.K. Mine waters: Acidic to circumneutral. Elements 2011, 7, 393–398. [Google Scholar] [CrossRef]
- Singer, P.C.; Stumm, W. Acidic Mine Drainage: The Rate-Determining Step. Science 1970, 167, 1121–1123. [Google Scholar] [CrossRef]
- Wilson, D.; Amos, R.T.; Blowes, D.W.; Langman, J.B.; Ptacek, C.J.; Smith, L.; Sego, D.C. Diavik waste rock project: A conceptual model for temperature and sulfide-content dependent geochemical evolution of waste rock—Laboratory scale. Appl. Geochem. 2018, 89, 160–172. [Google Scholar] [CrossRef]
- Amos, R.T.; Blowes, D.W.; Bailey, B.L.; Sego, D.C.; Smith, L.; Ritchie, A.I.M. Waste-rock hydrogeology and geochemistry. Appl. Geochem. 2015, 57, 140–156. [Google Scholar] [CrossRef]
- Langman, J.B.; Moore, M.L.; Ptacek, C.J.; Smith, L.; Sego, D.; Blowes, D.W. Diavik waste rock project: Evolution of mineral weathering, element release, and acid generation and neutralization during a five-year humidity cell experiment. Minerals 2014, 4, 257–278. [Google Scholar] [CrossRef] [Green Version]
- Bigham, J.M.; Nordstrom, D.K. Iron and Aluminum Hydroxysulfates from Acid Sulfate Waters. Rev. Minral. Geochem. 2000, 40, 351–393. [Google Scholar] [CrossRef]
- Plumlee, G.S. The environmental geology of mineral deposits. In The Environmental Geochemistry of Mineral Deposits, Part A: Processes, Techniques, and Health Issues; Plumlee, G.S., Logsdon, M.J., Eds.; Reviews in Economic Geology; Society of Economic Geologists: Littleton, CO, USA, 1999; Volume 6, pp. 71–116. [Google Scholar] [CrossRef]
- Jambor, J.L.; Nordstrom, D.K.; Alpers, C.N. Metal-sulfate salts from sulfide mineral oxidation. Rev. Minral. Geochem. 2000, 40, 302–350. [Google Scholar] [CrossRef]
- Hutchison, I.; Ellison, R. Mine Waste Management; Lewis Publishers: Boca Raton, FL, USA, 1992. [Google Scholar]
- McKibben, M.A.; Tallant, B.A.; del Angel, J.K. Kinetics of inorganic arsenopyrite oxidation in acidic aqueous solutions. Appl. Geochem. 2008, 23, 121–135. [Google Scholar] [CrossRef]
- Lapakko, K.A.; Engstrom, J.N.; Antonson, D.A. Effects of particle size on drainage quality from three lithologies. In Proceedings of the 7th International Conference on Acid Rock Drainage 2006, ICARD—Also Serves as the 23rd Annual Meetings of the American Society of Mining and Reclamation, St. Louis, MO, USA, 26–30 March 2006; Volume 2, pp. 1026–1050. [Google Scholar]
- Krauskopf, K.B.; Bird, D.K. Introduction to Geochemistry; McGraw-Hill: New York, NY, USA, 1995. [Google Scholar]
- Rimstidt, J.D.; Chermak, J.A.; Gagen, P.M. Rates of Reaction of Galena, Sphalerite, Chalcopyrite, and Arsenopyrite with Fe(III) in Acidic Solutions; ACS Symposium Series. In Environmental Geochemistry of Sulfide, Oxidation; Alpers, C.N., Blowes, D.W., Eds.; American Chemical Society: Washington, DC, USA, 1994; Volume 1, pp. 2–13. [Google Scholar] [CrossRef] [Green Version]
- Kwong, Y.T.J.; Swerhone, G.W.; Lawrence, J.R. Galvanic sulphide oxidation as a metal-leaching mechanism and its environmental implications. Geochem. Explor. Environ. Anal. 2003, 3, 337–343. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Alpers, C.N. Negative pH, efflorescent mineralogy, and consequences for environmental restoration at the Iron Mountain Superfund site, California. Proc. Natl. Acad. Sci. USA 1999, 96, 3455–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelou, V.P. Pyrite Oxidation and Its Control; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Evangelou, V.P.; Zhang, Y.L. A review: Pyrite oxidation mechanisms and acid mine drainage prevention. Crit. Rev. Environ. Sci. Technol. 1995, 25, 141–199. [Google Scholar] [CrossRef]
- Chopard, A.; Plante, B.; Benzaazoua, M.; Bouzahzah, H.; Marion, P. Geochemical investigation of the galvanic effects during oxidation of pyrite and base-metals sulfides. Chemosphere 2017, 166, 281–291. [Google Scholar] [CrossRef]
- Mehta, A.P.; Murr, L.E. Kinetic study of sulfide leaching by galvanic interaction between chalcopyrite, pyrite, and sphalerite in the presence of T. ferrooxidans (30 °C) and a thermophilic microorganism (55 °C). Biotechnol. Bioeng. 1982, 24, 919–940. [Google Scholar] [CrossRef]
- Majima, H. How oxidation affects selective flotation of complex sulphide ores. Can. Metall. Q. 1969, 8, 269–273. [Google Scholar] [CrossRef]
- Sidenko, N.V.; Sherriff, B.L. The attenuation of Ni, Zn and Cu, by secondary Fe phases of different crystallinity from surface and ground water of two sulfide mine tailings in Manitoba, Canada. Appl. Geochem. 2005, 20, 1180–1194. [Google Scholar] [CrossRef]
- Jambor, J.L. The environmental geochemistry of sulfide mine-wastes. In Mineralogy of Sulfide-Rich Tailings and Their Oxidation Products; Blowes, D.W., Jambor, J.L., Eds.; Mineralogical Association of Canada: Nepean, ON, Canada, 1994. [Google Scholar]
- Cook, N.J.; Ciobanu, C.L.; Pring, A.; Skinner, W.; Shimizu, M.; Danyushevsky, L.; Saini-Eidukat, B.; Melcher, F. Trace and minor elements in sphalerite: A LA-ICPMS study. Geochim. Cosmochim. Acta 2009, 73, 4761–4791. [Google Scholar] [CrossRef]
- U.S.EPA (Environmental Protection Agency). National Primary Drinking Water Regulations, EPA 816-F-09-004. Available online: https://www.epa.gov/sites/production/files/2016-06/documents/npwdr_complete_table.pdf (accessed on 9 October 2019).
- Peter, A.L.J.; Viraraghavan, T. Thallium: A review of public health and environmental concerns. Environ. Int. 2005, 31, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Nriagu, J. Revised hydrolysis constants for thallium(I) and thallium(III) and the environmental implications. J. Air Waste Manage. Assoc. 1998, 48, 151–156. [Google Scholar] [CrossRef]
- Dold, B. Basic concepts of environmental geochemistry of sulfide mine-waste. Mineralogía, geoquímica y geomicrobiología para el manejo ambiental de desechos mineros. XXIV Curso Latinoam. Metalog. 2005, 22, 1–36. [Google Scholar]
- Huminicki, D.M.C.; Rimstidt, J.D. Iron oxyhydroxide coating of pyrite for acid mine drainage control. Appl. Geochem. 2009, 24, 1626–1634. [Google Scholar] [CrossRef]
Figure 1.
Experimental design of small-scale test cells filled with sulfidic waste rock.

Figure 2.
Petrographical microscopic image of pyrite grains in the initial waste rock and after 52 and 103 weeks of leaching. A: Area selected for mapping (Figure 3); C: Area selected for mapping (Figure 3); 0-a-j, 52-a-j, and 103-a-l: LA-ICPMS quantitative element analysis with laser spot size 35 µm. Spot analysis of crystalline/massive pyrite (initial) is indicated by a smooth circle, and porous/fractured pyrite is indicated by a dashed circle.
Figure 2.
Petrographical microscopic image of pyrite grains in the initial waste rock and after 52 and 103 weeks of leaching. A: Area selected for mapping (Figure 3); C: Area selected for mapping (Figure 3); 0-a-j, 52-a-j, and 103-a-l: LA-ICPMS quantitative element analysis with laser spot size 35 µm. Spot analysis of crystalline/massive pyrite (initial) is indicated by a smooth circle, and porous/fractured pyrite is indicated by a dashed circle.

Figure 3.
Extracted element content in the waste rock before leaching (initial) and on separate occasions at year one and two (weeks 52 and 103), modified from Nyström et al. [5]. Steps I–IV in sequential extractions after Dold (2003). Water-soluble fraction (I), exchangeable fraction (II), Fe(III)oxyhydroxides (III), and Fe(III)oxides (IV). Content below the detection limit is only outlined. The residual fraction (e.g., primary silicates and sulfides) is calculated as the difference between the whole rock element content of the sample (NaOH fusion) and the sum of steps I–IV.
Figure 3.
Extracted element content in the waste rock before leaching (initial) and on separate occasions at year one and two (weeks 52 and 103), modified from Nyström et al. [5]. Steps I–IV in sequential extractions after Dold (2003). Water-soluble fraction (I), exchangeable fraction (II), Fe(III)oxyhydroxides (III), and Fe(III)oxides (IV). Content below the detection limit is only outlined. The residual fraction (e.g., primary silicates and sulfides) is calculated as the difference between the whole rock element content of the sample (NaOH fusion) and the sum of steps I–IV.

Figure 4.
Estimated accumulated content of selected elements released into the leachate during 103 weeks of leaching the waste rock. The element content was calculated based on element concentrations reported by Nyström et al. [5] multiplied with the leachate volume withdrawn. Filled markings are actual measurements in the leachate. In contrast, outlined markings are estimated as the average between the two most adjacent actual measurements. Weeks 52 and 103 are marked with dashed vertical lines.
Figure 4.
Estimated accumulated content of selected elements released into the leachate during 103 weeks of leaching the waste rock. The element content was calculated based on element concentrations reported by Nyström et al. [5] multiplied with the leachate volume withdrawn. Filled markings are actual measurements in the leachate. In contrast, outlined markings are estimated as the average between the two most adjacent actual measurements. Weeks 52 and 103 are marked with dashed vertical lines.

Figure 5.
(a) Geochemical calculations of saturation index of Gypsum, Jarosite, Goethite (wateq4f.dat thermodynamic database [17]), Schwertmannite [20] originating from [21], and 6-line ferrihydrite [19] in the leaching of pyrite-rich waste rock. Dashed lines mark values close to zero (±0.5) which indicate equilibrium between the solution and mineral phase. Petrographical microscopic images after leaching the waste rock for 52 weeks. (b) Inferred HFO overgrowing muscovite (sericite). (c) Inferred Gypsum and HFO formation in bulk (between mineral grains).
Figure 5.
(a) Geochemical calculations of saturation index of Gypsum, Jarosite, Goethite (wateq4f.dat thermodynamic database [17]), Schwertmannite [20] originating from [21], and 6-line ferrihydrite [19] in the leaching of pyrite-rich waste rock. Dashed lines mark values close to zero (±0.5) which indicate equilibrium between the solution and mineral phase. Petrographical microscopic images after leaching the waste rock for 52 weeks. (b) Inferred HFO overgrowing muscovite (sericite). (c) Inferred Gypsum and HFO formation in bulk (between mineral grains).

Figure 6.
LA-ICP-MS element distribution with time showing how the association of As, Cu Hg, Sb, Pb, Tl, and Zn change with depth in spots measured in the initial sulfidic waste rock (Figure 2).
Figure 6.
LA-ICP-MS element distribution with time showing how the association of As, Cu Hg, Sb, Pb, Tl, and Zn change with depth in spots measured in the initial sulfidic waste rock (Figure 2).

Figure 7.
Quantified LA-ICP-MS element distribution maps of an area of pyrite grains in the initial sulfidic waste rock (A) and after 103 weeks of leaching (B) showing the spatial distribution of As, Cu, Hg, Pb, Sb, Tl, and Zn. Non-pyrite minerals are shaded.
Figure 7.
Quantified LA-ICP-MS element distribution maps of an area of pyrite grains in the initial sulfidic waste rock (A) and after 103 weeks of leaching (B) showing the spatial distribution of As, Cu, Hg, Pb, Sb, Tl, and Zn. Non-pyrite minerals are shaded.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sequential extraction according to Dold (2003) adopted to copper-sulfide mine waste with the minerals preferentially dissolved in each step.
Table 1.
Sequential extraction according to Dold (2003) adopted to copper-sulfide mine waste with the minerals preferentially dissolved in each step.
| Fraction | Approach | Preferentially Dissolved Minerals |
|---|---|---|
| I Water soluble fraction | Deionized water leach (L/S 50, 1 h) | Hydrous metal sulfates, gypsum, etc. |
| II Exchangeable fraction | Ammonium acetate leach (pH 4.5, 2 h) | Adsorbed and exchangeable ions, calcite, etc. |
| III Fe(III) oxyhydroxides | Cold ammonium oxalate leach (pH 3, 1 h in darkness) | Easily reducible Fe–Mn–Al(hydr)oxides and hydroxysulfates |
| IV Fe(III) oxides | Hot ammonium oxalate leach (pH 3, 2 h, 80 °C) | Remaining Fe–Mn–Al(hydr)oxides |
Table 2.
Quantitative mineralogical composition of pyritic waste rock determined by quantitative automated mineralogy (QEMSCAN) based on a total of 38 waste rock cross-sections mounted onto four thin sections.
Table 2.
Quantitative mineralogical composition of pyritic waste rock determined by quantitative automated mineralogy (QEMSCAN) based on a total of 38 waste rock cross-sections mounted onto four thin sections.
| Mineral | Formula | 1 | 2 | 3 | 4 | Average | Stdv. |
|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | ||
| Pyrite | FeS2 | 74 | 62 | 63 | 65 | 66 | 5.3 |
| Quartz | SiO2 | 15 | 17 | 19 | 16 | 17 | 1.7 |
| Muscovite | KAl2(Si3Al)O10(OH)2 | 2 | 10 | 6 | 7 | 6 | 3 |
| Chlorite | (Mg,Fe,Li)6AlSi3O10(OH)8 | 3 | 3 | 4 | 5 | 4 | 0.8 |
| Calcite | CaCO3 | 2 | 1 | 1 | 0.1 | 1 | 0.9 |
| Dravite | NaMg3Al6(Si6O18)(BO3)3(OH)3OH | 0.2 | 0.4 | 1 | 0.5 | 0.4 | 0.2 |
| Kaolinite | Al2Si2O5(OH)4 | 0.1 | 0.4 | 0.3 | 0.3 | 0.3 | 0.1 |
| Albite | NaAlSi3O8 | 0.2 | 0.3 | 0.3 | 0.4 | 0.3 | 0.1 |
| Flourite | CaF2 | 0.3 | 0.01 | 0.1 | 0.1 | 0.1 | 0.1 |
| Oligoclase | (Na,Ca)[Al(Si,Al)Si2O8) | 0.04 | 0.1 | 0.1 | 0.2 | 0.09 | 0.05 |
| Apatite | Ca5(PO4)3(F,Cl,OH) | 0.01 | 0.1 | 0.01 | 0.03 | 0.03 | 0.02 |
| Anatase | TiO2 | 0.004 | 0.02 | 0.01 | 0.01 | 0.01 | 0.009 |
| Clinopyroxene | (Mg,Fe,Mn,Li,Ca,Na)(Al,Fe,Ti,Cr,V,Zr,Sc,Zn,Mg,Mn)(Si,Al)2O6 | 0.01 | 0.005 | 0.01 | 0.005 | 0.006 | 0.002 |
| Allanite | (Ce,Ca,Y,La)2(Al,Fe+3)3(SiO4)3(OH) | 0.005 | 0.01 | 0.01 | 0.005 | 0.006 | 0.002 |
| Anorthite | CaAl2Si2O8 | 0.003 | 0.001 | 0.004 | 0.002 | 0.003 | 0.001 |
| Sanidine | K(AlSi3O8) | 0.001 | 0.002 | 0.001 | 0.001 | 0.001 | 0.0006 |
| Chalcopyrite | CuFeS2 | 0.001 | 0.001 | 0.002 | 0.0004 | 0.001 | 0.0008 |
| Biotite | K(Mg,Fe)3(AlSi3O10)(F,OH)2 | 0.001 | 0.001 | 0.001 | 0.001 | 0.0009 | 0.0002 |
| Bournonite | PbCuSbS3 | 0.001 | - | - | 0.00001 | 0.0004 | 0.0006 |
| Sphalerite | (Zn,Fe)S | 0.001 | 0.0002 | 0.0001 | 0.0001 | 0.0003 | 0.0003 |
| Arsenopyrite | FeAsS | 0.001 | 0.00004 | 0.0001 | - | 0.0002 | 0.0003 |
| Pyrrhotite | Fe1−xS (x = 0 to 0.2) | 0.0004 | 0.0003 | 0.0003 | 0.00002 | 0.0002 | 0.002 |
Table 3.
Abundance of selected elements in the sulfidic waste rock. The waste rock is presented as the average of screening and results reported by Alakangas et al. [7] and Nyström et al. [5]. The waste rock is compared to minimum and maximum concentrations of rock (mainly ore) sampled in the area and average content in the average earth’s crust [33].
Table 3.
Abundance of selected elements in the sulfidic waste rock. The waste rock is presented as the average of screening and results reported by Alakangas et al. [7] and Nyström et al. [5]. The waste rock is compared to minimum and maximum concentrations of rock (mainly ore) sampled in the area and average content in the average earth’s crust [33].
| Element | Average | Mine Area | Average | |
|---|---|---|---|---|
| % | Waste Rock | Min. | Max. | Earth’s Crust |
| Al | 3 | 1 | 16 | 8 |
| Ca | 1 | 0.01 | 20 | 3 |
| Fe | 14 | 0.2 | 34 | 5 |
| K | 0.4 | 0.01 | 6 | 2.6 |
| Mg | 1 | 0.05 | 15 | 2.1 |
| Na | 0.3 | 0.007 | 4 | 2.4 |
| P | 0.01 | 0.0004 | 0.2 | 0.1 |
| Stotal | 27 | 0.01 | 39 | 0.05 |
| Si | 13 | 6 | 28 | 27 |
| Ti | 0.1 | 0.02 | 1 | 0.5 |
| mg/kg | ||||
| As | 193 | 1 | 18,000 | 2 |
| Ag | 1 | <0.1 | 32 | 0.07 |
| Au | <0.01 | <0.0005 | 0.8 | 0.004 |
| B | 5 | - | - | 10 |
| Ba | 88 | <0.5 | 1670 | 430 |
| Be | 0.2 | - | - | 3 |
| Bi | 0.2 | <0.1 | 42 | 0.2 |
| Br | <5 | - | - | 3 |
| Cd | 0.2 | <0.1 | 55 | 0.18 |
| Ce | 13 | 4 | 123 | 45 |
| Co | 2 | <1 | 121 | 25 |
| Cr | 51 | <5 | 2513 | 200 |
| Cs | 0.2 | <0.1 | 5 | 2 |
| Cu | 15 | 2 | 15,600 | 60 |
| Dy | 1 | 0.7 | 16 | 4.5 |
| Er | 1 | 0.5 | 11 | - |
| Eu | 1 | 0.2 | 5 | 1.2 |
| Ga | 5 | 3 | 43 | 17 |
| Gd | 1 | 0.8 | 12 | 7 |
| Ge | 0.4 | <1 | 4 | 15 |
| Hf | 1 | 0 | 10 | - |
| Hg | 13 | <0.01 | >200 | 0.08 |
| Ho | 0.2 | 0.2 | 4 | - |
| I | <0.5 | - | - | 0.5 |
| In | - | <0.2 | 4 | 0.1 |
| Ir | <0.01 | - | - | 0.001 |
| La | 6 | 1 | 52 | 6.5–100 |
| Li | 14 | - | - | 30 |
| Lu | 0.1 | 0.1 | 2 | 0.9 |
| Mn | 208 | 77 | 7357 | 900 |
| Mo | 3 | <0.5 | 34 | 2 |
| Nb | 0.2 | <1 | 13 | 20 |
| Nd | 6 | 3 | 53 | 25 |
| Ni | 1 | <0.1 | 864 | 80 |
| Os | <0.01 | - | - | - |
| Pb | 20 | <2 | 5181 | 16 |
| Pd | <0.1 | - | - | 0.01 |
| Pr | 2 | 0.6 | 14 | - |
| Pt | <0.01 | - | - | 0.005 |
| Rb | 6 | <0.5 | 105 | 120 |
| Re | <0.01 | - | - | 0.001 |
| Rh | <0.01 | - | - | - |
| Ru | <0.01 | - | - | - |
| Sb | 24 | 0.2 | 1327 | 0.15–1 |
| Sc | 6 | 1 | 66 | 5–22 |
| Se | <1 | <0.5 | 83 | 0.09 |
| Sm | 1 | 0.6 | 12 | 7 |
| Sn | 0.5 | <1 | 29 | 2.5 |
| Sr | 40 | <0.5 | 396 | 350 |
| Ta | <0.01 | <0.05 | 23 | 2 |
| Tb | 0.2 | 0.1 | 2 | - |
| Te | 0.1 | - | - | 0.00036–0.01 |
| Th | 1 | 0.6 | 11 | 10 |
| Tl | 26 | <0.1 | 75 | 1 |
| Tm | 0.1 | 0.1 | 2 | - |
| U | 2 | 0.5 | 24 | 3 |
| V | 7 | <5 | 212 | 150 |
| W | 1 | 0.1 | 34 | 1 |
| Y | 11 | 4 | 125 | 30 |
| Yb | 1 | 0.4 | 13 | 3 |
| Zn | 74 | 7 | 49,700 | 70 |
| Zr | 65 | 10 | 344 | 160 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nyström, E.; Thomas, H.; Wanhainen, C.; Alakangas, L. Occurrence and Release of Trace Elements in Pyrite-Rich Waste Rock. Minerals 2021, 11, 495. https://doi.org/10.3390/min11050495
AMA Style
Nyström E, Thomas H, Wanhainen C, Alakangas L. Occurrence and Release of Trace Elements in Pyrite-Rich Waste Rock. Minerals. 2021; 11(5):495. https://doi.org/10.3390/min11050495
Chicago/Turabian StyleNyström, Elsa, Helen Thomas, Christina Wanhainen, and Lena Alakangas. 2021. "Occurrence and Release of Trace Elements in Pyrite-Rich Waste Rock" Minerals 11, no. 5: 495. https://doi.org/10.3390/min11050495
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.