
Abstract
Background
In Helicobacter pylori (Hp)-uninfected individuals, diffuse-type gastric cancer (DGC) was reported as the most common type of cancer. However, the carcinogenic mechanism of Hp-uninfected sporadic DGC is largely unknown.
Methods
We performed whole-exome sequencing of Hp-uninfected DGCs and Hp-uninfected normal gastric mucosa. For advanced DGCs, external datasets were also analyzed.
Results
Eighteen patients (aged 29–78 years) with DGCs and nine normal subjects (28–77 years) were examined. The mutation burden in intramucosal DGCs (10–66 mutations per exome) from individuals aged 29–73 years was not very different from that in the normal gastric glands, which showed a constant mutation accumulation rate (0.33 mutations/exome/year). Unbiased dN/dS analysis showed that CDH1 somatic mutation was a driver mutation for intramucosal DGC. CDH1 mutation was more frequent in intramucosal DGCs (67%) than in advanced DGCs (27%). In contrast, TP53 mutation was more frequent in advanced DGCs (52%) than in intramucosal DGCs (0%). This discrepancy in mutations suggests that CDH1-mutated intramucosal DGCs make a relatively small contribution to advanced DGC formation. Among the 16 intramucosal DGCs (median size, 6.5 mm), 15 DGCs were pure signet ring cell carcinoma (SRCC) with reduced E-cadherin expression and a low proliferative capacity (median Ki-67 index, 2.4%). Five SRCCs reviewed endoscopically over 2–5 years showed no progression.
Conclusions
Impaired E-cadherin function due to CDH1 mutation was considered as an early carcinogenic event of Hp-uninfected intramucosal SRCC. Genetic and clinical analyses suggest that Hp-uninfected intramucosal SRCCs may be less likely to develop into advanced DGCs.
Similar content being viewed by others


Introduction
Epidemiological studies have reported that Helicobacter pylori (Hp) infection is the greatest risk factor for gastric cancer [1, 2]. Chronic gastritis caused by Hp infection was shown to promote oncogenic mutations [3, 4]. Although most gastric cancers arise in Hp-infected stomachs, gastric cancers in Hp-uninfected stomachs also occur, with their prevalence reported to account for 0.42–5.4% of all gastric cancers [5]. The prevalence of Hp infection is lower in developed countries than in developing countries [6]. In Japan, the prevalence of Hp infection and incidence of gastric cancer was previously high. However, in recent decades, its prevalence and mortality rate by gastric cancer have decreased [7,8,9]. The incidence of Hp-uninfected gastric cancers is expected to increase as the Hp infection rate declines. However, the molecular basis of gastric cancer in the Hp-uninfected stomach is largely unknown. Recent studies showed that somatic mutations accumulate in normal tissues with age [10,11,12,13,14]; however, mutation accumulation in the Hp-uninfected normal gastric mucosa has not been reported.
Histologically, gastric cancer is classified into intestinal-type gastric cancer and diffuse-type gastric cancer (DGC) [15]. DGCs include poorly differentiated adenocarcinomas (PDAs) and signet ring cell carcinomas (SRCCs). In Hp-uninfected patients, gastric cancers are most often found as intramucosal DGCs, which are typically ‘pure’ SRCCs that contain only SRCC cells and no PDA components [5, 16]. Hp-uninfected intramucosal DGC is smaller in size than Hp-infected intramucosal DGC [16, 17]. Additionally, T1 stage (UICC classification) DGCs were shown to be significantly less invasive in patients without Hp infection than in those with Hp infection [17].
Hereditary diffuse gastric cancer (HDGC) is mainly caused by a CDH1 germline mutation [18, 19]. E-cadherin, encoded by CDH1, is a cell adhesion molecule that regulates signaling pathways associated with cell proliferation, survival, invasion, and migration. Based on studies of HDGC, DGCs are thought to arise from SRCCs because of reduced cell adhesion associated with the loss of E-cadherin function [19]. Although somatic CDH1 mutations have been found in 29–56% of sporadic DGCs in previous genetic analyses, the mechanisms of sporadic DGC development remain unclear [20,21,22]. RHOA mutations and CLDN18-ARHGAP fusion have also been reported to cause DGC [21,22,23,24]. These results were mainly obtained in studies of advanced gastric cancer, whereas few studies have examined intramucosal cancer. Furthermore, previous studies either mainly consisted of Hp-infected patients or did not assess the Hp infection status. In the present study, we examined the clinicopathological and genetic features of Hp-uninfected DGCs. We performed whole-exome sequencing (WES) to investigate the genetic alterations in early and advanced stage DGCs. These genetic alterations were then compared with those in the Hp-uninfected normal gastric mucosa.
Materials and methods
Patient and sample collection
Tumor samples, paired blood samples, and clinical data were collected from patients diagnosed with Hp-uninfected DGC and enrolled in the Kyoto University Hospital and Kansai Electric Power Hospital between January 2016 and March 2020. As normal controls, we collected biopsied gastric tissues and peripheral blood from nine subjects without Hp-infection or gastric cancer who underwent diagnostic endoscopy for upper gastrointestinal symptoms. All patients and normal subjects provided written informed consent to participate in the study. The study protocol conformed to the ethical guidelines of the Declaration of Helsinki and was approved by the ethics committee of Kyoto University Hospital and institutional board of Kansai Electric Power Hospital.
Certified pathologists (S.M. and T.S.) histologically examined the surgically or endoscopically-resected or biopsied specimens according to the third English edition of Japanese classification of gastric carcinoma and Lauren’s classification [15, 25].
Hp infection status evaluation
Diagnostic criteria for Hp-uninfected gastric cancer have not been established. These criteria must be strict to rule out naturally Hp-eradicated cases [5]. Patients were defined as Hp-uninfected when they met all of the following criteria: (i) no history of Hp eradication therapy; (ii) negative results in the Hp infection test (serum anti-Hp antibody < 3 U/mL and/or negative urea breath test result); (iii) absence of Hp bacterial body in normal gastric mucosa identified by immunohistochemistry with an anti-Hp antibody (Dako, Glostrup, Denmark), Giemsa staining, or hematoxylin and eosin (H&E) staining of formalin-fixed paraffin-embedded (FFPE) sections; and (iv) no endoscopic findings of mucosal atrophy.
Immunohistochemistry
Immunohistochemistry analysis was performed on FFPE specimens using primary antibodies against E-cadherin (clone NCH-38, Dianova, Hamburg, Germany) and Ki-67 (clone MIB-1, Dako). The proportion of tumor cells positive for Ki-67 was measured at the site with the highest number of labeled nuclei. Investigators were blinded to the tumor genotypes.
Tumor-DNA extraction
Ten-micrometer-thick sections were sliced from the FFPE tissue onto membrane frame slides (Leica, Wetzlar, Germany). Laser capture microdissection of tumor cells was performed using a Leica LMD 7000 instrument. To perform WES, genomic DNA was extracted from the microdissected tumor cells and matched peripheral blood using the GeneRead DNA FFPE Kit and the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), respectively.
Gastric single gland isolation
We performed WES on Hp-uninfected normal gastric epithelia for comparison with Hp-uninfected DGCs. Normal gastric epithelium is composed of single-layer columnar cells that are compacted into numerous small replication units known as glands. Each gland is replenished by stem cells located at the neck or base of the gland [26]. To evaluate the precise somatic mutation in the Hp-uninfected normal gastric epithelia, genomic DNA extracted from fresh single glands was subjected to WES as previously described [11]. Briefly, two biopsies of normal gastric mucosa were obtained from the fundic gland area, at the gastric angle, for each Hp-uninfected subject without a history of gastric cancer. A total of 18 samples were obtained from nine subjects. Gastric epithelia were manually dissociated from the lamina propria after treating the gastric mucosa with 20 mM EDTA in PBS at 4 °C for 20 min. Subsequently, single glands were picked up under a stereomicroscope. Genomic DNA isolated from each gland was split into two aliquots, each of which was independently subjected to whole-genome amplification (WGA) using the REPLI-g Single Cell Kit (Qiagen). Both amplified DNA samples were subjected to WES independently. Variants commonly detected in the two split samples were considered as true somatic mutations.
Whole-exome sequencing
Whole-exome sequecing libraries were prepared using SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) or xGen Exome Research Panel (Integrated DNA Technologies, Coralville, IA, USA), followed by sequencing of enriched exon fragments using a HiSeq 2500 or NovaSeq 6000 system (Illumina, San Diego, CA, USA) in 100–150-bp paired-end mode as previously described [27]. The target depth was 100 × the actual depth was 108 × on average. Sequencing reads were aligned to the human reference genome (GRCh37), and mutation calling was performed using the Genomon2 pipeline (v.2.6) as previously described [11]. Candidate mutations were adopted using the Empirical Bayesian Mutation Calling (EBCall) algorithm with the following filtering criteria: (i) a sufficient number of reads (total reads ≥ 8 and variant reads ≥ 3); (ii) variant allele frequencies (VAFs) ≥ 0.05 (for FFPE samples), ≥ 0.25 (for single-gland WGA samples), and < 0.02 (for germline control); (iii) a strand ratio not equal to 0 or 1; and (iv) p value by EBCall ≤ 10−3.5. Putative germline variants were also excluded by comparing VAFs with matched controls using Fisher’s exact test (p ≤ 10−1). Candidate mutations in tumor samples were validated by PCR-based amplicon deep sequencing [27]. Because of the limited amount of DNA available, validation was performed with three samples, and the true positive rate was 95% (39 of 41).
Driver genes were investigated based on dN/dS, which is the ratio of the number of nonsynonymous substitutions per nonsynonymous site to the number of synonymous substitutions per synonymous site, using dNdScv [28]. We also adopted 69 driver genes previously reported in comprehensive genetic analyses of gastric cancer [21,22,23,24] (Table S1). Because mutations in the driver gene had a high prior probability of being true mutations, we included the driver gene mutations found by loosening the filter criteria: (i) VAFs > 0.04; (ii) p value by EB call ≤ 10−3; and (iii) a strand ratio = 0–1. Copy number abnormalities were evaluated using WES data with our in-house pipeline ‘CNACS’ [12].
External dataset
Because we collected only two cases of advanced DGC, somatic mutation data identified by MuTect and clinical information from patients with DGC were downloaded from The Cancer Genome Atlas (TCGA) data portal [23]. Thirty-seven DGC cases from TCGA were reported as Hp-negative, and all had T2–T4 tumors (UICC TNM classification 6th edition). As hypermutated tumors were considered genetically different from our samples, tumors with more than 12 mutations/Mb were excluded from the comparative data [23]. We also used 30 Hp-negative advanced DGC data from Hong Kong [22], all of which were DGCs without microsatellite instability. In total, data for 60 Hp-uninfected advanced DGCs (two from our cohort, 28 from TCGA, and 30 from Hong Kong dataset) were collected and compared with data for nine intramucosal DGCs from our cohort. The Hong Kong dataset could not be used for mutation number and mutational signature analyses because of a lack of information about synonymous mutations. Next, 282 single-nucleotide variants (SNVs) from 11 DGC samples, 260 SNVs from 18 normal glands, and 2596 SNVs from 28 DGCs from TCGA were allocated to 65 ‘the Catalogue Of Somatic Mutations In Cancer’ (COSMIC) single base substitution signatures (SBS) using SigProfilerExtractor [29].
Statistical analysis
All tests were two-tailed, and p < 0.05 was considered as significant. The linearity of the number of mutations in the normal gastric glands, intramucosal DGCs, and advanced DGCs with age was evaluated based on Pearson’s correlation coefficient in a linear regression model that assumed a zero intercept, because the number of somatic mutations in an exome region at 0 years of age was assumed to be nearly 0, as previously described [11]. The Mann–Whitney U test was used to compare normal glands, intramucosal DGCs, and advanced DGCs. Driver mutation rates between intramucosal and advanced DGCs were compared by Fisher’s exact test with Benjamini–Hochberg adjustment. q < 0.05 was considered to indicate significance. All statistical analyses were performed using R software (version 3.6.3).
Results
Clinical information
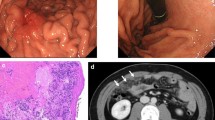
The clinical information of 18 patients with Hp-uninfected DGCs (median age 57 years; range 29–78) and nine subjects without Hp infection (median age 44 years; range 28–77) was collected (Table 1). Sixty-seven percent of patients with DGC had a smoking history, and 33% were current smokers (Table 2). Fifty percent of patients with DGC had alcohol consumption habit. Among the 18 patients, 16 were diagnosed with intramucosal DGC (median age 60 years; range 29–73) and two with advanced DGC. Sixteen intramucosal DGCs were found as flat pale areas upon endoscopy, all of which were resected endoscopically and were found to contain an SRCC component. One DGC localized in the gastric cardia consisted of a mixture of SRCC and PDA components (Fig. 1a and lower panels of Fig. 1b), and the remaining 15 DGCs localized near the gastric angle were ‘pure’ SRCCs scattered around the neck of the gland (Fig. 1a and upper panels of b). These intramucosal ‘pure’ SRCCs had a low proliferation capacity, with a median Ki-67 labeling index of 2.4% (range 0–15.4%). Five cases with intramucosal ‘pure’ SRCC showed no progression over 2–5 years according to endoscopic image review (Fig. 2).

Clinicopathological features of 18 Hp-uninfected DGCs. a Tumor locations and pathological features. Sixteen cases of intramucosal DGC and two cases of advanced DGC were sampled. The red dots represent intramucosal DGCs. Fifteen of sixteen intramucosal DGCs were concentrated near the gastric angle and were 'pure' SRCCs with reduced E-cadherin immunoreactivity. One cardiac intramucosal DGC consisted of SRCC and PDA components with maintained E-cadherin immunoreactivity. Green ovals represent advanced DGCs. Advanced DGCs consisted of SRCC and PDA components with reduced E-cadherin immunoreactivity. b Representative endoscopic and histological images of intramucosal DGC cases. Upper panels: images of case 5. A pale flat lesion was observed at the anterior wall of the gastric antrum, close to the gastric angle, upon endoscopy (yellow arrowhead). H&E staining of the endoscopic submucosal dissection specimen showed that SRCCs were confined to the proliferative zone of the mucosa. Immunohistochemistry for E-cadherin showed weak immunoreactivity in SRCCs. Immunohistochemistry for Ki-67 showed only a few SRCCs positive for nuclear staining. Lower panels: images of case 13. A pale flat lesion was observed at the greater curvature of the gastric cardia upon endoscopy (yellow arrowhead). H&E staining of the endoscopic submucosal dissection specimen showed SRCCs and PDAs in the mucosa. Immunohistochemistry for E-cadherin showed almost the same immunoreactivity in carcinoma cells as in surrounding epithelial cells. Immunohistochemistry for Ki-67 showed that all carcinoma cells were negative for nuclear staining. Scale bar indicates 50 µm

Changes in endoscopic findings in Hp-uninfected intramucosal signet ring cell carcinoma over time. Top panels: endoscopic images of Case 8. A biopsy from a tiny pale area at the greater curvature of the gastric angle revealed an SRCC in the year 2018. After biopsy, the lesion became indistinct. In 2020, the lesion became visible, and the size of the lesion was almost the same as that observed in 2018. The second, third, fourth and fifth panels: endoscopic images of Cases 10, 16, 17, and 18, respectively. Upon retrospective review of the endoscopic images, SRCCs were found that remained unchanged for 5, 3, 2, and 3 years, respectively. Yellow arrowheads show the lesions. The number in the lower right-hand corner represents the year in which the image was taken
Genomic analysis of intramucosal DGCs
Tumor cells were dissected by laser microdissection to increase the tumor cell content because the intramucosal DGCs were small (median 6.5 mm; range 3–14 mm) and their tumor cells were scattered around the necks of glands. Small tumors with low densities of signet ring cells did not yield adequate amounts of DNA to perform WES. Accordingly, WES was performed for only nine intramucosal DGCs (Table 2). The median amount of extracted DNA was 67 ng (range 10–309 ng). A total of 239 mutations (median 20 mutations/exome; range 10–66) were found (Table S2). The most recurrently mutated gene was CDH1; CDH1 mutations were found in six of nine intramucosal DGCs (67%) and were considered as positively selected by dN/dS analysis (q < 0.001). All six cases with CDH1 mutations had ‘pure’ SRCCs, which were near the gastric angle. For the other genes, dN/dS analysis showed no significant results (q > 0.05). However, a search for 69 previously reported driver genes revealed mutations in three genes, RNF43, TGFBR2, and FAT4 (Fig. 3a, Tables S1–S3). All nine cases of Hp-uninfected intramucosal DGC had two or fewer driver mutations.

Mutational landscape and CDH1 mutation sites. a Mutational landscape of Hp-uninfected nine intramucosal DGCs and two advanced DGCs. Tumor invasion depth, location, histological type, and E-cadherin immunoreactivity are indicated. LOH loss of heterozygosity. b CDH1 mutation sites. Alterations in our sequenced tumor samples are plotted above the CDH1 protein. Somatic mutations in sporadic DGCs that have been repeatedly reported in previous studies are plotted below the CDH1 protein [Refs. 20, 21, 24, 33]. Sig signal peptide, Precursor precursor sequence, EC extracellular domain, TM transmembrane domain, Cytoplasmic cytoplasmic domain. # reported as a germline mutation of HDGC
CDH1 mutation is generally known as a loss-of-function mutation [30]. A missense mutation at L581R and loss of heterozygosity were found in Case 2 (Fig. 3a). A missense mutation at L581 was previously reported in a sporadic DGC [20]. Two-hit CDH1 mutations, including truncating mutations, were observed in Cases 5 and 6 (Fig. 3a). Cases 3 and 5 harbored a mutation at I250 (L249_T253del, I250N, respectively), which was reported as a mutation hotspot in sporadic DGCs and resulted in impaired cellular aggregation in vitro [21] (Fig. 3b). Hotspot splice site mutations (c.531 + 2 T > A, c.687 + 1_687 + 4del) reported in sporadic DGCs were observed in Cases 4 and 7 [21, 24] (Fig. 3b). These splice site mutations are thought to induce exon truncations in extracellular domain 1 to prevent the homodimerization of E-cadherin according to computer models [21]. Case 13, the only case with PDA and SRCC located at the cardia, did not harbor CDH1 mutations. Among the three cases without CDH1 mutations, we detected a frameshift RNF43 mutation (A146fs) in Case 10, but we detected no driver mutations in Cases 12 and 13. We also searched for germline mutations in CDH1, CTNNA1, PALB2, BRCA1, and RAD51C, all of which had been reported as causative genes of HDGC using blood DNA [19, 31, 32]. None of the nine subjects possessed pathogenic germline mutations in these genes. These results indicate that somatic CDH1 mutations can be considered as an early event in intramucosal SRCC carcinogenesis. E-cadherin expression was immunohistochemically reduced in all eight cases with intramucosal ‘pure’ SRCCs near the gastric angle but was maintained in DGC without CDH1 mutations at the gastric cardia (Fig. 1a, b). These data suggest that reduced E-cadherin expression contributes to SRCC development near the gastric angle, whereas the one case of PDA/SRCC at the gastric cardia may have been caused by different mechanisms.
Analysis of advanced DGCs
Genomic DNA extracted from laser capture microdissected SRCC cells in the mucosal part of two advanced DGCs was subjected to WES. For technical reasons, some PDA cells were included. A TP53 missense mutation at R248W and loss of heterozygosity was found in Case 11 (Fig. 3a, Table S4). Four driver mutations in CDH1, TGFBR2, ARID1A, and RHOA were found in Case 14 (Fig. 3a, Tables S3, S4). The CDH1 mutation at D402V has been reported in sporadic DGCs [20, 33] (Fig. 3b). Neither of the two cases had pathogenic germline mutations in the HDGC-causative genes described above.
To explore the differences in gene alternations between Hp-uninfected intramucosal and advanced DGCs, we examined nine intramucosal DGCs and 60 advanced DGCs, including two from our cohort and 58 from external datasets (Table S5). CDH1 mutations were more frequent in intramucosal DGCs (6 of 9, 67%) than in advanced DGCs (16 of 60, 27%) (p = 0.02, q = 0.11, Fig. 4a). In contrast, TP53 mutations were significantly more frequent in advanced DGCs (31 of 60, 52%) than in intramucosal DGCs (0 of 9, 0%) (p < 0.01, q = 0.03). CDH1 and TP53 co-mutated DGC occurred in only 10% (6 of 60) of advanced DGC (Fig. 4b). CDH1-mutated/TP53 wild-type advanced DGC accounted for 17% (10 of 60) and TP53-mutated/CDH1 wild-type advanced DGC accounted for 42% (25 of 60) of total advanced DGCs. The difference of CDH1 and TP53 mutation frequencies between intramucosal and advanced DGCs suggests that Hp-uninfected advanced DGCs may be more likely develop from precursor lesions other than CDH1-mutated intramucosal DGCs.

Driver gene mutation frequencies in Hp-uninfected intramucosal and advanced DGCs. a Comparison of mutation frequencies in driver genes between Hp-uninfected intramucosal DGCs (n = 9) and advanced DGCs (n = 60). p and q values were provided for CDH1 and TP53 calculated by two-tailed Fisher’s exact test with Benjamini–Hochberg adjustment. There were no significant differences in other genes. b Detailed CDH1/TP53 mutation profiles for Hp-uninfected intramucosal and advanced DGCs
Comparison of Hp-uninfected normal gastric mucosa, intramucosal DGC, and advanced DGC
To investigate the mechanism of Hp-uninfected intramucosal DGC development, we compared the mutations and mutational signatures between intramucosal DGCs and normal gastric glands. WES also indicated that mutations accumulated with age at a frequency of 0.33 mutations/exome/year in the normal gastric gland (Fig. 5a, Table S6). The number of mutations in intramucosal DGCs was not significantly higher than that in normal glands when the age of the patient was considered (p = 0.40, Fig. 5a). In contrast, advanced DGCs had a significantly higher number of mutations than intramucosal DGCs and normal glands (p = 0.001, p < 0.001, respectively, Fig. 5a).

Mutation number and mutational signature analyses of Hp-uninfected normal gastric glands, intramucosal DGCs, and advanced DGCs. a Correlation between age and number of mutations in Hp-uninfected normal glands, intramucosal DGCs and advanced DGCs. The number of mutations in single glands from normal gastric epithelium without Hp infection was plotted against patient age (n = 18, blue dots). A regression line (blue dotted line) assuming an intercept of zero is shown, with R2 and coefficient values. Orange and gray dots indicate the number of mutations in intramucosal and advanced DGCs respectively. The two-tailed Mann–Whitney U test for comparison between normal glands and intramucosal DGCs showed no significant difference (p = 0.40). In contrast, advanced DGCs had a significantly higher number of mutations than intramucosal DGCs and normal glands (p = 0.001, p < 0.001, respectively). b Relative contribution of COSMIC SBSs in Hp-uninfected normal glands, intramucosal DGCs, and advanced DGCs. c Proportion of mutations allocated to SBS 5/40 in intramucosal DGCs and normal glands. SBS 5/40 in intramucosal DGCs was relatively higher than that in normal glands (p = 0.07, two-tailed Mann–Whitney U test). d Correlation between age and number of mutations allocated to SBS 5/40 in normal glands (blue dots) and intramucosal DGCs (orange dots). Blue dotted line shows regression line assuming an intercept of zero for normal glands. Intramucosal DGCs showed a relatively larger number of mutations than normal glands (p = 0.08, two-tailed Mann–Whitney U test)
Next, we performed mutational signature analysis, as different mutational processes may contribute to the accumulation of mutations in a cell, with each imprinting a mutational signature on the cell genome. COSMIC SBSs 1, 5, and 40 accounted for the majorities of normal glands and intramucosal DGCs (Fig. 5b). SBS 1 is known as an age-related signature [34]. The underlying mechanism of SBS 1 is likely deamination of 5-methylcytosine at CpG sites, leading to a C > T transition. The relative contributions of SBS 1 did not significantly differ between the normal gland and intramucosal DGC (p = 0.40). In addition, the number of mutations allocated to SBS 1 did not differ between the normal gland and intramucosal DGC when considering the age of the patient (p = 0.92). SBS 5 is characterized by C > T and T > C transitions [34]. SBS 40 is similar to SBS 5, making it difficult to estimate their separate contributions [13, 29]. Therefore, we counted SBS 5 and SBS 40 together (designated as SBS 5/40). SBS 5/40 also accumulates with age, although the underlying mutational processes are not well understood [34]. The contribution of SBS 5/40 tended to be higher in intramucosal DGCs than in normal glands (p = 0.07, Fig. 5c). This trend did not change when the number of mutations allocated to SBS 5/40 was compared while considering the age of the patient (p = 0.08, Fig. 5d). This difference in mutational signature spectra suggests that intramucosal DGC and normal glands undergo different mutational processes despite similar numbers of mutations.
To further investigate the mechanism underlying the progression from intramucosal DGC to advanced DGC, we compared the mutational signatures. The contribution of SBS 5/40 was lower and that of SBS 17b was higher in advanced DGCs than in intramucosal DGCs (Fig. 5b). Although the etiology of SBS 17b is not well understood, its possible link to damage inflicted by reactive oxygen species has been reported [35]. This result suggests that other or additional mutational processes are involved in the development of advanced DGCs compared to intramucosal DGCs.
Discussion
In the present study, we found that 67% (6 of 9) of Hp-uninfected intramucosal DGCs harbored CDH1 somatic mutations, and the six DGCs with CDH1 somatic mutations were histologically ‘pure’ SRCCs without PDA components. Hp-uninfected intramucosal DGCs harbored similar numbers of mutations as normal gastric glands and few driver mutations other than those in CDH1. These results suggest that CDH1 somatic mutations are driver mutations for the development of Hp-uninfected ‘pure’ SRCCs.
The median Ki-67 index of ‘pure’ SRCCs was 2.4%, which is similar to that in a previous report, indicating their low proliferative capacity [16]. Interestingly, five cases with ‘pure’ SRCCs whose endoscopic images could be reviewed showed no progression over 2–5 years. These results suggest that Hp-uninfected ‘pure’ SRCCs have an indolent feature that makes them less likely to become invasive. Yorita et al. also reported in a retrospective study that Hp-uninfected early SRCCs were less likely to be invasive cancers [17]. Furthermore, all 15 ‘pure’ SRCCs, including those that were not genetically analyzed, were located near the gastric angle and characterized by reduced E-cadherin expression. In animal studies, the loss of E-cadherin function due to Cdh1 deficiency resulted in the development of SRCC-like atypical cells, but it was not sufficient for invasive cancer formation [26, 30]. Factors such as chronic stimulation by Helicobacter infection or nitroso compounds and Trp53 mutations are required for the invasion of SRCC in Cdh1 knockout mice [26, 30, 36, 37]. The present study showed that 10% of advanced DGCs harbored both CDH1 and TP53 mutations in contrast to none of the CDH1-mutated intramucosal SRCCs harboring TP53 mutation. These results suggest that some CDH1-mutated intramucosal SRCCs become advanced DGCs with the addition of TP53 mutations, as shown in animal experiments [26, 36]. This hypothesis is supported by a study showing that p53 is not aberrantly expressed in intramucosal HDGCs, whereas its expression is altered in invasive HDGCs [38]. However, the discrepancy in the CDH1 and TP53 mutation frequencies between intramucosal and advanced DGC suggests that CDH1-mutated intramucosal DGCs make a relatively small contribution to advanced DGC formation in the Hp-uninfected stomach and that unknown TP53-mutated precancerous lesions might exist.
In addition to Hp infection, smoking has been reported to increase the risk of DGC compared to in non-smokers [39, 40]. Previous studies reported that the prevalence of smoking was high in patients with Hp-uninfected DGCs [41, 42]. However, SBS 4, the signature associated with smoking, was not observed in the present study (Fig. 5b), which is consistent with the findings of a recent genetic study of DGC [43]. Further analysis is required to clarify the carcinogenic effect of smoking on DGC development. Recent studies reported alcohol-related mutagenesis in gastric cancers in patients with alcohol consumption habit and ALDH2-defective alleles [12, 43]. However, SBS 16, an alcohol-related mutational signature, was not detected in our cohort (Fig. 5b), possibly because of the small number of mutations detected by WES analysis (Table 2, Fig. 3a).
We confirmed that somatic mutations accumulate with age at a frequency of 0.33 mutation/exome/year in the Hp-uninfected normal gastric epithelia. This mutation frequency is lower than that in the normal colon, as previously determined by WES [11]. The predominant mutational signature of normal gastric epithelia without Hp infection was SBS 5/40, followed by SBS 1. This result differs from that observed for the normal colon and small intestine, in which SBS 1 is most predominant [11, 14].
This study had several limitations. First, the cohort size was small. Second, the size of intramucosal DGC lesion in this study was small. The small lesion size may have contributed to the very slow disease progression, such as that observed for SRCC foci found in HDGC [44]. Further studies are needed to evaluate larger-sized early DGCs. Third, even cases without CDH1 mutation showed reduced E-cadherin expression. We could not determine the mechanism by which E-cadherin expression was attenuated despite the absence of CDH1 mutations and copy number alternations. Although we performed CDH1 promoter methylation analysis, we failed to obtain informative results. The methylation rates in intramucosal DGCs were generally low, although they tended to be higher than those in the adjacent normal mucosa (data not shown). Recent genetic analysis of Hp-uninfected DGC reported that CDH1 mutations were found in only one of seven cases, in contrast to the high prevalence of CDH1 mutations observed in our study [42]. The low amount of input DNA (10 ng in every case) and the long period of paraffin embedding (median 6 years) may have affected the quality of the analysis, resulting in a low frequency of detection of CDH1 mutations [45].
In conclusion, we demonstrated that CDH1 somatic mutations are positively selected as driver mutations in Hp-uninfected sporadic intramucosal SRCCs and that Hp-uninfected intramucosal SRCCs may progress slowly and be less likely to develop into advanced DGCs.
References
Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–31.
Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med. 1991;325:1132–6.
Shimizu T, Marusawa H, Matsumoto Y, Inuzuka T, Ikeda A, Fujii Y, et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology. 2014;147:407–17.
Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–6.
Yamamoto Y, Fujisaki J, Omae M, Hirasawa T, Igarashi M. Helicobacter pylori-negative gastric cancer: characteristics and endoscopic findings. Dig Endosc. 2015;27:551–61.
Zamani M, Ebrahimtabar F, Zamani V, Miller WH, Alizadeh-Navaei R, Shokri-Shirvani J, et al. Systematic review with meta-analysis: the worldwide prevalence of Helicobacter pylori infection. Aliment Pharmacol Ther. 2018;47:868–76.
Kamada T, Haruma K, Ito M, Inoue K, Manabe N, Matsumoto H, et al. Time trends in Helicobacter pylori infection and atrophic gastritis over 40 years in Japan. Helicobacter. 2015;20:192–8.
Inoue M. Changing epidemiology of Helicobacter pylori in Japan. Gastric Cancer. 2017;20:3–7.
Bertuccio P, Chatenoud L, Levi F, Praud D, Ferlay J, Negri E, et al. Recent patterns in gastric cancer: a global overview. Int J Cancer. 2009;125:666–73.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Kakiuchi N, Yoshida K, Uchino M, Kihara T, Akaki K, Inoue Y, et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature. 2020;577:260–5.
Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–7.
Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020;580:640–6.
Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–4.
Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49.
Horiuchi Y, Fujisaki J, Yamamoto N, Shimizu T, Miyamoto Y, Tomida H, et al. Biological behavior of the intramucosal Helicobacter pylori-negative undifferentiated-type early gastric cancer: comparison with Helicobacter pylori-positive early gastric cancer. Gastric Cancer. 2016;19:160–5.
Yorita N, Ito M, Boda T, Kotachi T, Nagasaki N, Abuduwaili M, et al. Potential of Helicobacter pylori-uninfected signet ring cell carcinoma to invade the submucosal layer. J Gastroenterol Hepatol. 2019;34:1955–62.
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–5.
van der Post RS, Vogelaar IP, Carneiro F, Guilford P, Huntsman D, Hoogerbrugge N, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet. 2015;52:361–74.
Machado JC, Oliveira C, Carvalho R, Soares P, Berx G, Caldas C, et al. E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene. 2001;20:1525–8.
Cho SY, Park JW, Liu Y, Park YS, Kim JH, Yang H, et al. Sporadic early-onset diffuse gastric cancers have high frequency of somatic CDH1 alterations, but low frequency of somatic RHOA mutations compared with late-onset cancers. Gastroenterology. 2017;153:536–49.
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–82.
Network CGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–7.
JGC Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer. 2011;14:101–12.
Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW, et al. Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell. 2015;28:800–14.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9.
Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171:1029–41.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Mimata A, Fukamachi H, Eishi Y, Yuasa Y. Loss of E-cadherin in mouse gastric epithelial cells induces signet ring-like cells, a possible precursor lesion of diffuse gastric cancer. Cancer Sci. 2011;102:942–50.
Majewski IJ, Kluijt I, Cats A, Scerri TS, de Jong D, Kluin RJ, et al. An α-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol. 2013;229:621–9.
Sahasrabudhe R, Lott P, Bohorquez M, Toal T, Estrada AP, Suarez JJ, et al. Germline mutations in PALB2, BRCA1, and RAD51C, which regulate DNA recombination repair, in patients with gastric cancer. Gastroenterology. 2017;152:983–6.
Kuboki Y, Yamashita S, Niwa T, Ushijima T, Nagatsuma A, Kuwata T, et al. Comprehensive analyses using next-generation sequencing and immunohistochemistry enable precise treatment in advanced gastric cancer. Ann Oncol. 2016;27:127–33.
Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47:1402–7.
Tomkova M, Tomek J, Kriaucionis S, Schuster-Böckler B. Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Biol. 2018;19:129. https://doi.org/10.1186/s13059-018-1509-y.
Shimada S, Mimata A, Sekine M, Mogushi K, Akiyama Y, Fukamachi H, et al. Synergistic tumour suppressor activity of E-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut. 2012;61:344–53.
Humar B, Blair V, Charlton A, More H, Martin I, Guilford P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009;69:2050–6.
van der Post RS, Gullo I, Oliveira C, Tang LH, Grabsch HI, O’Donovan M, et al. Histopathological, molecular, and genetic profile of hereditary diffuse gastric cancer: current knowledge and challenges for the future. Adv Exp Med Biol. 2016;908:371–91.
Ye W, Ekström AM, Hansson LE, Bergström R, Nyrén O. Tobacco, alcohol and the risk of gastric cancer by sub-site and histologic type. Int J Cancer. 1999;83:223–9.
Koizumi Y, Tsubono Y, Nakaya N, Kuriyama S, Shibuya D, Matsuoka H, et al. Cigarette smoking and the risk of gastric cancer: a pooled analysis of two prospective studies in Japan. Int J Cancer. 2004;112:1049–55.
Horiuchi Y, Fujisaki J, Ishizuka N, Omae M, Ishiyama A, Yoshio T, et al. Study on clinical factors involved in Helicobacter pylori-uninfected undifferentiated type early gastric cancer. Digestion. 2017;96:213–9.
Kiso M, Urabe Y, Ito M, Masuda K, Boda T, Kotachi T, et al. Clinical and genomic characteristics of mucosal signet-ring cell carcinoma in Helicobacter pylori-uninfected stomach. BMC Gastroenterol. 2020;20:243. https://doi.org/10.1186/s12876-020-01387-9.
Suzuki A, Katoh H, Komura D, Kakiuchi M, Tagashira A, Yamamoto S, et al. Defined lifestyle and germline factors predispose Asian populations to gastric cancer. Sci Adv. 2020. https://doi.org/10.1126/sciadv.aav9778.
Guilford P, Humar B, Blair V. Hereditary diffuse gastric cancer: translation of CDH1 germline mutations into clinical practice. Gastric Cancer. 2010;13:1–10.
Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin Chem. 2015;61:64–71.
Acknowledgements
This research was supported by AMED under Grant Number JP20gm1110011 [S.O.], JSPS Scientific Research on Innovative Areas under Grant Number 15H05909 [S.O.], and JSPS and MEXT KAKENHI under Grand Numbers 17K09381 [S.M.], 17K09380 [T.S.], 20H03513 and 20K21543 [N.K.]. The authors thank the Endoscopy Unit, Kyoto University Hospital, for endoscopic sampling; the Center for Anatomical, Pathological and Forensic Medical Research, Kyoto University Graduate School of Medicine, for preparing microscope slides; and the Division of Breast Surgery, Department of Surgery, Kyoto University Graduate School of Medicine, for laser capture microdissection. We would also like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Contributions
MN, NK, SM and TF conceived the study. MN, TF, AY, YK and SU collected and provided samples and clinical information. SM and TS performed histological analysis. MN, NK, TH, YT and YY conceived experiments and analyzed and interpreted the data. MN prepared the figures and drafted the manuscript, which was then extensively edited by NK, SM and TC. All authors were involved in writing the paper and had final approval of the submitted versions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nikaido, M., Kakiuchi, N., Miyamoto, S. et al. Indolent feature of Helicobacter pylori-uninfected intramucosal signet ring cell carcinomas with CDH1 mutations. Gastric Cancer 24, 1102–1114 (2021). https://doi.org/10.1007/s10120-021-01191-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-021-01191-8