
Abstract
Aims
Phyllosphere bacteria play critical roles in plant growth promotion, disease suppression and global nutrient cycling but remain understudied.
Methods
In this project, we examined the bacterial community on the phyllosphere of eight diverse lines of Brassica napus for ten weeks in Saskatoon, Saskatchewan Canada.
Results
The bacterial community was shaped largely by plant growth stage with distinct communities present before and after flowering. Bacterial diversity before flowering had 111 core members with high functional potential, with the peak of diversity being reached during flowering. After flowering, bacterial diversity dropped quickly and sharply to 16 members of the core community, suggesting that the plant did not support the same functional potential anymore. B. napus line had little effect on the larger community, but appeared to have more of an effect on the rare bacteria.
Conclusions
Our work suggests that the dominant bacterial community is driven by plant growth stage, whereas differences in plant line seemed to affect rare bacteria. The role of these rare bacteria in plant health remains unresolved.
Similar content being viewed by others


Introduction
The phyllosphere is one of the largest microbial habitats globally yet remains understudied (Vorholt 2012). The phyllosphere can encompass all the aerial parts of the plant (Vorholt 2012), but most commonly is referred to as the surface and interior of the leaves (Redford et al. 2010; Vacher et al. 2016). Phyllosphere microorganisms provide benefits to the plant, such as increased nutrient cycling, disease suppression, and growth promotion among many others. Fürnkranz et al. (2008) found that some species of tropical plants had higher rates of beneficial nitrogen fixation by diazotrophic phyllosphere bacteria. Vogel et al. (2016) and Jarvis et al. (2015) both found that the pre-existing phyllosphere bacterial communities combined with plant genetics, impacted the reaction to and the extent of disease on plants that had been inoculated with known pathogens. Phyllosphere microbial communities also play important roles in promoting plant growth (Batool et al. 2016), and the diversity and abundance of these communities can impact the levels of insect herbivory (Humphrey et al. 2014). Finally, phyllosphere bacteria are likely an important driver in the global methane cycle and other global nutrient cycling processes (Iguchi et al. 2015).
Despite the importance of phyllosphere microbial communities for overall plant and ecosystem functioning, they remain poorly described, particularly in agricultural systems, which account for 37% (1385 mil. ha) of the global land use in 2017 (FAOSTAT 2017). With the global population predicted to reach 9 billion by 2050, agricultural intensification will increase (Glibert et al.2010). Producing more food on decreasing arable land and in more erratic climate conditions poses a unique and difficult problem for crop breeders and farmers. Breeding crops to have a more robust microbiome may be a sustainable way to improve crop yield without additional inputs or an increase in cultivated land (Ryan et al. 2009). However, before crops can be successfully bred to optimize microbiome contributions, we must first understand the diversity and dynamics of the microbiome of crop plants.
Canola (Brassica napus) is produced for its high-quality oil, and increasingly, to be used as high-quality animal feed and for the production of biofuels. Canada is the leading producer of canola, with an estimated 3 million tons produced in 2018 (AFFC 2019). The importance and large-scale production of canola makes it a good target for microbiome studies. Canola also has large nitrogen requirements. Using microbiome manipulation to increase nitrogen use efficiency could help make canola a more sustainable crop. Additionally, the use of genetically modified crops, including canola, is widely banned in Europe. Breeding canola with improved microbiomes could help lessen the disease pressure placed on canola or rapeseed crops that cannot be modified genetically to ameliorate some of this pressure. Work on the microbiome of canola and oilseed crops has focused primarily on the roots and rhizosphere (Copeland et al. 2015; Cordero et al. 2020; Gopalakrishnan et al. 2015; Glaeser et al. 2020), or on cultured microbial isolates (Romero et al. 2019) or pathogens from Brassica species, and not on the microbiome as a whole (Wassermann et al. 2017). In some cases, the interaction of canola root- and rhizosphere-associated microbiomes with other crops was considered (Hilton et al. 2018; Schlatter et al. 2019). To date, only one study has examined the canola phyllosphere microbiome (Copeland et al. 2015) observing differences compared to common bean and soybean. They further observed that the canola phyllosphere microbiome was affected by rainfall and became increasingly different from the rhizosphere community as the growing season progressed.
Not only does the composition of the phyllosphere remain understudied but what impacts the assembly of these communities has also been largely ignored. Many of the studies on the canola microbiome have both limited sampling dates and canola lines. In order to successfully breed B. napus to have a more robust microbiome we must first gain an understanding of how the microbiome develops and changes throughout the growing season for the whole plant, not just in the rhizosphere. In this study, we examined the bacterial phyllosphere microbiome of eight different lines of B. napus in Saskatchewan over a ten-week period to determine the composition of the phyllosphere and what factors are important for the structuring of bacterial communities on leaf surfaces. We hypothesized that the phyllosphere bacterial community composition would vary with developmental stage and B. napus line.
Methods
Eight lines of B. napus (Mason et al. 2017; Clarke et al. 2016) were seeded on May 28 or 29, 2017 in the field at the Agriculture and Agri-Food Canada (AAFC) research farm outside of Saskatoon, Saskatchewan, Canada (52.1718° N, 106.5052° W) which lies in the dark brown soil zone (Chernozem). The plots were seeded using a 6 row, double disc drill with 1284 B. napus seeds applied per plot (6.1 m long by 1.8 m wide). A granular blend of 20–8–0-20 fertilizer was applied on May 23 at the rate of 139 kg ha−1. These lines of Brassica napus are part of the AAFC canola breeding program and thus were not commercial canola varieties, but rather different lines created by nested associating mapping, referred to as NAM lines (Clarke et al. 2016). They ranged in seed origin and color, fiber content, erucic acid content and seed glucosinolate levels (SI Table 1) meaning that some of them were not technically canola which requires a low erucic acid content, but all lines were varying varieties of B. napus. Differences in these traits will result in differences in oil quality and are related to phenological development. Bazghaleh et al. (2020) have described the experimental design extensively, but briefly, the site consisted of three replicate blocks with each B. napus line seeded randomly within each block, but not repeated within a block. All lines were planted on May 28, 2017. The site received 127.9 mm of precipitation throughout the growing season with a mean air temperature of 16.4 °C. Several leaf samples were collected from each of the eight lines in each block every week for ten weeks beginning on June 20, 2017 until August 22, 2017. Plants on the edges of the block were avoided as these leaves were visibly dusty and thus would likely have a different microbiome than most leaves on the interior of the rows. Leaves that were clearly diseased, had extensive insect damage or were senescing as the plant matured were also avoided, as these leaves did not represent the majority of the leaves present and would again bias the microbiome results. During flowering, uppermost leaves were avoided as these leaves had a thick layer of petals on them and differentiating the leaf microbiome from the flower microbiome would have been impossible. However, some petal contamination was impossible to avoid, but was bypassed when feasible. Finally, during the later stages of seed development and ripening, leaves that were mostly senesced were avoided as dead tissue would cause large changes in the microbiome. Leaf samples, consisting of only one or two leaves were placed into sterile whirl-pak bags (Nasco, Wisconsin, USA) and placed onto ice. Leaf samples from the same NAM line, but from different blocks were not combined and plants were not destructively sampled as only a single leaf or two were sampled. One sample from each NAM line in each block was collected (24 samples) as well as one randomly selected duplicate sample within block (line was not considered here), leading to the collection of 27 samples collected weekly, or 270 samples over the entire growing season. Samples were then returned to the lab and stored at −80 °C until further processing.
Frozen, brittle leaves were crumbled manually in the whirl-pak and a 0.05 g subsample was taken and extracted using Qiagen PowerPlant extraction kit (Hilden, Germany) following manufacturer instructions. After extraction, DNA was tested for quantity and quality following the standard Qubit protocol (Thermo Fisher Scientific, Waltham Massachusetts). Template DNA was standardized to 4 ng μl−1 prior to amplification. Bacterial diversity was assessed by amplifying the V4 region of the bacterial 16S rRNA using the primer set 515F with Illumina adapters (5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGYCAGCMGCCGCGGTAA 3′) and the 806R (5′ - GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG GGA CTA CCG GGG TAT CT - 3′) (Walters et al. 2015). The PCR reaction mix consisted of 7 μl Invitrogen Platinum SuperFi PCR master mix (Thermo Fisher Scientific, Waltham, Massachusetts), 0.1 μl of each primer (10 μM stock), 3 μl (5 μM stock) plastid peptide nucleic acid blocker (pPNA) and 2 ul (5 μM stock) mitochondrial peptide nucleic acid blocker (mPNA) (PNA Bio, California, USA) and 10.3 μl nuclease free water, and 2 μl of the standardized template DNA. PNAs were included to block the amplification of host DNA, plant mitochondria and chloroplasts, which are a common contaminant from plant tissues (Ray and Nordén 2000, Von Wintzingerode et al. 2000). The PCR conditions were 95 °C for 5 min as an initial denaturization, followed by 95 °C for 30 s., 78 °C for 10 s., 54 °C for 45 s., 72 °C for 60 s. for 35 cycles, and a final elongation of 72 °C for 7 min. Negative controls and PCR duplicates were included. PCR product was purified to eliminate primers and impurities using 1:1 ratio of Nucleomag NGS clean-up and size select (D-mark Biosciences, Scarborough, Ontario). Randomly selected duplicates were included during DNA extraction, amplification, and sequencing stages adding in 56 duplicates, bringing the total sample size to 326. After purification, samples were indexed following the Illumina protocol (Illumina, San Diego, California), purified again to remove excess index primers, quantified and standardized to 4 nM, and pooled. Pooled libraries were then sequenced using the Illumina MiSeq platform using V3 chemistry.
A total of 326 samples were sequenced resulting in 10,839,325 reads with an average of 18,186 reads per sample. Sequences were imported into QIIME2 v 2019.1 (Bolyen et al. 2018) and primers were removed using cutadapt (Martin 2011). Reads were then processed into amplicon sequence variants (ASVs) and chimeras were removed using Deblur (Amir et al. 2017) resulting in 1968 ASVs. ASVs were then classified using a 515/806 trained Greengenes database classifier (Desantis et al. 2006). Host mitochondria and chloroplasts were removed after classification. Host DNA ranged from 6% to 100% of the read in each sample with an average of 32% across samples. The abundance and taxonomy artifacts produced in QIIME2 were exported to BIOM format (McDonald et al. 2012) for processing in R v. 3.5.3 (R Core Team 2018). Where available, species level classification was used although the authors recognize that there are issues (Johnson et al. 2019), this is standard practice at the time of writing. If a species level classification was not available, the ASV was marked as Unclassified. This was also done for higher levels of taxonomy.
To select the core microbiome (Shade and Handelsman 2012; Lundberg et al. 2013; Vandenkoornhuyse et al. 2015; Shade and Stopnisek 2019), each ASV was converted from number of reads to presence or absence. No singletons or doubletons formed part of the core, with all members being both prevalent (present every week during sampling) and abundant (> 0.05% of all reads). The ASVs that were present across all lines and weeks were selected as the Core and represents 15 ASVs, which fall into four different classifications (Table 1). Upon this conversion, it was noted that there appeared to be a shift in community composition after flowering. The alpha diversity indices for the weeks before and after flowering were similar despite there being a shift in community composition. To investigate if there was a shift in composition, that was not being reflected accurately in the diverisity indices, the community was subset into (i) ASVs present in all weeks up to and including flowering (Flowering) and (ii) those present in all weeks after flowering (Pod), using the same prescence and absence matrix that was used to select the core that was present across all ten weeks. In order for an ASV to be selected for either the Flowering or Pod communities, the ASV had to be present consistently every week either before or after flowering. There were 111 ASVs present before and during flowering (Flowering) and 16 present post flowering (Pod), a small porportion of the original 1968 ASVs. This separation into Flowering and Pod allowed for the conservation of the shift in community composition that would not have been captured by examining the entirety of the sampling period together, nor by looking at diversity metrics alone. The Flowering community has distinct ASVs from that of the Pod community as many of the ASVs present during and before the flowering stage disappeared once the plants matured into seed and pod development. However, for analysis both of these communites do inlcude the original 15 Core ASVs, as this small subset of ASVs were present all ten weeks. This resulted in Flowering consisting of 126 ASVs and Pod having 31 ASVs. Unweighted UniFrac distance matrices were performed on the entire data set of 1968 ASVs (Total) and well as the combined Core, Flowering and Pod communities (Expanded Core).
Alpha diversity was calculated using the package vegan in R (Oksanen et al. 2017). All diversity measures calculate the variety of organisms in a community based on combinations of species richness and evenness (Kim et al. 2017). The Shannon-Weaver index, Abundance-based Coverage Estimate (ACE) and the Simpson Index were chosen because they represent a mix of approaches to diversity. The Shannon-Weaver index places more emphasis on species richness whereas the Simpson index places more emphasis on species evenness (Kim et al. 2017). Unlike the previous two indices, the ACE index takes into consideration the abundance of the species present and for this study most closely reflected the observed diversity. Shannon-Weaver index, ACE (Chao and Lee 1992), Simpson index and species richness (Jost 2007) estimates were calculated and showed similar trends, with peak diversity occurring across all lines during week 4 of sampling and declining steadily with plant maturity (Table 2). However, both the Shannon-Weaver and the Simpson indices showed an initially very high level of diversity, but this is likely an over estimation due to a high abundance of rare ASVs present in the first few weeks.
The BBCH-scale is a scale used to uniformly identify and quantify the phenological stages of plant development, with scales developed for species specific development (Lancashire et al. 1991). Despite differences in crop species as well as varietal differences, all plants have the same growth stages of leaf development, formation of side shoots, stem elongation, harvestable vegetative plant part development, inflorescence emergence, flowering, seed development, ripening and senescence. All B. napus lines were assigned BBCH weekly using the Canola Council of Canada BBCH guide (Canola Council of Canada 2020a, b) and were averaged. This was done because despite identical planting times, the eight B. napus lines did exhibit some differences in plant development. Sampling week 1 and 2 took place during the leaf development stage for most B. napus lines sampled, with bolting happening during week 3 (Fig. 1). Peak flowering was reached for most lines during sampling week 4 (Flowering) with seed development occurring in the following two weeks (Pod). The last four weeks of sampling were characterized by ripening of the B. napus seed pods (Pod).

Abundance-based Coverage Estimate (ACE) average of the bacterial community on the leaf surface against the average BBCH of all 8 lines of B. napus across the ten sampling weeks. Each point is the average ACE for that week for the community microbiome (1968 ASVs (amplicon sequence variants) with error bars representing the standard error of the estimate. The BBCH range represents the range of BBCH observed across the eight different lines for that sampling week. Change in BBCH stage is indicated by changes in background color with the name of the stage indicated at the top of the colored bar
Permutational analysis of variance (PERMANOVA) was performed using the adonis function in the vegan package in R (Oksanen et al. 2017). The weighted and unweighted UniFrac (Lozupone and Knight 2005) distances were calculated using the phyloseq package (McMurdie and Holmes 2013), for the entire community (Total), the Expanded Core (Core, Flower and Pod communities) as well as the communities present before and during flowering (Flower) and the communities present after flowering (Pod). UniFrac distances were used instead of Bray-Curtis or other similar methods, to maintain the phylogenetic signals as there was a shift in community composition from the community present pre-flowering and post-flowering. Other distance matrices that do not contain phylogenetic information would not accurately capture this shift.
Distance-based redundancy analysis (dbRDA) (Legendre and Andersson 1999) was performed using the capscale function in the vegan package in R (Oksanen et al. 2017). The weighted and unweighted UniFrac matrices for both the Total and the Expanded Core communities were constrained by NAM line and BBCH growth stage, as well as the interaction, and these constraints were then analyzed using the anova function (base R). Given that block was found to not be significant for any of the communities in the PERMANOVA results it was not included in the dbRDAs.
To isolate the effect of B. napus NAM line more thoroughly, a dbRDA was performed constraining each UniFrac matrix by growth stage only. This was done to investigate whether NAM line influenced the community when it was not outweighed by the importance of growth stage. The residuals of this model were extracted, and an additional dbRDAs were performed constraining by NAM line. By doing this we accounted for the effect of growth stage without the influence of NAM line, then after this variation was accounted for, we could isolate the additional variation caused by NAM line alone without the large signal of growth stage overshadowing the effect of NAM line.
Results
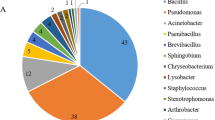
The leaf bacterial community was dominated by a small subset of the total ASVs present. The leaf microbial community (Fig. 2) of 1968 ASVs was primarily Proteobacteria (79%), followed by Bacteroidetes (7%), Acidobacteria (6%) and Firmicutes (6%). The Proteobacteria were primarily Gammaproteobacteria (74%), followed by Betaproteobacteria (20%) and Alphaproteobacteria (4.7%). However, despite 1968 ASVs being present, the majority of the community was dominated by a few species. The Expanded Core microbiome of 136 ASVs were primarily Pseudomonas virdiflava (Table 1) (12.9% of the Total, 31.9% Core), followed by Pantoea sp. (10.6% Total, 12.5% Core), Massilia sp. (5% Total, 13.1% Core) and Stenotrophomonas retroflexus (2.9% Total, 5.6% Core) (Fig. 2). However, it should be noted that these identifications are tentative due to the difficulty of assigning taxonomy using only one region of the bacterial genome.

Relative abundance of genera that composed the Core as well as the communities present during the Flowering and Pod stages (Expanded Core). Many of the ASVs (amplicon sequence variants) in the Expanded Core were classified into the same genus or even species, but due to the nature of ASVs, they remained separate. For this graphic, the ASVs present in the Expanded Core have been grouped into genus instead of showing all 136 individual ASVs in the Expanded Core. Colors are grouped as genus and the weekly range of BBCH observed is present on the x axis
Bacterial diversity was highest during week four, or the flowering period for both the observed diversity (the number of ASVs present) and abundance-based coverage estimator (ACE) (Table 2 and Fig. 1). The highest diversity for the Shannon index and inverse Simpson was observed during the first sampling week (Table 2), however there was a sharp decline in both of these indices during the second sampling week with both reaching a peak during the flowering period. The initially high diversity found during week one was likely due to the plants being recently emerged from the soil and thus still being colonized by primarily by soil bacteria. After flowering, all the diversity indices declined steadily to levels similar to, or lower than those observed during the leaf development stages. There was a sharp decrease in diversity between weeks six and seven (Fig. 2 and Table 2) which could have been caused by low precipitation. Diversity increased again during weeks seven to nine, only to decrease again at week ten. The overall decline in diversity after flowering could have been caused by the gradual leaf senescence that occurs with B. napus during seed production.
PERMANOVA results indicated that stage was the primary factor shaping the community structure. The PERMANOVA of the Total weighted UniFrac distances (Table 3) showed that plant development stage (BBCH) significantly affected the community composition (p = 0.026, R2 = 0.029) and that NAM line was nearly significant (p = 0.06, R2 = 0.021) (Table 3). There were no significant terms for the PERMANOVA of the Total Unweighted UniFrac Distances (Table 3). For the Expanded Core, stage was significant in the Weighted Unifrac distances (p = 0.018, R2 = 0.026). In the unweighted Flower community, there was only a marginally significant interaction between NAM line, stage and block (Table 3). Stage was significant for both the Weighted (p = 0.001, R2 = 0.039) and Unweighted (p = 0.001, R2 = 0.052) UniFrac distances of the Pod community. Additionally, NAM line was significant (p = 0.045, R2 = 0.048) for the weighted UniFrac distances in the Pod community (Table 3).
Given the significant, but low, explanatory values given by the PERMANOVAs, db-RDAs were done to further isolate the effects driving community composition while maintaining the phylogenetic relationships provided by the UniFrac distance matrices. Similar to the PERMANOVAs, the db-RDAs indicated that growth stage was the dominate factor shaping the bacterial community. BBCH stage significantly (p < 0.001) affected Total bacterial community structure for both weighted and unweighted UniFrac dbRDA analyses (Fig. 3). NAM line was significant only for the unweighted UniFrac analysis of the Total community (p = 0.048; Table 3 and Fig. 3b). There was a marginally significant NAM line by growth stage interaction for the unweighted Total community (p = 0.058; Table 3) and for the weighted, Expanded Core community (p = 0.030; Table 3), likely caused by slight differences in plant development times between lines. In the dbRDA models of the Total community, BBCH stage and NAM line explained 28% of the variance in the weighted Unifrac analysis (Fig. 3a) and 24% of the variance in the unweighted UniFrac analysis (Fig. 3b). The slight decrease in explanatory power between the weighted and unweighted analysis could have been caused by the bacterial community being largely dominated by a small number of ASVs, which would not have been reflected in the unweighted distance matrix. When only the Expanded Core was analysed using dbRDA, this increased to 41% (SI Fig. 1a) and 32% (SI Fig. 1b) for the weighted and unweighted community analyses (Table 3). This increase in explanatory power is likely caused by the reduction in the number of ASVs included in the analysis (1968 ASVs in the Total to 136 ASVs in the Expanded Core). Despite there being a large number of rare ASVs, they were not abundant thus reducing the noise for the analysis. For the Flowering community, stage was significant for both the weighted (p = 0.002) and unweighted (p = 0.001) UniFrac matrices, but neither NAM line nor the interaction of NAM line and stage were significant for either (Table 3). For the Pod Development community, stage was significant for both the weighted (p = 0.003) and the unweighted (p = 0.001) UniFrac matrices, but again, neither NAM line nor the interaction was significant (Table 3).

Distance-based redundancy analysis (dbRDA) of the weighted (28% of the variation) UniFrac distances (a) and unweighted (24% of the variation) UniFrac distances (b) of the Total bacterial community, across the entire 10-week sampling period, constrained by BBCH (p = 0.001) stage x B. napus line (NAM) (not significant) (biplots). Points are colored based on B. napus growth stage and correspond with the colors in Fig. 1
For the Total community dbRDA, BBCH stage alone explained 19% of the variation observed for the weighted UniFrac matrix (Fig. 3a) and 12% of the variation for the unweighted UniFrac matrix (Fig. 3b). NAM line was observed to have slight differences in development time and visual appearance of the leaves (observations from the field) however these differences had little effect on the bacterial communities present on the leaves. After isolating the residuals of each of the above models, NAM line explained only 2% of the variation observed and was not significant for the weighted UniFrac distance residuals. Similarly, for the unweighted UniFrac distance residuals, NAM line explained 3% (p = 0.1) of the variation. For the Expanded Core community, growth stage alone explained 28% of the variation observed for the weighted UniFrac matrix (Fig. 3a) and 19% for the unweighted matrix (Fig. 3b). NAM line explained 3% for the weighted and unweighted Expanded Core residuals and was not significant. The leaf is an inhospitable environment which can change drastically with plant growth stage, thus the small differences in NAM line were likely insignificant to shaping the bacterial community when compared to the much larger force of plant growth stage.
Discussion
B. napus growth stage is the dominant factor shaping the bacterial community present on the leaves, throughout the growing season with a distinct community present before and after flowering. The distinct community is evidenced by the Flowering community with 111 ASVs present every week before flowering, with most of these disappearing after flowering, leaving only 16 ASVs present in the Pod community. Additionally, significant differences in both PERMANOVAs and dbRDAs were found when comparing the weighted UniFrac to the unweighted UniFrac distances, with BBCH stage being significant for both the total community and the Expanded Core. Differences between weighted UniFrac, which uses phylogenetic relationships as well as abundance, and unweighted UniFrac, which does not use abundance, indicate that both community structure, and evenness change throughout the sampling period (Lozupone et al. 2007). If both community structure and evenness were not significantly impacted by BBCH growth stage, there would be no significant differences between the weighted and unweighted UniFrac distances.
The dominance of growth stage appears to outweigh the importance of different B. napus lines on shaping the bacterial community despite differences in NAM line as the Expanded Core remained abundant and constant and only the composition of the rare ASVs seemed to vary. Furthermore, NAM line significantly impacted the community structure only a limited number of times (Table 3). It is unclear if this is a B. napus specific finding or perhaps a phyllosphere specific finding. Crop variety has been shown to impact rhizosphere communities (Edwards et al. 2015, Coleman-Derr et al. 2016, Dombrowski et al. 2017). Additionally, Bokulich et al. (2014) found that microbial communities in grape must varied with grape cultivar. However, crop line has variable impacts on phyllosphere communities. Singh et al. (2019) found that grape cultivar impacted the microbial community present in the phyllosphere. Conversely, Johnston-Monje et al. (2016) found that maize genotype had no effect on microbial communities in the phyllosphere. Most importantly, Copeland et al. (2015) did not find that B. napus line impacted the microbial communities in the phyllosphere. The lack of impact of line on bacterial community structure could be due to thick, waxy cuticle present on B. napus leaves. The thick cuticle likely makes the B. napus leaf an inhospitable environment for bacteria with low availability of plant derived carbon (Vorholt 2012; Vacher et al. 2016). More work is needed to characterize the phyllosphere and impact of line on B. napus as well as other crops.
Copeland et al. (2015) found that the canola phyllosphere was dominated by a few high abundant genera with many rare ASVs, with decreasing diversity as the growing season progressed. However, they examined only sampling date, but not plant growth stage, so while the trends are similar, it is difficult to extrapolate further. The dbRDAs, as well as the PERMANOVA results suggest that perhaps B. napus line plays a larger role in structuring the rare ASVs present on the leaves, but not the Expanded Core microbiome which was comprised of relatively abundant bacteria. This is especially true for the Pod community which showed a significant effect of NAM line on the unweighted UniFrac distances, but not in the weighted UniFrac distances. Since the unweighted UniFrac distances do not incorporate abundance, simply presence or absence of an ASV, this indicates that there might be a larger influence of line on the rarer ASVs, which are not given as much weight when using the abundance weighted UniFrac distance matrix. However, there appears to be no consistent patterns in what rare ASVs are present throughout the sampling season, unlike the Expanded Core community, that was generally highly abundant, relative to other taxa. This lack of both consistency and abundance suggests that perhaps these rare ASVs are transitory inhabitants of the leaves that may be selected for by more nuanced biochemical or morphological characteristics that are line specific, such as cuticle thickness or unmeasured secondary metabolites. In other words, the composition of the rare ASVs is likely determined by the stochastic effects of random replication, death and immigration, whereas the Expanded Core bacterial community is likely selected primarily by plant growth stage, making it a more deterministic process.
Changes in the plant bacterial microbiome with plant growth stage are well documented for rhizosphere soils (Smalla et al. 2001, Wagner et al. 2014, 2016, Copeland et al. 2015, Hilton et al. 2018) so it is not surprising that growth stage changes were found in B. napus leaves. Additionally, it has been well documented that plants undergo large physiological and biochemical changes during flowering and fruit production (Nitsch 1965, Mohan Ram and Rao 1984, Shu et al. 2010). B. napus leaves have a thick, waxy cuticle which thickens with age. This thickening of the cuticle would make access to plant derived carbon more limited as the plant ages, which could be the cause of the shifts observed during the growing season. Furthermore, during flowering, the petals are quickly shed, many of which land on the leaf. This additional and large source of labile carbon could be one of the factors accounting for the large increase in bacterial diversity and abundance seen around the time of flowering. After flowering, as the seeds begin to develop and ripen, many of the different lines started to show signs of leaf senescence and death, in some cases shedding their leaves entirely. This slow leaf senescence coupled with the thickened waxy cuticle likely drives the rapid decrease in bacterial diversity seen after flowering. While both the PERMANOVA results and the db-RDAs have relatively small R2, their cumulative effect explains quite a bit of the variation. It is well established that environmental variables or unmeasured variables explain most of the variation seen in microbial communities and it is not uncommon to have low R2 (Redford et al. 2010, Wagner et al. 2014, Coleman-Derr et al. 2016, Leff et al. 2017,), therefore these results are valid despite the relatively low explanatory power of each variable.
The most prevalent non-pathogenic ASVs in the phyllosphere appear to be well suited for life in this difficult environment. Stenotrophomonas retroflexus forms biofilms, which could aid in the colonization of the leaf surface and protect the bacteria from desiccation and UV stress (Ren et al. 2015, Lui et al. 2017). Oxalobacteraceae massilia exists in the rhizosphere, root surface and seed coat of cucumber plants, so it’s not unexpected to find it on the leaf surface as well (Ofek et al. 2012). Additionally, this species varies with plant development stage in cucumber plants so given that we see large growth stage patterns in B. napus leaves, we might expect to see a similar pattern here (Ofek et al. 2012). Enterobacteriaceae Pantoea is a functionally diverse genera with plant growth promoting abilities, nitrogen fixing capabilities, can produce antibiotics and might be an opportunistic pathogen (Walterson and Stavrinides 2015; Coutinho and Venter 2009). Many species of Pantoea are also pigmented, which also likely increases their ability to withstand UV stress, making them well suited for life in the phyllosphere (Andrews and Harris 2000). Furthermore, Pantoea spp. live in other extreme environments such as oil sands (Mitter et al. 2017) and seeds (Feng et al. 2006) indicating that they can live in difficult environments such as the phyllosphere. However, given the uncertainty of assigning taxonomy based on 16S rRNA sequencing alone, a follow-up, culture dependent study to determine the identity and functionality of these ASVs would be needed to confirm these potential functions in the B. napus phyllosphere.
The most common ASV found was Pseudomonas virdiflava. This is a pathogenic species that causes foliar blights, root rots, and stem necrosis (Marina et al. 2008; Sarris et al. 2012). Furthermore, this species can cause disease in a wide variety of cultivated plants, including cantaloupe, celery and tomatoes. Typical pathology for infection caused by Pseudomonas virdiflava is the wilting and yellowing of leaves, which was seen in the B. napus leaves, but the plants did not have high disease levels, and this could have been normal leaf senescence. Culture dependent methods would need to be employed to verify if the observed wilting and yellowing leaves observed in the field was indeed caused by Pseudomonas virdiflava as its presence alone does not indicate the cause of disease.
The initially very high estimates of diversity during the first sampling week (Table 1) was likely caused by the plants being relatively small during the first sampling week as they were still in the leaf development stage, with two to six leaves. These plants had only recently emerged and may have been predominantly colonized by bacteria originating from the soil. The large number of unique or low abundance ASVs found during the first sampling week quickly disappeared in subsequent weeks as the leave matured (Fig. 2) and may have been selected against by the harsh conditions on the leaf surface. Both the Shannon-Weaver index and the Simpson index include estimates of observed diversity as well evenness but not abundance into the estimates, which likely lead to the over estimation of initial diversity as rare species are given equal weight (Kim et al. 2017). We did not see the same initial trend in the ACE metric likely because the ACE metric uses both evenness and abundance of rare ASVs to estimate the likelihood of a particular ASV to be present or not (Kim et al. 2017). A similar phenomenon was observed by Copeland et al. (2015) where the Shannon diversity index was very high initially and the community composition was similar to the soil, but as the growing season progressed, the diversity index decreased, and the leaves developed communities that were distinct from the soil.
Increasing bacterial diversity between the initial sampling week and the peak at flowering could have been caused by a number of factors. It is well established that while phyllosphere communities are separate from rhizosphere communities they do have considerable overlap in community composition (Wagner et al. 2014; Copeland et al. 2015; Wagner et al. 2016). This increase in diversity leading up to flowering could stem from the deposition of soil dust from nearby farming activities. Vokou et al. (2012) found air samples and the phyllosphere of nine different species shared a similar composition, suggesting that aerial deposition is a likely source of bacterial diversity on plants. Aerial deposition of soil dust would also explain the overlap in community composition between the rhizosphere and phyllosphere seen in other studies as the rhizosphere draws its community from the surrounding bulk soil which would also be the source of the dust. Additionally, this increase in diversity could be caused by visiting pollinators or insect herbivory. Humphrey et al. (2014) found that insect herbivory was positively correlated with bacterial diversity in the phyllosphere. The most likely explanation for the increasing diversity is a combination of aerial soil deposition, insect drivers and the plants selecting for a beneficial community.
Pre-flowering, the bacterial community on the leaves had greater diversity members (Fig. 2, SI Table 2), compared to the Pod community. These functionally rich bacteria included members of the Bradyrhizobiaceae which are well documented for their widespread symbiosis with plants and their ability to fix nitrogen (Marcondes de Souza et al. 2014; Gopalakrishnan et al. 2015). There were other nitrogen-associated bacteria present pre-flowering as well, such as Nitrosovibrio tenuis which oxidized ammonium (Harms et al. 1976) and Streptomyces mirabilis which produces the enzyme nitroreductase and has shown antifungal and antibacterial activity (Bordoloi et al. 2001; Yang et al. 2012). Other functionally useful bacteria present on the leaves pre-flowering that disappear after flowering included Burkholderia bryophila which has been shown to have anti-fungal activity, carbon monoxide reduction capabilities and plant growth promoting properties (Vandamme et al. 2007; Weber and King 2012) which would be useful to the B. napus plant as many of its major pathogens are fungal (Canola Council of Canada 2020a, b). Methylobacterium were also present on the leaves prior to flowering. This species can use both methane and methanol as a carbon source, both of which are byproducts of plant metabolism (Hanson and Hanson 1996; Nisbet et al. 2009; Dorokhov et al. 2018). Furthermore, Methylobacterium has the ability to form biofilms, making it well adapted for life in the phyllosphere (Hanson and Hanson 1996).
This bacterial diversity largely disappears after flowering (Fig. 2, SI Table 2), with the majority of the core species found post flowering simply being additional AVSs of the core bacteria present throughout the entire growing season and a large dip in diversity seen between weeks six and seven. This decrease in diversity (Fig. 2 and Table 2) could have been caused by weeks five and six having relatively low precipitation and hotter temperatures (unpublished site data). The recovery of some diversity in weeks eight and nine was likely caused by these weeks experiencing much cooler and wetter conditions, allowing the bacteria present the moisture needed to rebound. The phyllosphere is a harsh environment that can fluctuate rapidly in temperature, moisture and ultraviolet radiation levels, all of which will have an impact on the bacterial communities living on the surface of these leaves (Kinkel 1997; Vorholt 2012; Vacher et al. 2016). However, this diversity was not maintained during week ten as most of the leaves on the plants were undergoing senescence at this point. Despite this one anomalous large dip in diversity, the overall trend of decreasing diversity stands with one exception. Firmicute Exiguobacterium was not found in the pre-flowering core community and was abundant in the Pod community. Exiguobacterium is not widely studied creating large gaps in knowledge about this genus, however it has been shown to grow in difficult environments (Vishnivetskaya et al. 2009) which could be the plant preparing for transfer to the seed (Shade et al. 2017) or the community adapting to the quickly senescing leaves.
Plant growth stage, but not B. napus line, was the dominant factor in shaping the bacterial phyllosphere across all eight lines of B. napus sampled. There was a small, but abundant Core community present throughout the growing season, regardless of plant growth stage with rarer ASVs changing rapidly and seemingly without pattern. Initially, there was very high diversity, but this was likely caused by colonization by soil bacteria that quickly selected against due to the difficult nature of life in the phyllosphere. During the development stages, the bacterial community retained some of this diversity which included a higher potential for functional diversity. The bacterial community reached peak diversity during flowering and rapidly became much less diverse during the pod development and ripening stage, consisting primarily of ASVs from a small number of genera. These swift changes in both diversity, abundant and rare ASV composition suggests that while the plant has some control over the community composition, likely based on growth stage, there is also a probable large stochastic component to the community assembly processes. Community assembly process on the phyllosphere warrant further investigation as research into harnessing the microbiome for plant health intensifies. This work shows that the bacterial community present will change drastically throughout the growing season, and that multiple sampling points are necessary to get an accurate look at the microbiome and how it interacts with the plant as it develops.
Data availability
All raw sequence files can be found at the National Center for Biotechnology Information (NCBI) under Bioproject PRJNA 635907.
References
Amir A, Daniel M, Navas-Molina J, Kopylova E, Morton J, Xu ZZ, Eric K, Thompson L, Hyde E, Gonzalez A, Knight R (2017) Deblur rapidly resolves single-nucleotide community sequence patterns. Am Soc Microbiol 2:1–7
Andrews JH, Harris RF (2000) The ecology and biogeography of microorganisms on plant surfaces. Annu Rev Phytopathol 38:145–180
Bartoli C, Berge O, Monteil CL, Guilbaud C, Balestra GM, Varvaro L, Jones C, Dangl JL, Baltrus DA, Sands DC, Morris CE (2014) The Pseudomonas viridiflava phylogroups in the P.syringae species complex are characterized by genetic variability and phenotypic plasticity of pathogenicity-related traits. Environ Microbiol 16:2301–2315
Batool F, Rehman Y, Hasnain S (2016) Phylloplane associated plant bacteria of commercially superior wheat varieties exhibit superior plant growth promoting abilities. Front Life Sci 9:313–322
Bazghaleh N, Mamet SD, Bell JK, Moreira ZM, Taye ZM, Williams S, Arcand M, Lamb EG, Shirtliffe S, Vail S, Siciliano SD, Helgason B (2020) An intensive multilocation temporal dataset of fungal communities in the root and rhizosphere of Brassica napus. Data Brief 30:105467
Bokulich NA, Thorngate JH, Richardson PM, Mills DA (2014) Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc Natl Acad Sci U S A 111:E139–E148
Bolyen E et al. (2018) QIIME 2 : Reproducible , interactive , scalable , and extensible microbiome data science. PeerJ. doi:https://doi.org/10.7287/peerj.preprints.27295
Bordoloi GN, Kumari B, Guha A, Bordoloi M, Yadav RNS, Roy MK, Bora TC (2001) Isolation and structure elucidation of a new antifungal and antibacterial antibiotic produced by Streptomyces sp. 201. Bioscience. Biotechnol Biochem 65:1856–1858
Canola Council of Canada. (2020a)-05-05. https://www.canolacouncil.org/canola-encyclopedia/diseases/
Canola Council of Canada, (2020b)-05-25. https://www.canolacouncil.org/canola-encyclopedia/crop-development/growth-stages/
Chao A, Lee S-M (1992) Estimating the number of classes via SampleCoverage. J Am Stat Assoc 87(417):210–217
Clarke WE, Higgins EE, Plieske J, Wieseke R, Sidebottom C, Khedikar Y et al (2016) A high - density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single - locus markers in the allotetraploid genome. Theor Appl Genet 129(10):1887–1899
Coleman-Derr D, Desgarennes D, Fonseca-Garcia C, Gross S, Clingenpeel S, Woyke T, North G, Visel A, Partida-Martinez LP, Tringe SG (2016) Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol 209(2):798–811
Copeland JK, Yuan L, Layeghifard M, Wang PW, Guttman DS (2015) Seasonal community succession of the phyllosphere microbiome. Mol Plant-Microbe Interact 28:274–285
Cordero J, de Freitas JR, Germida JJ (2020) Bacterial microbiome associated with the rhizosphere and root interior of crops in Saskatchewan, Canada. Can J Microbiol 66:71–85
Coutinho TA, Venter SN (2009) Pantoea ananatis: an unconventional plant pathogen. Mol Plant Pathol 10(3):325–335
Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Dombrowski N, Schlaeppi K, Agler MT, Hacquard S, Kemen E, Garrido-Oter R, Wunder J, Coupland G, Schulze-Lefert P (2017) Root microbiota dynamics of perennial Arabis alpina are dependent on soil residence time but independent of flowering time. ISME J 11(1):43–55
Dorokhov YL, Sheshukova EV, Komarova TV (2018) Methanol in plant life. Front Plant Sci 871:1–6
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V (2015) Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci 112(8):E911–E920
FAOSTAT (2017) FAOSTAT, Food and Agriculture Organization of the United Nations. http://www.fao.org/faostat
Feng Y, Shen D, Song W (2006) Rice endophyte Pantoea agglomerans YS19 promotes host plant growth and affects allocations of host photosynthates. J Appl Microbiol 100:938–945
Fürnkranz M, Wanek W, Richter A, Abell G, Rasche F, Sessitsch A (2008) Nitrogen fixation by phyllosphere bacteria associated with higher plants and their colonizing epiphytes of a tropical lowland rainforest of Costa Rica. ISME J 2:561–570
Glaeser SP, Gabur I, Haghighi H, Bartz JO, Kämpfer P, Snowdon R, Obermeier C (2020) Endophytic bacterial communities of oilseed rape associate with genotype-specific resistance against Verticillium longisporum. FEMS Microbiol Ecol 96:1–15
Gopalakrishnan S, Sathya A, Vijayabharathi R, Varshney RK, Gowda CLL, Krishnamurthy L (2015) Plant growth promoting rhizobia: challenges and opportunities. 3 Biotech 5:355–377
Hanson RS, Hanson TE (1996) Methanotrophic bacteria. Microbiol Rev 60:439–471
Harms H, Koops HP, Wehrmann H (1976) An ammonia-oxidizing bacterium. Nitrosovibrio tenuis nov gen nov sp Archives of Microbiology 108:105–111
Hilton S, Bennett AJ, Chandler D, Mills P, Bending GD (2018) Preceding crop and seasonal effects influence fungal, bacterial and nematode diversity in wheat and oilseed rape rhizosphere and soil. Appl Soil Ecol 126:34–46
Hu FP, Young JM, Fletcher MJ (1998) Preliminary description of biocidal (syringomycin) activity in fluorescent plant pathogenic Pseudomonas species. J Appl Microbiol 85:365–371
Humphrey PT, Nguyen TT, Villalobos MM, Whiteman NK (2014) Diversity and abundance of phyllosphere bacteria are linked to insect herbivory. Mol Ecol 23:1497–1515
Iguchi H, Yurimoto H, Sakai Y (2015) Interactions of Methylotrophs with Plants and Other Heterotrophic Bacteria. Microorganisms 3(2):137–151
Jarvis KG, White JR, Grim CJ, Ewing L, Ottesen AR, Beaubrun JJ-G, Pettengill JB, Brown E, Hanes DE (2015) Cilantro microbiome before and after nonselective pre-enrichment for Salmonella using 16S rRNA and metagenomic sequencing. BMC Microbiol 15:160
Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E, Weinstock GM (2019) Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 10(1):1–11
Johnston-Monje D, Lundberg DS, Lazarovits G, Reis VM, Raizada MN (2016) Bacterial populations in juvenile maize rhizospheres originate from both seed and soil. Plant Soil 405(1–2):337–355
Jost L (2007) Partitioning diversity into independent alpha and beta components. Ecology 88:2427–2439
Kim BR, Shin J, Guevarra RB, Lee JH, Kim DW, Seol KH, Lee JH, Kim HB, Isaacson RE (2017) Deciphering diversity indices for a better understanding of microbial communities. J Microbiol Biotechnol 27:2089–2093
Kinkel LL (1997) Microbial population dynamics on leaves. Annu Rev Phytopathol 35:327–347
Lancashire PD, Bleiholder H, Van Den Boom T, Langelüddeke P, Stauss R, Weber E, Witzenberger A (1991) A uniform decimal code for growth stages of crops and weeds. Ann Appl Biol 119:561–601
Leff JW, Lynch RC, Kane NC, Fierer N (2017) Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol 214(1):412–423
Legendre P, Andersson MJ (1999) Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69:1–24
Liu W, Russel J, Røder HL, Madsen JS, Burmølle M, Sørensen SJ (2017) Low-abundant species facilitates specific spatial organization that promotes multispecies biofilm formation. Environ Microbiol 19(7):2893–2905
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative B diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73(5):1576–1585
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL (2013) Defining the core Arabidopsis thaliana root microbiome. Nature 488(7409):86–90
Marcondes de Souza JA, Carareto Alves LM, de Mello Varani A, de Macedo Lemos EG (2014) The Family Bradyrhizobiaceae BT - The Prokaryotes: Alphaproteobacteria and Betaproteobacteria (E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, & F. Thompson (eds.); pp. 135–154). Springer, Berlin Heidelberg
Marina M, Maiale SJ, Rossi FR, Romero MF, Rivas EI, Gárriz A, Ruiz OA, Pieckenstain FL (2008) Apoplastic polyamine oxidation plays different roles in local responses of tobacco to infection by the necrotrophic fungus Sclerotinia sclerotiorum and the biotrophic bacterium Pseudomonas viridiflava. Plant Physiol 147(4):2164–2178
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17:10–12
Mason AS, Higgins EE, Snowdon RJ, Batley J, Stein A, Werner C, Parkin IAP (2017) A user guide to the Brassica 60K Illumina Infinium ™ SNP genotyping array. Theor Appl Genet 130(4):621–633
McDonald D, Clemente JC, Kuczynski J, Rideout JR, Stombaugh J, Wendel D et al (2012) The biological observation matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. GigaScience 464(1):1–6
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217
Mitter EK, de Freitas JR, Germida JJ (2017) Bacterial root microbiome of plants growing in oil sands reclamation covers. Front Microbiol 8:1–14
Mohan Ram HY, Rao IVR (1984) Physiology of flower bud growth and opening. Proc: Plant Sci 93(3):253–274
Nisbet RER, Fisher R, Nimmo RH, Bendall DS, Crill PM, Gallego-Sala AV, Hornibrook ERC, López-Juez E, Lowry D, Nisbet PBR, Shuckburgh EF, Sriskantharajah S, Howe CJ, Nisbet EG (2009) Emission of methane from plants. Proc R Soc B Biol Sci 276:1347–1354
Nitsch JP (1965) Physiology of flower and fruit development BT - Differenzierung und Entwicklung / Differentiation and Development (A. Lang (ed.); pp. 1537–1647). Springer, Berlin Heidelberg
Ofek M, Hadar Y, Minz D (2012) Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS One 7:e40117
Oksanen, Jari, Pierre Legendre, Bob O’Hara, M Henry H Stevens, Maintainer Jari Oksanen, and MASS Suggests. (2007). “The Vegan Package.” Community Ecology Package 10: 631–37
R Core Team (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria URL https://www.R-project.org/
Ray A, Nordén B (2000) Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB J 14:1041–1060
Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N (2010) The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol 12(11):2885–2893
Ren D, Madsen JS, Sørensen SJ, Burmølle M (2015) High prevalence of biofilm synergy among bacterial soil isolates in cocultures indicates bacterial interspecific cooperation. ISME J 9:81–89
Romero FM, Rossi FR, Gárriz A, Carrasco P, Ruíz OA (2019) A bacterial endophyte from apoplast fluids protects canola plants from different phytopathogens via antibiosis and induction of host resistance. Phytopathology 109(3):375–383
Ryan PR, Dessaux Y, Thomashow LS, Weller DM (2009) Rhizosphere engineering and management for sustainable agriculture. Plant Soil 321(1–2):363–383
Sarris PF, Trantas EA, Mpalantinaki E, Ververidis F, Goumas DE (2012) Pseudomonas viridiflava, a multi host plant pathogen with significant genetic variation at the molecular level. PLoS One 7:e36090
Schlatter DC, Hansen JC, Schillinger WF, Sullivan TS, Paulitz TC (2019) Common and unique rhizosphere microbial communities of wheat and canola in a semiarid Mediterranean environment. Appl Soil Ecol 144:170–181
Shade A, Handelsman J (2012) Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol 14(1):4–12
Shade A, Stopnisek N (2019) Abundance-occupancy distributions to prioritize plant core microbiome membership. Curr Opin Microbiol 49:50–58
Shade A, Jacques MA, Barret M (2017) Ecological patterns of seed microbiome diversity, transmission, and assembly. Curr Opin Microbiol 37:15–22
Shu Z, Shi Y, Qian H, Tao Y, Tang D (2010) Distinct respiration and physiological changes during flower development and senescence in two freesia cultivars. HortScience 45(7):1088–1092
Singh P, Santoni S, Weber A, This P, Péros JP (2019) Understanding the phyllosphere microbiome assemblage in grape species (Vitaceae) with amplicon sequence data structures. Sci Rep 9(1):1–11
Smalla K, Wieland G, Buchner A, Zock A, Parzy J, Kaiser S, Roskot N, Heuer H, Berg G (2001) Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl Environ Microbiol 67:4742–4751
Vacher C, Hampe A, Porté AJ, Sauer U, Compant S, Morris CE (2016) The Phyllosphere: microbial jungle at the plant–climate Interface. Annu Rev Ecol Evol Syst 47:1–24
Vandamme P, Opelt K, Knöchel N, Berg C, Schönmann S, De Brandt E, Eberl L, Falsen E, Berg G (2007) Burkholderia bryophila sp. nov. and Burkholderia megapolitana sp. nov., moss-associated species with antifungal and plant-growth-promoting properties. Int J Syst Evol Microbiol 57:2228–2235
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A (2015) The importance of the microbiome of the plant holobiont. New Phytol 206(4):1196–1206. https://doi.org/10.1111/nph.13312
Vishnivetskaya TA, Kathariou S, Tiedje JM (2009) The Exiguobacterium genus: biodiversity and biogeography. Extremophiles 13:541–555
Vogel C, Bodenhausen N, Gruissem W, Vorholt JA (2016) The Arabidopsis leaf transcriptome reveals distinct but also overlapping responses to colonization by phyllosphere commensals and pathogen infection with impact on plant health. New Phytol 212:192–207
Vokou D, Vareli K, Zarali E, Karamanoli K, Constantinidou HIA, Monokrousos N, Halley JM, Sainis I (2012) Exploring biodiversity in the bacterial Community of the Mediterranean Phyllosphere and its relationship with airborne Bacteria. Microb Ecol 64(3):714–724
Von Wintzingerode F, Landt O, Ehrlich A, Göbel UB (2000) Peptide nucleic acid-mediated PCR clamping as a useful supplement in the determination of microbial diversity. Appl Environ Microbiol 66:549–557
Vorholt J a (2012) Microbial life in the phyllosphere. Nat Rev Microbiol 10:828–840
Wagner MR, Lundberg DS, Coleman-Derr D, Tringe SG, Dangl JL, Mitchell-Olds T (2014) Natural soil microbes alter flowering phenology and the intensity of selection on flowering time in a wild Arabidopsis relative. Ecol Lett 17:717–726
Wagner MR, Lundberg DS, del Rio TG, Tringe SG, Dangl JL, Mitchell-Olds T (2016) Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun 7:1–15
Walters W, Hyde ER, Berg-lyons D, Ackermann G, Humphrey G, Parada A, Gilbert JA, Jansson JK (2015) Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1:1–10
Walterson AM, Stavrinides J (2015) Pantoea: insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol Rev 39:968–984
Wassermann B, Rybakova D, Müller C, Berg G (2017) Harnessing the microbiomes of Brassica vegetables for health issues. Sci Rep 7:1–12
Weber CF, King GM (2012) The phylogenetic distribution and ecological role of carbon monoxide oxidation in the genus Burkholderia. FEMS Microbiol Ecol 79(1):167–175
Yang J, Xie B, Bai J, Yang Q (2012) Purification and characterization of a nitroreductase from the soil bacterium Streptomyces mirabilis. Process Biochem 47:720–724
Acknowledgements
We would like to acknowledge Kira Blomquist for assistance in field collections and Alix Schebel and Zayda Morales for laboratory assistance. We acknowledge that this work took place on Treaty 6 Territory, homeland of the Métis. We gratefully acknowledge Dr. Sally Vail and the team of field technical staff at Saskatoon who managed the field trial.
Code availability
Funding
This work is supported by a grant from the Plant Phenotyping and Imaging Research Centre (P2IRC). P2IRC is a digital agriculture research center funded by the Canada First Research Excellence Fund (CFREF) from the Natural Sciences and Engineering Research Council (NSERC), managed by the Global Institute for Food Security (GIFS), and located at the University of Saskatchewan (U of S).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest
Additional information
Responsible Editor: Tim S. George.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bell, J.K., Helgason, B. & Siciliano, S.D. Brassica napus phyllosphere bacterial composition changes with growth stage. Plant Soil 464, 501–516 (2021). https://doi.org/10.1007/s11104-021-04965-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-021-04965-2