
Abstract
Epidemiological evidence establishes obesity as an independent risk factor for increased susceptibility and severity to viral respiratory pneumonias associated with H1N1 influenza and SARS-CoV-2 pandemics. Given the global obesity prevalence, a better understanding of the mechanisms behind obese susceptibility to infection is imperative. Altered immune cell metabolism and function are often perceived as a key causative factor of dysregulated inflammation. However, the contribution of adipocytes, the dominantly altered cell type in obesity with broad inflammatory properties, to infectious disease pathogenesis remains largely ignored. Thus, skewing of adipocyte-intrinsic cellular metabolism may lead to the development of pathogenic inflammatory adipocytes, which shape the overall immune responses by contributing to either premature immunosenescence, delayed hyperinflammation, or cytokine storm in infections. In this review, we discuss the underappreciated contribution of adipocyte cellular metabolism and adipocyte-produced mediators on immune system modulation and how such interplay may modify disease susceptibility and pathogenesis of influenza and SARS-CoV-2 infections in obese individuals.
Similar content being viewed by others

Introduction
The obesity pandemic continues unabated, afflicting >500 million people worldwide. The U.S. has the highest mean adult body mass index (BMI) among high-income countries,1 with 42.4% of adults considered obese (BMI 30-35).2 It is anticipated that roughly half of the U.S. population will be obese by 2030, with morbid obesity (BMI > 35) impacting an estimated 24% of Americans.3 Obesity promotes chronic low-grade systemic and tissue inflammation that is central to the pathogenesis of various metabolic diseases including type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), cardiovascular disease (CVD), and various cancers.4 Although commonly overlooked, obesity is also linked with elevated susceptibility and risk of developing serious complications to viral pneumonias.5 Epidemiological evidence from the 2009 H1N1 influenza pandemic established obesity as an independent risk factor for increased disease severity and mortality,6 including pulmonary and cardiovascular damage.7 Similar clinical observations in obese individuals are reported in SARS-CoV-2 (colloquially known as COVID-19) pandemic. Despite the clinical significance, the key cellular and molecular mechanisms underlying obese susceptibility to infection are not well defined.
Immune cells are the conventional source of inflammatory milieu in obesity. Intuitively, immune cell contributions to obesity-associated susceptibility to infections is the most well-examined to date. Alterations in the systemic and tissue inflammatory milieu impact cellular metabolic pathways and inflammatory vigor.8 Obesity-associated dysregulation of immune responses, inflammatory vigor,9 and adipose tissue (AT) immune cell infiltration and function10,11,12 play major roles in infectious disease pathogenesis.13,14 However, this dogmatic view tends to overlook the contribution of the primary cell type in AT: the adipocyte. Adipocytes, akin to immune cells, can impact the immune system locally and systemically15,16 via underappreciated utility of multiple immune-modulatory pathways.17,18 In this context, adipocyte cellular metabolism and inflammatory phenotype are interconnected.19 Thus, the role of adipocyte cellular metabolism and inflammatory/pathologic capacity in instructing immune responsiveness to viral pathogens is of potential significance.
Here we briefly summarize the mounting clinical evidence supporting the premise that obesity is an independent risk factor for infectious disease morbidity and mortality, with emphasis on influenza and SARS-CoV-2. While the impact of obesity on immune cells in influenza has been comprehensively reviewed,13 such studies in SARS-CoV-2 are nascent. Importantly, the focus on adipocyte inflammatory contributions in the context of either infection is finite. Hence, by drawing parallels between influenza and SARS-CoV-2 outcomes in obesity, here we emphasize the need for additional studies to improve our understanding of how obesity shapes outcomes of viral pneumonias. Thus, in light of the dominant impact of obesity on adipocyte function, we discuss the understudied immune and inflammatory capacities of adipocytes, the methods in which they communicate with the immune system, and the known pathologies to which these responses contribute. We then highlight the contribution of adipocyte cellular metabolism to immune system dysfunctions. Lastly, we invoke the idea of “pathogenic adipocytes,” an adipocyte population with altered metabolic and inflammatory states which potentially arises in obesity and detrimentally impacts immune responses to infection.
Epidemiological evidence
Clinical studies have recently begun to explore the role of obesity in infection. Although few studies have invoked a protective role,20,21 the majority of epidemiological data points toward the detrimental effects of obesity on infection susceptibility and disease severity.5,14,22,23 The variety of pertinent infections is broad, and ranges from pandemic-causing strains of pathogens to common community- and hospital-acquired infections (Table 1). For example, obese individuals have higher incidences of community-acquired skin, fungal foot, orofacial, acute respiratory (e.g., pertussis), gastrointestinal (e.g., H. pylori), and urinary tract infections (particularly in obese pregnant and post-partum women).22,24,25 Obese individuals also have an increased likelihood of contracting nosocomial infections, such as post-surgical site and catheter-acquired infections, which are attributed to metabolic factors such as hyperglycemia and diabetes.22,26 Further, while in critical care, obese individuals are at an elevated risk of developing and succumbing to life-threatening conditions (e.g., invasive candidiasis and E. coli bloodstream infections).27,28 However, while obese individuals have an increased risk of developing sepsis from infection,29 they are less likely to die from it.30 While most of these studies have been conducted in adults, congruent effects are observed in adolescents.31
Obesity’s impact on disease severity isn’t limited to common infections. While frequently linked to worsened disease severity during seasonal influenza A virus infections,32 obesity was also established as an independent risk factor for severe pulmonary disease and mortality during the 2009 H1N1 influenza pandemic.6 Subsequently, obesity was associated with elevated disease severity in coronavirus-induced outbreaks including Severe Acute Respiratory Syndrome (SARS)33 and Middle East Respiratory Syndrome (MERS).34 Most recently, systematic reviews of Coronavirus Disease 2019 (SARS-CoV-2; COVID-19) cases revealed that obese individuals are at an increased risk for hospitalization, respiratory failure, ICU admission, mechanical intubation, and mortality.35,36,37,38 Both H1N1 influenza and SARS-CoV-2 viral shedding is also prolonged in obese individuals,32,39 highlighting the negative impact obesity can have on efforts to control spread of pandemic strains of disease. Further, elevated risk of progression to severe pneumonia due to SARS-CoV-2 and influenza is coupled with obesity-associated metabolic sequelae, including T2D,40,41 CVD,7,42 and NAFLD.43,44,45 Given the overlaps between disease susceptibility and pathogenesis in either influenza and SARS-CoV-2 infection, inciting the discussion of potential obesity-impacted cellular and molecular mechanisms could promote the discovery and development of novel therapeutics.
Adipocytes: altered by obesity
Traditionally, adipocytes play a key role in energy homeostasis.46 However, recent insights have broadened our knowledge of adipocytes as important endocrine and inflammatory cells47 with immune cell-like inflammatory properties.18 In fact, it is now well appreciated that adipocytes can take either beneficial or detrimental fates in disease.17 Adipocytes can sense and respond to bacterial, fungal, or viral components via expression of various TLRs,48 and adipocytes can present antigens to surrounding immune cells via MHC Class II expression.49 Adipocyte-derived secretory chemokines and acute-phase reactants, such as SAA3-α1 acid glycoprotein, CRP-relative pentraxin-3, and lipocalins can amplify local immune cell inflammatory cytokine expression.50 The role of adipocytes in systemic inflammation was emphasized during the “fatless” study, in which inactivation of adipocytes resulted in a strongly reduced septic LPS response.51 Adipocyte-secreted factors (e.g., adiponectin, leptin) impact systemic and tissue function and cellular metabolism resulting in altered immune cell responsiveness to external stimuli52 and AT inflammation.17 Adiponectin activity is viewed as anti-inflammatory, as it inhibits neutrophil ROS production, promotes macrophage IL-10 secretion, limits TLR-mediated NF-kB activation, and reduces Th1 T-cell polarization.53,54 Conversely, leptin is viewed as a pro-inflammatory mediator, as it enhances neutrophil ROS production and ICAM-1 expression, promotes macrophage proinflammatory cytokine (e.g., TNF, IL-6, IL-12) production, and skews T-cell polarization toward Th1 and Th17 subtypes.55
However, this adipocyte-immune cell interplay is modified by obesity. Systemically, obesity’s impact on various immune cells, summarized in Table 2, has been reviewed.56 In brief, obesity increases numbers and inflammatory capacity of circulating monocytes, macrophages, neutrophils, and Th1/Th17 CD4+ T cells.57,58,59 In contrast, the effector functions of eosinophils, Natural Killer (NK) cells, Dendritic Cells (DC), CD8+ T cells, and B cells are decreased.60,61,62,63,64,65 Within AT, obesity-associated adipocyte hypertrophy and hyperplasia leads to extensive AT remodeling that includes impaired AT angiogenesis, extracellular matrix protein deposition, and hypoxia-induced adipocyte pyroptosis.66 In response to pyroptotic adipocytes, recruited AT monocytes and macrophages increase AT inflammation.67,68 Further, direct communication between adipocytes and AT-resident T cells via MHC-II49 and macrophages via IL-1β69 instigate AT inflammation. This ultimately results in the surviving hypertrophic adipocytes having decreased adiponectin70 and increased leptin secretion.71,72 Obesity also impairs adiponectin exocytosis73 and decreases adiponectin receptor surface expression,74 while increased adipocyte leptin secretion is associated with changes in insulin regulation75 and IL-6 expression in AT.76 Despite such advances, further studies are needed to establish direct molecular mechanisms underlying adiponectin and leptin secretion in obesity. Adipocyte release of free fatty acids (FFA) is also enhanced in the obese state. Increased FFA levels promote inflammatory processes within monocytes and macrophages.77 In contrast, excessive exposure to FFA can decrease the cytotoxic capabilities of NK cells78 and cytotoxic T cells.79
Recent reports also show that adipocytes secrete various proinflammatory mediators (e.g., IL-6, TNF, IL-1β) in response to external stimuli and that such responsiveness is further exacerbated in obesity.19 Locally, adipocyte IL-6 production regulates macrophage infiltration into AT.80 Systemically, IL-6 is elevated in the serum of obese individuals,81 with one-third of circulating IL-6 calculated to originate from AT.82 These interactions ultimately result in macrophages, neutrophils, and Th1/Th17 CD4+ T-cell skewing toward proinflammatory states and induction of AT inflammation.69,83,84 In contrast to their circulating counterparts, tissue NK, DC, CD8+ T, memory T, and B cells also gain proinflammatory phenotypes within AT.83,85,86 Interestingly, reduction of eosinophils within AT in obesity87 is linked with overall AT inflammation, as these cells regulate AT inflammation88 and promote the development of anti-inflammatory macrophages.89 Together, these findings invoke the importance of adipocyte contributions to an AT inflammatory microenvironment and immune cells function in obesity. How adipocyte-altered immune cell function within AT shapes the overall responses to an infection remains unanswered.
Obesity and the immune system
Both adipocyte and immune cell inflammatory output contribute to increased systemic proinflammatory cytokine (e.g., IL-6, TNF, C-reactive protein) levels in obese individuals.11,82 However, the mechanisms connecting chronic low-grade inflammation and immune dysfunction in obesity remain undefined. Other disease states associated with chronic, low-grade inflammation such as aging90 and some cancers91 are linked with immune cell exhaustion and decline in immune cell function. This impaired immunological state robustly impacts host’s responses to pathogens and vaccine efficacies.92 Hence, obesity-associated chronic inflammation may similarly favor immune cell exhaustion/tolerance resulting in a premature immunosenescent-like state and dampened response to pathogens. Notably, obesity leads to splenic NK senescence, resembling changes seen in aging.93 Further, plasma from obese individuals is sufficient to induce senescence in cytotoxic T cells,94 and increased leptin levels (hallmark of obesity) induce B cell senescense.95 Altered immune responses of these cells to influenza in obesity96 may lead to impaired control of viral replication and increased disease severity. Whether obesity-induced senescence also impacts cellular regulatory process of inflammatory cascades97,98 in adipocytes should be examined, given their ability to directly respond to infection.99 If so, it is plausible that during influenza infection, immune cell senescence might contribute to increased viral load, whereupon dysregulated and continuous hyperinflammation by non-hematopoietic cells shapes inflammation-induced lung damage13 and impairs wound healing.100
In contrast, obesity-associated chronic immune activation could lead to an increased propensity of developing a dysregulated hyperinflammatory state that results in a “cytokine storm.” Cytokine storm is typically associated with severe or fatal immune reactions to pathogens or therapeutic interventions.101,102 Thus, obesity-mediated increases of inflammatory propensity in immune cells outlined in Table 2 might contribute to increased likelihood of developing cytokine storm. Obese mice which received stimulatory immunotherapy (anti-CD40/IL-2) develop lethal cytokine storm at an elevated rate.103 Similarly, obese mice, compared to lean counterparts, had exacerbated cytokine storm in response to Francisella tularensis infection.104 One potential mechanism lies within the ability of type I interferons (IFNs) to promote cytokine storm and viral-induced sepsis.105 Type I IFNs are independently induced by both obesity and viral infection.106,107 Further, type I IFN priming of immune cell108,109,110 or adipocyte19 amplifies their inflammatory vigor. Thus, additional studies focused on how obesity- and infection-elevated type I IFNs impact cellular inflammatory propensity may reveal novel molecular pathways by which obesity skews systemic inflammation toward a cytokine storm-susceptible state.
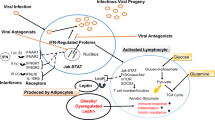
While immunosenescence, delayed hyperinflammatory responses, and cytokine storm theories in obesity seem diametrically opposed, they are not necessarily mutually exclusive. The presence of a delayed immune response potentially bridges initial lack of responsiveness and an ultimately excessive response. As established earlier, not all immune cells are impacted in the same manner by obesity, and these effects may differ depending on organ or microenvironment where the immune cell resides. Increased susceptibility to infection in obese individuals may be dependent on the complex interplay between premature immunosenescence, delayed hyperinflammation by non-immune cells, and combined immune and non-immune cell-dependent cytokine storm. It is also important to consider the contribution of adipocyte inflammatory capacities on the immune system. Adipocytes respond to the elevated circulating endotoxemia present in obese individuals111,112 and communicate with immune cells.113 Thus, it is likely that preferential inflammatory skewing of adipocytes contributes to these aforementioned immunomodulatory effects of obesity via their secreted factors that in unison shape AT and systemic inflammation (Fig. 1). Whether these effects are pathogen-specific remains to be defined. These observations collectively invoke a fine nuance between obesity-associated chronic inflammation and immune cell inflammatory modulation—something that requires in-depth investigation and may be linked to adipocyte function in obesity and infection.

Adipocyte-secreted factors (e.g., Adiponectin, Leptin, Type I IFNs, and IL-6) contribute to homeostatic immune responses and appropriate immune defense mechanisms against infectious agents resulting in pathogen clearance in the lean state. Obesity-associated chronic inflammation leads to expansion in adipocyte number and size. These alterations in adipose tissue unlocks a proinflammatory skewing of adipocyte phenotype and leads to dysregulated secretion of adipocyte-produced mediators (increased in red, decreased in blue). Hence, obesity-dependent changes in adipocyte function can contribute to the immune system being in a state of: (1) Immunosenescence (suppressed immune response against pathogens); (2) Delayed immune inflammation (reduced pathogen clearance and compensatory exacerbated adipocyte inflammation); and (3) “Cytokine storm” (uncontrolled proinflammatory response and tissue damage). Overall, each altered inflammatory state in obesity could individually or synergistically shape immune responses, culminating in worsened disease pathology and increased morbidity and mortality.
Adipocytes and Infection: Pathogenic Links
The cellular and molecular mechanisms underlining the low-grade inflammatory state in obese individuals are not fully understood. However, alterations in cellular metabolism might provide a pathogenic link, as they are critical drivers of immune cell inflammatory function.114,115,116 Various studies have emphasized T cell and macrophage capabilities to sense environmental cues, quickly changing and adapting their cellular metabolic pathways to meet necessary energy demands and reshape their inflammatory/immune functions.117,118,119,120,121,122,123 Further, the contribution of AT macrophages and T cells pro‐inflammatory states to the pathogenesis of obesity-associated metabolic derangements is well established.84,120,121,124 Findings that in vivo alterations of multiple cellular metabolic pathways (e.g., glycolysis, TCA, OXPHOS) ameliorate inflammatory disease severity120,125,126 corroborate this postulated interplay between immune cell metabolism, inflammatory phenotype, and systemic inflammation. Thus, how obesity-associated metabolic alterations of immune cells and adipocytes influence infectious disease pathogenesis should be examined.
Sensing of Type I IFNs produced in response to viral infections shapes cellular function of T cells and macrophages via engagement of the ubiquitously expressed IFNAR receptors. Subsequent activation of the Janus kinase–signal transducer and activator of transcription (JAK–STAT) signaling pathway results in amplification of proinflammatory/antiviral gene expression.127 The ability of a type I IFNs to divergently impact immune cell function has been linked with altered disease pathology. For example, acute type I IFN production enhances DC maturation and monocyte recruitment and activation to sites of infection, which contributes to T-cell activation and establishment of anti-viral responses.128 In contrast, macrophages exposed to IFNβ show suppressed expression of the IFNγ receptor leading to lower IFNγ responsiveness and suppressed phagocytotic effects.129 A recent report suggests that type I IFN sensing by adipocytes resulted in approximately 30% gene expression profile conversion to that found in macrophages and was sufficient to alter adipocyte glycolysis and unlock adipocyte inflammatory potential,19 emphasizing the intimate relationship between these two processes. Additional recent reviews have also highlighted the relevance of immunometabolism in the function of other immune cells.8,130 Thus, the interplay between AT resident/infiltrating immune cells and adipocytes might constitute a key process in shaping cellular metabolism and inflammatory output of both cell types. Importantly, obesity-driven changes in metabolic pathways lead to altered inflammatory phenotypes in immune cells and adipocytes. These findings have led to an appreciation that distinct bioenergetics profiles not only guide immune cell pathogenic functions,131,132,133 but may similarly favor induction of a “pathogenic adipocyte” population—something that could be particularly deleterious to immune responses in an obesity-altered state. Whereas the roles of multiple metabolic pathways (e.g., glycolysis, OXPHOS, TCA cycle, pentose phosphate pathway, fatty acid oxidation/synthesis) in inflammation, cell proliferation, phenotype, and metabolic derangements have been examined in immune cells (Table 3), analysis of these pathways in adipocytes, and specifically obese-adipocytes, are highly limited (Table 4). Thus, given the availability of resources employed in interrogation of metabolic mechanisms underlying immune cell-driven disease pathogenesis, extension of their application to adipocytes is feasible and needed.
Various immune mediators can modulate inflammatory capacity, tissue healing, and viral pathogenesis, including type I IFN (e.g., IFNα, IFNβ) and type III IFNs (e.g., IFNλ).134 While both are necessary for regulation of inflammatory responses, type I IFNs are more often associated with worse disease immunopathology.135,136 Given the relevance of type I and type III IFNs in viral infections, it is not surprising that pathogen manipulation of these axes might play a role in disease pathogenesis.137,138 In fact, early activation of the Type I IFN axis through IFNAR/STAT signaling lead to decreased viral load and mild disease severity in SARS-CoV-2 infections139 and diminished viral replication and diversity in influenza infections in obese mice.140 In contrast, delayed activation of IFNs, but not other proinflammatory cytokines, resulted in poor disease progression during initial stages of SARS-CoV-2 infection.141 This initial delay results in elevated type I IFN responses in late stages of infection142 and increased and persistent viral load, inflammation, and severe disease outcome.139,143,144 Further, neutralizing anti-IFN auto-antibodies145 or inborn errors of type I IFN signaling146,147 were found in subjects that developed severe SARS-CoV-2-associated pneumonia. However, despite such advances, the role of type I IFNs in severe SARS-CoV-2 pneumonia is still controversial and requires further study,148 with most recent reports highlighting that SARS-CoV-2-induced type I IFN dysregulation is cell specific.149 Given that type I IFNs also impact adipocyte metabolism and inflammatory vigor,19 the ultimate effect these changes have on inflammation in obesity and viral infection should be investigated. As adipocytes are not “professional immune cells”, delayed and unchecked adipocyte inflammation might be a major contributor to the excessive inflammatory responses seen in obese individuals.
Few studies have examined the role of adipocyte-secreted factors in modulating immunosenescence leading to adverse infectious outcomes in obesity. Specifically, while obesity-associated chronic inflammation impairs cytokine signaling in hematopoietic stem cells, adiponectin treatment of obese mice restores proper stem cell proliferation and pathogen clearance.150 This suggests that adiponectin’s protective role may restrict basal inflammation via modulation of immune cell progenitor function. These protective effects could be diminished in obesity due to reduced adiponectin levels and negative correlation between adiponectin levels and immune cell sensitivity to LPS.151 The literature also supports the theory that the alteration of adiponectin secretion in obesity increases propensity for cytokine storm, with a focus on respiratory disease. Adiponectin promotes proliferation and wound repair of human bronchial epithelial cells152 and limits excessive lung inflammatory responses to invasive Aspergillosis in lean mice.153 Intuitively, an association between decreased adiponectin levels in obesity and immune cell hyperresponsiveness to influenza A has been suggested,154 highlighting the potential role of obesity-altered adipocytes.
Another key adipocyte-secreted mediator in disease pathogenesis is IL-6. The overproduction of IL-6 is an important risk factor for worse outcomes in H1N1 influenza,155 SARS,156 MERS,157 and SARS-CoV-2.158,159 Tocilizumab-dependent neutralization of IL-6 as a SARS-CoV-2 therapeutic is not fully elucidated. Randomized control trials of Tocilizumab therapy report both a reduction in likelihood of mechanical ventilation or death in patients with severe SARS-CoV-2 pneumonia160,161 and lack of a protective effect.162 These findings reveal that while IL-6 is linked with severe SARS-CoV-2-associated pneumonia, additional studies are needed to determine the best therapeutic approach to IL-6 inhibition, with potential requirement for cell-specific inhibition.149 Notably, adipocyte, but not AT macrophage, IL-6 secretion is the key regulator of WAT inflammation.80 Further, IL-6 secreted by adipocytes in response to inflammatory stimuli exacerbates responses to LPS-induced endotoxemia.163 Thus, adipocyte-derived IL-6 might play a role in the severity of SARS-CoV-2 and influenza-induced pneumonia seen in obese individuals.
Other infectious models also highlight the detrimental contribution of elevated leptin to inflammatory outcomes in obesity. Clinically, elevated plasma leptin levels are correlated with systemic inflammation in obese individuals,164 and leptin signaling through STAT3 promotes early inflammation associated with worse clinical disease during C. difficile infections.165 Experimentally, elevated basal leptin expression in obese mice correlates with increased parasite burdens in visceral leishmaniasis166 and exacerbated cytokine storm leading to increased mortality in response to Francisella tularensis infection.104 Elevated leptin levels contribute to lung tissue damage during H1N1 influenza infection via an IL-6 inducing mechanism.167 LCMV induces a pathologic memory T-cell response in the WAT that leads to adipocyte damage and reduced survival in obese mice. Intriguingly, the culpable memory T-cell population within the WAT is transcriptionally distinct from other tissues.86 Given that leptin limits proliferation and responsiveness of regulatory T cells while having opposite effects on effector T cells,168 inflammation-skewed adipocytes secreting excess amounts of leptin might play a role in the development of the culpable hyperinflammatory memory T cells, a phenotype that might extend to viral pneumonias in obesity.
Adipocyte inflammation: future implications
Susceptibility to severe infection isn’t limited to the innate immune response to a pathogen. Obesity is also associated with decreased efficacy of vaccines. Inadequate levels of protective anti-Hepatitis B antibodies have been reported in obese individuals.169 Significant declines in protective levels of antibodies after immunization have been reported in obese individuals that received either the Hepatitis A or rabies vaccination.170 Vaccinated obese adults have twice the risk of influenza or influenza-like illness despite equal serological response to vaccination.171 Similar trends are seen in obese pediatric patients that receive tetanus vaccines.172 While these reports are limited in the breadth of vaccines affected by obesity, it is evident that the obese state negatively alters the adaptive immune system, and that bodyweight should be a considered variable during clinical trials of vaccines, most pertinently the SARS-CoV-2 vaccine. However, although direct mechanisms behind this observation remain unclear, contribution of AT inflammation in this setting should be considered. B cells that reside within AT are skewed toward a pro-inflammatory state during obesity, a state that limits their effector function and capabilities to produce antibodies.173,174 Obesity also decreases the numbers and diminishes activation of T follicular helper cells in peripheral lymphoid organs,175 potentially impacting proper development of mature B Cells. Finally, metabolism plays an important regulatory role in shaping the humoral immune response.176 Together, these observations invoke following questions: Does adipocyte inflammation play a role in B cell phenotypes within AT? Can adipocyte inflammatory capacity impact T and B cell metabolism and function in AT or in peripheral immune organs via secreted factors? If so, what impact does adipocyte inflammation have on vaccine efficacy, and importantly the efficacy of novel vaccines for pandemic strains of influenza and SARS-CoV-2?
The adipocyte is an important regulator of immune system homeostasis. During normal metabolic conditions, adipocytes modulate immune cells via their secreted mediators. The effect of obesity on adipocytes impacts the immune system’s capacity to respond to infection. On the other side of the spectrum, malnourished and underweight individuals are also more susceptible to infectious disease severity. While this is attributed to immune dysfunction due to immune cell nutrient starvation, recent advances suggest that decreased circulating levels of adipocyte-produced leptin play a significant role,55 and allude to adipocyte nutrient starvation impacting adipocyte-immune cell communication. Further, the suppression of murine adipocyte inflammation promotes metabolic disease.177 These findings suggest a basal level of adipocyte inflammation is critical to mount a proper immune response. Thus, being in a metabolic state that significantly alters adipocyte inflammatory capacity in either direction could be detrimental. Systematic reviews have revealed a “J-shaped curve”-distribution between BMI and risk of all-infection mortality178 and, notably, influenza-associated pneumonia,179 mirroring the relationship between obesity and all-cause mortality.180,181 While being slightly overweight (BMI 25-29.9) has been reported to be protective from infection-associated disease severity, further progression in increasing BMI (obese – BMI 30–34.9, morbidly obese – BMI 35 + ) increases susceptibility to severe infection.180 This is possibly attributed to obesity-associated inflammation having a detrimental effect on adipocyte function, either mitigating their protective capabilities and altering the immune response or driving protective traits (e.g., inflammatory response) to a point where they are harmful toward the host. If adipocytes are critical mediators of inflammatory responses, then maintaining a proper balance of AT inflammation is important, as being on either side of this “J-shaped curve” is unfavorable to host defenses against pathogens. Together, these observations invoke the following salient questions that address the immune contribution of adipocyte inflammation: Is there an ideal level of adipocyte inflammation that enhances immune responses to infection? How do we define the threshold where adipocyte inflammatory effects become detrimental to the host? Does activation of specific metabolic pathways in adipocytes regulate such threshold? Is adipocyte inflammatory capacity negatively impacting obese individuals combatting viral pneumonias such as influenza or SARS-CoV-2?
Concluding remarks
In summary, multiple pandemics (e.g., 2009 H1N1 Influenza, SARS-CoV-2) reveal obesity as an important risk factor for increased disease morbidity and mortality. Considered alongside the global trend toward a more obese population, it is imperative to develop a better understanding of the mechanisms behind obese susceptibility to infection. In this review, we highlighted a new and exciting area of long-neglected research: adipocyte metabolism and inflammation in viral pneumonias. Being skewed toward a proinflammatory state, alterations in adipocyte-intrinsic cellular metabolism impacts adipocyte-immune cell crosstalk via changes in adiponectin, leptin, and IL-6, as well as other pro-inflammatory mediators (e.g., TNF, IL-1, type I IFNs, etc.) is dysregulated during obesity. Published reports demonstrate the contributions of “pathogenic immune cells”131,132,133,182 and “sick fat”17 to inflammatory and metabolic disease. As not all obese individuals have the same susceptibility to metabolic disease due to the existence of healthy vs unhealthy WAT inflammation183 and obesity drives differentiation of pathogenic subsets of inflammatory Th17 cells,184 further studies examining adipocyte function in obesity and infection might reveal that a “pathogenic adipocyte” phenotype similarly contributes to disease severity (Fig. 2a). Importantly, as with immune cells, such adipocytes could have unique transcriptional signatures and functions that would serve as potential biomarkers for increased risk of infection severity in obesity. Through their inflammatory capabilities, “pathogenic adipocytes” may represent a critical underappreciated link between obesity, immune responses, and infection pathogenesis (Fig. 2b). Thus, defining the interconnected metabolic and inflammatory mechanisms that regulate adipocyte’s phenotype and contribution to obesity-associated immunomodulation and increased susceptibility to infection may provide a robust platform for discovery of novel biomarkers and therapeutic targets to reduce the clinical burden of infectious disease in an increasingly obese population worldwide.

a Normal cellular metabolism regulates homeostatic cytokine production in healthy adipocytes. In contrast, altered cellular metabolism favors induction of a “pathogenic adipocyte” population that exhibit dysregulated cytokine and adipokine production which in turn amplifies disease severity. This pathogenic state could arise in obesity due to the altered metabolic and inflammatory microenvironment of adipose tissue. b Adipocytes contribute to homeostatic immune cell responses and pathogen clearance in the lean state. Obesity impacts adipocyte inflammatory phenotype, which could result in induction of pathogenic adipocytes. This results in dysregulated immune cell responses and culminates as altered pathogen clearance.
References
Finucane, M. M. et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 377, 557–567 (2011).
Hales, C. M., Carroll, M. D., Fryar, C. D. & Ogden, C. L. Prevalence of obesity and severe obesity among adults: United States, 2017-2018. NCHS. Data Brief. 360, 1–7 (2020).
Ward, Z. J. et al. Projected U.S. state-level prevalence of adult obesity and severe obesity. N. Engl. J. Med. 381, 2440–2450 (2019).
Whitlock, G. et al. Body-mass index and cause-specific mortality in 900000 adults: collaborative analyses of 57 prospective studies. Lancet 373, 1083–1096 (2009).
Falagas, M. E. & Kompoti, M. Obesity and infection. Lancet Infect. Dis. 6, 438–446 (2006).
Van Kerkhove, M. D. et al. Risk factors for severe outcomes following 2009 influenza A (H1N1) infection: a global pooled analysis. PLoS Med 8, e1001053 (2011).
Siegers J. Y., et al. A high fat diet increases influenza A virus-associated cardiovascular damage. J. Infectious Dis. 222, 820–831 (2020).
O’Neill, L. A., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Ghigliotti, G. et al. Adipose tissue immune response: novel triggers and consequences for chronic inflammatory conditions. Inflammation 37, 1337–1353 (2014).
Coppack, S. W. Pro-inflammatory cytokines and adipose tissue. Proc. Nutr. Soc. 60, 349–356 (2001).
Gregor, M. F. & Hotamisligil, G. S. Inflammatory mechanisms in obesity. Annu Rev. Immunol. 29, 415–445 (2011).
Lu, J., Zhao, J., Meng, H. & Zhang, X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front Immunol. 10, 1173 (2019).
Honce, R. & Schultz-Cherry, S. Impact of obesity on influenza a virus pathogenesis, immune response, and evolution. Front Immunol. 10, 1071 (2019).
Karlsson, E. A. & Beck, M. A. The burden of obesity on infectious disease. Exp. Biol. Med (Maywood) 235, 1412–1424 (2010).
Martí, A., Marcos, A. & Martínez, J. A. Obesity and immune function relationships. Obes. Rev. 2, 131–140 (2002).
Desruisseaux, M. S., Nagajyothi, Trujillo, M. E., Tanowitz, H. B. & Scherer, P. E. Adipocyte, adipose tissue, and infectious disease. Infect. Immun. 75, 1066–1078 (2007).
Barchetta, I., Cimini, F. A., Ciccarelli, G., Baroni, M. G. & Cavallo, M. G. Sick fat: the good and the bad of old and new circulating markers of adipose tissue inflammation. J. Endocrinol. Invest. 42, 1257–1272 (2019).
Chan, C. C., Damen, M., Alarcon, P. C., Sanchez-Gurmaches, J. & Divanovic, S. Inflammation and immunity: from an adipocyte’s perspective. J. Interferon Cytokine Res 39, 459–471 (2019).
Chan, C. C. et al. Type I interferon sensing unlocks dormant adipocyte inflammatory potential. Nat. Commun. 11, 2745 (2020).
King, P. et al. Impact of obesity on outcomes for patients hospitalised with pneumonia. Eur. Respir. J. 41, 929–934 (2013).
Singanayagam, A., Singanayagam, A. & Chalmers, J. D. Obesity is associated with improved survival in community-acquired pneumonia. Eur. Respir. J. 42, 180–187 (2013).
Huttunen, R. & Syrjanen, J. Obesity and the risk and outcome of infection. Int J. Obes. (Lond.) 37, 333–340 (2013).
Hegde, V. & Dhurandhar, N. V. Microbes and obesity-interrelationship between infection, adipose tissue and the immune system. Clin. Microbiol Infect. 19, 314–320 (2013).
Liu, B. C. et al. Pertussis in older adults: prospective study of risk factors and morbidity. Clin. Infect. Dis. 55, 1450–1456 (2012).
Baik, I., Curhan, G. C., Rimm, E. B., Bendich, A., Willett, W. C. & Fawzi, W. W. A prospective study of age and lifestyle factors in relation to community-acquired pneumonia in US men and women. Arch. Intern. Med. 160, 3082–3088 (2000).
Campbell, K. A. C., Hasan, C., Hutzler, S., Bosco, L. & Risk, J. A. Factors for developing staphylococcus aureus nasal colonization in spine and arthroplasty surgery. Bull. Hospital Jt. Dis. 73, 276–281 (2015).
Barber, K. E., Wagner, J. L., Miller, J. M., Lewis, E. A. & Stover, K. R. Impact of obesity in patients with candida bloodstream infections: a retrospective cohort study. Infect. Dis. Ther. 9, 175–183 (2020).
Paulsen, J. et al. Associations of obesity and lifestyle with the risk and mortality of bloodstream infection in a general population: a 15-year follow-up of 64 027 individuals in the HUNT Study. Int J. Epidemiol. 46, 1573–1581 (2017).
Wang, H. E., Griffin, R., Judd, S., Shapiro, N. I. & Safford, M. M. Obesity and risk of sepsis: a population-based cohort study. Obes. (Silver Spring) 21, E762–E769 (2013).
Kuperman E. F., Showalter, J. W., Lehman, E. B., Leib, A. E., Kraschnewski, J. L. The impact of obesity on sepsis mortality: a retrospective review. BMC Inf. Dis. 13, 377 (2013).
Twig, G. et al. Body mass index and infectious disease mortality in midlife in a cohort of 2.3 million adolescents. Int J. Obes. (Lond.) 42, 801–807 (2018).
Maier, H. E. et al. Obesity increases the duration of influenza A virus shedding in adults. J. Infect. Dis. 218, 1378–1382 (2018).
Moser, J. S. et al. Underweight, overweight, and obesity as independent risk factors for hospitalization in adults and children from influenza and other respiratory viruses. Influenza Other Respir. Viruses 13, 3–9 (2019).
Badawi, A. & Ryoo, S. G. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS-CoV): a systematic review and meta-analysis. Int J. Infect. Dis. 49, 129–133 (2016).
Popkin B. M., et al. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. Obes. Rev. 21, e13128 (2020).
Lighter J., et al. Obesity in patients younger than 60 years is a risk factor for Covid-19 hospital admission. Clin. Infect. Dis. 71, 896–897 (2020).
Goyal P., et al. Clinical characteristics of COVID-19 in New York City. N. Engl. J. Med. 382, 2372–2374 (2020).
Richardson S., et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City Area. JAMA. 323, 2052–2059 (2020).
Moriconi, D. et al. Obesity prolongs the hospital stay in patients affected by COVID-19, and may impact on SARS-COV-2 shedding. Obes. Res Clin. Pr. 14, 205–209 (2020).
Gupta, R., Ghosh, A., Singh, A. K. & Misra, A. Clinical considerations for patients with diabetes in times of COVID-19 epidemic. Diabetes Metab. Syndr. 14, 211–212 (2020).
Allard, R., Leclerc, P., Tremblay, C. & Tannenbaum, T. N. Diabetes and the severity of pandemic influenza A (H1N1) infection. Diabetes Care 33, 1491–1493 (2010).
Madjid, M., Safavi-Naeini, P., Solomon, S. D. & Vardeny, O. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. 5, 831–840 (2020).
Portincasa, P., Krawczyk, M., Smyk, W., Lammert, F. & Di Ciaula, A. COVID-19 and non-alcoholic fatty liver disease: Two intersecting pandemics. Eur. J. Clin. Invest 50, e13338 (2020).
Ji D., et al. Non-alcoholic fatty liver diseases in patients with COVID-19: A retrospective study. J. Hepatol. 2020.
Papic, N. et al. Liver involvement during influenza infection: perspective on the 2009 influenza pandemic. Influenza Other Respir. Viruses 6, e2–e5 (2012).
Chouchani, E. T. & Kajimura, S. Metabolic adaptation and maladaptation in adipose tissue. Nat. Metab. 1, 189–200 (2019).
Halberg, N., Wernstedt-Asterholm, I. & Scherer, P. E. The adipocyte as an endocrine cell. Endocrinol. Metab. Clin. North Am. 37, 753–768 (2008). x-xi.
Lin, Y. et al. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J. Biol. Chem. 275, 24255–24263 (2000).
Deng, T. et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. 17, 411–422 (2013).
Berg, A. H., Lin, Y., Lisanti, M. P. & Scherer, P. E. Adipocyte differentiation induces dynamic changes in NF-kappaB expression and activity. Am. J. Physiol. Endocrinol. Metab. 287, E1178–E1188 (2004).
Pajvani, U. B. et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat. Med 11, 797–803 (2005).
Francisco, V. et al. Obesity, fat mass and immune system: role for leptin. Front Physiol. 9, 640 (2018).
Robinson K., Prins J., Venkatesh B. Clinical review: Adiponectin biology and its role in inflammation and critical illness. Critical Care 15, 221 (2011).
Surendar, J. et al. Adiponectin limits IFN-gamma and IL-17 producing CD4 T cells in obesity by restraining cell intrinsic glycolysis. Front Immunol. 10, 2555 (2019).
Maurya, R., Bhattacharya, P., Dey, R. & Nakhasi, H. L. Leptin functions in infectious diseases. Front Immunol. 9, 2741 (2018).
Agrawal M., Kern P. A., Nikolajczyk B. S. The Immune System in Obesity: Developing Paradigms Amidst Inconvenient Truths. Current Diab. Rep. 17, 87 (2017).
Friedrich, K. et al. Perturbation of the monocyte compartment in human obesity. Front Immunol. 10, 1874 (2019).
Huang, J. Y. et al. Neutrophil elastase regulates emergency myelopoiesis preceding systemic inflammation in diet-induced obesity. J. Biol. Chem. 292, 4770–4776 (2017).
Roberts, H. M. et al. Impact of bariatric surgical intervention on peripheral blood neutrophil (PBN) function in obesity. Obes. Surg. 28, 1611–1621 (2018).
McLaughlin, T., Ackerman, S. E., Shen, L. & Engleman, E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Invest 127, 5–13 (2017).
O’Shea D., Hogan A. E. Dysregulation of natural killer cells in obesity. Cancers (Basel) 11, 573 (2019).
Pizzolla, A. et al. High fat diet inhibits dendritic cell and t cell response to allergens but does not impair inhalational respiratory tolerance. PLoS One 11, e0160407 (2016).
James, B. R. et al. Diet-induced obesity alters dendritic cell function in the presence and absence of tumor growth. J. Immunol. 189, 1311–1321 (2012).
Frasca, D. et al. Obesity decreases B cell responses in young and elderly individuals. Obesity 24, 615–625 (2016).
Kado, T., Nawaz, A., Takikawa, A., Usui, I. & Tobe, K. Linkage of CD8(+) T cell exhaustion with high-fat diet-induced tumourigenesis. Sci. Rep. 9, 12284 (2019).
Giordano, A. et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res 54, 2423–2436 (2013).
Strissel, K. J. et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56, 2910–2918 (2007).
Grant, R. W. & Dixit, V. D. Adipose tissue as an immunological organ. Obes. (Silver Spring) 23, 512–518 (2015).
Watanabe Y., Nagai Y., Takatsu K. Bidirectional crosstalk between neutrophils and adipocytes promotes adipose tissue inflammation. J. Immun. 204 (2020).
Drolet, R. et al. Fat depot-specific impact of visceral obesity on adipocyte adiponectin release in women. Obes. (Silver Spring) 17, 424–430 (2009).
Jernas, M. et al. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. 20, 1540–1542 (2006).
Skurk, T., Alberti-Huber, C., Herder, C. & Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 92, 1023–1033 (2007).
Musovic, S. & Olofsson, C. S. Adrenergic stimulation of adiponectin secretion in visceral mouse adipocytes is blunted in high-fat diet induced obesity. Sci. Rep. 9, 10680 (2019).
Achari A. E., Jain S. K. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int. J. Mol. Sci. 18, 1321 (2017).
Havel, P. J. Role of adipose tissue in body-weight regulation: mechanisms regulating leptin production and energy balance. Proc. Nutr. Soc. 59, 359–371 (2000).
Wueest, S. & Konrad, D. The role of adipocyte-specific IL-6-type cytokine signaling in FFA and leptin release. Adipocyte 7, 226–228 (2018).
Tripathy, D. et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 52, 2882–2887 (2003).
Niavarani, S. R. et al. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer 19, 823 (2019).
Kriebs, A. Free fatty acids clog up cytotoxic machinery. Nat. Rev. Endocrinol. 15, 3 (2018).
Han, M. S. et al. Regulation of adipose tissue inflammation by interleukin 6. Proc. Natl Acad. Sci. USA 117, 2751–2760 (2020).
de Heredia, F. P., Gomez-Martinez, S., & Marcos, A. Obesity, inflammation and the immune system. Proc. Nutr. Soc. 71, 332–338 (2012).
Fontana, L., Eagon, J. C., Trujillo, M. E., Scherer, P. E. & Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 56, 1010–1013 (2007).
Elgazar-Carmon, V., Rudich, A., Hadad, N. & Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res 49, 1894–1903 (2008).
Russo, L. & Lumeng, C. N. Properties and functions of adipose tissue macrophages in obesity. Immunology 155, 407–417 (2018).
Nishimura, S. et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med 15, 914–920 (2009).
Misumi, I. et al. Obesity expands a distinct population of T cells in adipose tissue and increases vulnerability to infection. Cell Rep. 27, 514–524 (2019). e515.
Ferrante, A. W. Jr The immune cells in adipose tissue. Diabetes Obes. Metab. 15, 34–38 (2013).
Brigger D. et al. Eosinophils regulate adipose tissue inflammation and sustain physical and immunological fitness in old age. Nat. Metab. 2, 688–702 (2020).
Wu, D. et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247 (2011).
Trim, W., Turner, J. E. & Thompson, D. Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol. 9, 169 (2018).
Jiang, Y., Li, Y. & Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 6, e1792 (2015).
Crooke, S. N., Ovsyannikova, I. G., Poland, G. A. & Kennedy, R. B. Immunosenescence and human vaccine immune responses. Immun. Ageing 16, 25 (2019).
Gheorghe, A. et al. Oxidative stress and immunosenescence in spleen of obese mice can be reversed by 2-hydroxyoleic acid. Exp. Physiol. 102, 533–544 (2017).
Parisi, M. M. et al. Immunosenescence Induced by Plasma from Individuals with Obesity Caused Cell Signaling Dysfunction and Inflammation. Obes. (Silver Spring) 25, 1523–1531 (2017).
Frasca, D., Diaz, A., Romero, M. & Blomberg, B. B. Leptin induces immunosenescence in human B cells. Cell Immunol. 348, 103994 (2020).
Smith, A. G., Sheridan, P. A., Harp, J. B. & Beck, M. A. Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J. Nutr. 137, 1236–1243 (2007).
Liu, J. & Cao, X. Cellular and molecular regulation of innate inflammatory responses. Cell Mol. Immunol. 13, 711–721 (2016).
Porritt, R. A. & Hertzog, P. J. Dynamic control of type I IFN signaling by an integrated network of negative regulators. Trends Immunol. 36, 150–160 (2015).
Bouwman, J. J., Visseren, F. L., Bouter, K. P. & Diepersloot, R. J. Infection-induced inflammatory response of adipocytes in vitro. Int J. Obes. (Lond.) 32, 892–901 (2008).
O’Brien, K. B. et al. Impaired wound healing predisposes obese mice to severe influenza virus infection. J. Infect. Dis. 205, 252–261 (2012).
Tisoncik, J. R. et al. Into the eye of the cytokine storm. Microbiol Mol. Biol. Rev. 76, 16–32 (2012).
Guo, X. J. & Thomas, P. G. New fronts emerge in the influenza cytokine storm. Semin Immunopathol. 39, 541–550 (2017).
Mirsoian, A. et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J. Exp. Med 211, 2373–2383 (2014).
Ramos Muniz, M. G. et al. Obesity exacerbates the cytokine storm elicited by francisella tularensis infection of females and is associated with increased mortality. Biomed. Res Int 2018, 3412732 (2018).
Lin, G. L., McGinley, J. P., Drysdale, S. B. & Pollard, A. J. Epidemiology and immune pathogenesis of viral sepsis. Front Immunol. 9, 2147 (2018).
Diebold, S. S. et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424, 324–328 (2003).
Killip, M. J., Fodor, E. & Randall, R. E. Influenza virus activation of the interferon system. Virus Res 209, 11–22 (2015).
Cappelletti, M. et al. Type I interferons regulate susceptibility to inflammation-induced preterm birth. JCI Insight 2, e91288 (2017).
Richard, K., Vogel, S. N. & Perkins, D. J. Type I interferon licenses enhanced innate recognition and transcriptional responses to Franciscella tularensis live vaccine strain. Innate Immun. 22, 363–372 (2016).
Kim, S. et al. Self-priming determines high type I IFN production by plasmacytoid dendritic cells. Eur. J. Immunol. 44, 807–818 (2014).
Clemente-Postigo, M. et al. Metabolic endotoxemia promotes adipose dysfunction and inflammation in human obesity. Am. J. Physiol. Endocrinol. Metab. 316, E319–E332 (2019).
Cani, P. D. et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 (2007).
Choe, S. S., Huh, J. Y., Hwang, I. J., Kim, J. I. & Kim, J. B. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol. (Lausanne) 7, 30 (2016).
Gerriets, V. A. et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest 125, 194–207 (2015).
Johnson, M. O. et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795 (2018). e1719.
Cham, C. M. & Gajewski, T. F. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 174, 4670–4677 (2005).
Mills, E. L. & O’Neill, L. A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 46, 13–21 (2016).
Palsson-McDermott, E. M. & O’Neill, L. A. J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res 30, 300–314 (2020).
Stienstra, R., Netea-Maier, R. T., Riksen, N. P., Joosten, L. A. B. & Netea, M. G. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. 26, 142–156 (2017).
Geltink, R. I. K., Kyle, R. L. & Pearce, E. L. Unraveling the complex interplay between T cell metabolism and function. Annu Rev. Immunol. 36, 461–488 (2018).
Green, W. D. & Beck, M. A. Obesity altered T cell metabolism and the response to infection. Curr. Opin. Immunol. 46, 1–7 (2017).
Pucino, V. et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4(+) T cell metabolic rewiring. Cell Metab. 30, 1055–1074 (2019). e1058.
Gubser, P. M. et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 14, 1064–1072 (2013).
Castoldi, A., Naffah de Souza, C., Camara, N. O. & Moraes-Vieira, P. M. The macrophage switch in obesity development. Front Immunol. 6, 637 (2015).
Nguyen, H. D. et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J. Clin. Invest 126, 1337–1352 (2016).
Angiari, S. et al. Pharmacological activation of pyruvate kinase M2 Inhibits CD4(+) T cell pathogenicity and suppresses autoimmunity. Cell Metab. 31, 391–405 (2020). e398.
Malmgaard, L. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res 24, 439–454 (2004).
Lee, A. J. & Ashkar, A. A. The dual nature of Type I and Type II interferons. Front Immunol. 9, 2061 (2018).
Eshleman, E. M., Delgado, C., Kearney, S. J., Friedman, R. S. & Lenz, L. L. Down regulation of macrophage IFNGR1 exacerbates systemic L. monocytogenes infection. PLoS Pathog. 13, e1006388 (2017).
Buck, M. D., Sowell, R. T., Kaech, S. M. & Pearce, E. L. Metabolic instruction of immunity. Cell 169, 570–586 (2017).
Tesmer, L. A., Lundy, S. K., Sarkar, S. & Fox, D. A. Th17 cells in human disease. Immunol. Rev. 223, 87–113 (2008).
Gaublomme, J. T. et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412 (2015).
Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Mesev, E. V., LeDesma, R. A. & Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol 4, 914–924 (2019).
Major, J. et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369, 6504 (2020).
Davidson, S., Maini, M. K. & Wack, A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J. Interferon Cytokine Res 35, 252–264 (2015).
Lim Y. X., Ng Y. L., Tam J. P., Liu D. X. Human coronaviruses: a review of virus-host interactions. Diseases 4, 26 (2016).
Gale, M. Jr & Sen, G. C. Viral evasion of the interferon system. J. Interferon Cytokine Res 29, 475–476 (2009).
Park, A. & Iwasaki, A. Type I and Type III interferons - induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27, 870–878 (2020).
Honce R., et al. Obesity-related microenvironment promotes emergence of virulent influenza virus strains. mBio. 11, e03341–19 (2020).
Trouillet-Assant, S. et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 146, 206–208 (2020). e203.
Lee J. S., et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immun. 5, eabd1554 (2020).
Channappanavar, R., Zhao, J. & Perlman, S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res 59, 118–128 (2014).
Zhao, X. et al. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc. Natl Acad. Sci. USA 111, 6756–6761 (2014).
Bastard P., et al Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science. 23, 370 (2020).
Zhang Q., et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 370, eabd4570 (2020).
Pairo-Castineira E., et al. Genetic mechanisms of critical illness in Covid-19. Nature. 591, 92–98 (2020).
Acharya, D., Liu, G. & Gack, M. U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 20, 397–398 (2020).
Yao, C. et al. Cell-type-specific immune dysregulation in severely Ill COVID-19 Patients. Cell Rep. 34, 108590 (2021).
Masamoto, Y. et al. Adiponectin enhances antibacterial activity of hematopoietic cells by suppressing bone marrow inflammation. Immunity 44, 1422–1433 (2016).
Zacharioudaki, V. et al. Adiponectin promotes endotoxin tolerance in macrophages by inducing IRAK-M expression. J. Immunol. 182, 6444–6451 (2009).
Zhu, X. L. et al. Adipokine adiponectin is a potential protector to human bronchial epithelial cell for regulating proliferation, wound repair and apoptosis: comparison with leptin and resistin. Peptides 40, 34–41 (2013).
Amarsaikhan, N., Tsoggerel, A., Hug, C. & Templeton, S. P. The metabolic cytokine adiponectin inhibits inflammatory lung pathology in invasive aspergillosis. J. Immunol. 203, 956–963 (2019).
Tsatsanis, C., Margioris, A. N. & Kontoyiannis, D. P. Association between H1N1 infection severity and obesity-adiponectin as a potential etiologic factor. J. Infect. Dis. 202, 459–460 (2010).
Yu, X. et al. Intensive cytokine induction in pandemic H1N1 influenza virus infection accompanied by robust production of IL-10 and IL-6. PLoS One 6, e28680 (2011).
Hsueh, P. C. P. et al. Patient data, early SARS epidemic, Taiwan. Emerg. Infect. Dis. 10, 489–493 (2004).
Hong, K. H. et al. Predictors of mortality in middle east respiratory syndrome (MERS). Thorax 73, 286–289 (2018).
Lei C., et al Analysis of clinical characteristics of 29 cases of new coronavirus pneumonia in 2019. Chin. J. Tuberculosis Resp. Dis. 43, E005 (2020).
Arunachalam, P. S. et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369, 1210–1220 (2020).
Guaraldi G., et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol. 2, e474–e484 (2020).
Salama, C. et al. Tocilizumab in patients hospitalized with covid-19 pneumonia. N. Engl. J. Med. 384, 20–30 (2021).
Stone J. H., et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 383, 2333–2344 (2020).
Pini, M., Castellanos, K. J., Rhodes, D. H. & Fantuzzi, G. Obesity and IL-6 interact in modulating the response to endotoxemia in mice. Cytokine 61, 71–77 (2013).
Shi, D. et al. Association between plasma leptin level and systemic inflammatory markers in patients with aggressive periodontitis. Chin. Med J. (Engl.) 128, 528–532 (2015).
Madan, R. & Petri, W. A. Jr Role of obesity and adipose tissue-derived cytokine leptin during Clostridium difficile infection. Anaerobe 34, 182–186 (2015).
Sarnaglia, G. D. et al. Diet-induced obesity promotes systemic inflammation and increased susceptibility to murine visceral leishmaniasis. Parasitology 143, 1647–1655 (2016).
Zhang, A. J. X. et al. Leptin mediates the pathogenesis of severe 2009 pandemic influenza A(H1N1) infection associated with cytokine dysregulation in mice with diet-induced obesity. J. Infect. Dis. 207, 1270–1280 (2013).
De Rosa, V. et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity 26, 241–255 (2007).
Weber, D. J., Rutala, W. A., Samsa, G. P., Santimaw, J. E. & Lemon, S. M. Obesity as a predictor of poor antibody response to hepatitis B plasma vaccine. JAMA 254, 3187–3189 (1985).
Painter, S. D., Ovsyannikova, I. G. & Poland, G. A. The weight of obesity on the human immune response to vaccination. Vaccine 33, 4422–4429 (2015).
Green, W. D. & Beck, M. A. Obesity impairs the adaptive immune response to influenza virus. Ann. Am. Thorac. Soc. 14, S406–S409 (2017).
Eliakim, A., Schwindt, C., Zaldivar, F., Casali, P. & Cooper, D. M. Reduced tetanus antibody titers in overweight children. Autoimmunity 39, 137–141 (2006).
Frasca, D., Diaz, A., Romero, M., Vazquez, T. & Blomberg, B. B. Obesity induces pro-inflammatory B cells and impairs B cell function in old mice. Mech. Ageing Dev. 162, 91–99 (2017).
Shaikh, S. R., Haas, K. M., Beck, M. A. & Teague, H. The effects of diet-induced obesity on B cell function. Clin. Exp. Immunol. 179, 90–99 (2015).
Esteves de Oliveira, E. et al. Obesity affects peripheral lymphoid organs immune response in murine asthma model. Immunology 157, 268–279 (2019).
Boothby, M. & Rickert, R. C. Metabolic Regulation of the Immune Humoral Response. Immunity 46, 743–755 (2017).
Zhu, Q. et al. Suppressing adipocyte inflammation promotes insulin resistance in mice. Mol. Metab. 39, 101010 (2020).
Yang, W. S. et al. The association between body mass index and the risk of hospitalization and mortality due to infection: a prospective cohort study. Open Forum Infect. Dis. 8, ofaa545 (2021).
Phung, D. T., Wang, Z., Rutherford, S., Huang, C. & Chu, C. Body mass index and risk of pneumonia: a systematic review and meta-analysis. Obes. Rev. 14, 839–857 (2013).
Bhaskaran, K., dos-Santos-Silva, I., Leon, D. A., Douglas, I. J. & Smeeth, L. Association of BMI with overall and cause-specific mortality: a population-based cohort study of 3·6 million adults in the UK. The Lancet Diabetes &. Endocrinology 6, 944–953 (2018).
Berrington de Gonzalez A., et al. Body-Mass Index and Mortality among 1.46 Million White Adults. N. Engl. J. Med. 363 (2010).
Ramesh, R. et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med 211, 89–104 (2014).
Crewe, C., An, Y. A. & Scherer, P. E. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest 127, 74–82 (2017).
Endo, Y. et al. Obesity Drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep. 12, 1042–1055 (2015).
Lauterbach, M. A. & Wunderlich, F. T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflug. Arch. 469, 385–396 (2017).
Talukdar, S. et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med 18, 1407–1412 (2012).
Wensveen, F. M. et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 16, 376–385 (2015).
Bertola, A. et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 61, 2238–2247 (2012).
Adler, B. J., Green, D. E., Pagnotti, G. M., Chan, M. E. & Rubin, C. T. High fat diet rapidly suppresses B lymphopoiesis by disrupting the supportive capacity of the bone marrow niche. PLoS One 9, e90639 (2014).
van der Weerd, Kea Morbidly obese human subjects have increased peripheral blood CD4+ t cells with skewing toward a Treg and Th2-dominated phenotype. Diabetes 61, 401–408 (2012).
Zhao, Y. et al. CD4(+) T cells in obesity and obesity-associated diseases. Cell Immunol. 332, 1–6 (2018).
Oren, R., Farnham, A. E., Saito, K., Milofsky, E. & Karnovsky, M. L. Metabolic patterns in three types of phagocytizing cells. J. Cell Biol. 17, 487–501 (1963).
Newsholme, P., Curi, R., Gordon, S. & Newsholme, E. A. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochemical J. 239, 121–125 (1986).
O’Neill, L. A. & Hardie, D. G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 (2013).
Michelucci, A. et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl Acad. Sci. USA 110, 7820–7825 (2013).
Koo, S. J., Szczesny, B., Wan, X., Putluri, N. & Garg, N. J. Pentose phosphate shunt modulates reactive oxygen species and nitric oxide production controlling trypanosoma cruzi in macrophages. Front Immunol. 9, 202 (2018).
Blagih, J. et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54 (2015).
Buck, M. D., O’Sullivan, D. & Pearce, E. L. T cell metabolism drives immunity. J. Exp. Med 212, 1345–1360 (2015).
Everts, B. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332 (2014).
Keppel, M. P., Saucier, N., Mah, A. Y., Vogel, T. P. & Cooper, M. A. Activation-specific metabolic requirements for NK Cell IFN-gamma production. J. Immunol. 194, 1954–1962 (2015).
Wang, Z. et al. Glycolysis and oxidative phosphorylation play critical roles in natural killer cell receptor-mediated natural killer cell functions. Front Immunol. 11, 202 (2020).
Injarabian L., Devin A., Ransac S., Marteyn B. S. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int. J. Mol. Sci. 21, 287 (2019).
Azevedo, E. P. et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J. Biol. Chem. 290, 22174–22183 (2015).
Doughty, C. A. et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 107, 4458–4465 (2006).
Waters, L. R., Ahsan, F. M., Wolf, D. M., Shirihai, O. & Teitell, M. A. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience 5, 99–109 (2018).
Malandrino, M. I. et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 308, E756–E769 (2015).
Winther, S. et al. Restricting glycolysis impairs brown adipocyte glucose and oxygen consumption. Am. J. Physiol. Endocrinol. Metab. 314, E214–E223 (2018).
Acknowledgements
This study was supported, in part, by National Institutes of Health (NIH) R01DK099222 (to S.D.); NIH R21AI139829 (associated with S.D.); American Diabetes Association (ADA) 1-18-IBS-100 (to S.D.); Department of Defense (DoD) W81XWH2010392 (to S.D.); and T32GM063483-14 (associated with P.C.A.).
Author information
Authors and Affiliations
Contributions
P.C.A., M.S.M.A.D, and S.D. participated in manuscript conceptualization literature review and data interpretation (#denotes equal contribution). P.C.A., M.S.M.A.D., R.M., G.S.D., P.S., S.S.W., and S.D. participated in manuscript writing, editing, and concept/data discussion. S.D. obtained the funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. S.D. is a consultant for Janssen Research & Development.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alarcon, P.C., Damen, M.S., Madan, R. et al. Adipocyte inflammation and pathogenesis of viral pneumonias: an overlooked contribution. Mucosal Immunol 14, 1224–1234 (2021). https://doi.org/10.1038/s41385-021-00404-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-021-00404-8
This article is cited by
-
The impact of obesity in hospitalized patients with COVID-19: a retrospective cohort study
Diabetology & Metabolic Syndrome (2024)
