
Abstract
Shallow aquifers are vulnerable to natural geogenic processes as well as anthropogenic influences, and this is especially apparent in desert regions. Within arid and hyperarid climates, evaporation is a controlling hydrologic process leads to an important increase in the concentration of dissolved minerals of both surface water and groundwater. In groundwater, this increase is not only dependent on shallow water table depth, but also on the hydraulic properties of sediments present within the unsaturated zone of the aquifer itself. The main objective of this research is to investigate possible mechanisms that might influence water quality changes under seasonal conditions in shallow aquifers situated within the Saharan desert region of Algeria. In this work, we focus on observed changes in hydrogeochemical characteristics, and the possible responsible processes. Under arid conditions, high water mineralization results in hypersaline water or brine solution formation within shallow aquifers. Due to active physico-chemical mechanisms such as Na+/Ca2+ ion exchange, the successive precipitation of calcite, gypsum, mirabilite or blœdite and halite is induced. Biological processes were also observed as prevalent; evidenced by large measured variations in CO2 load concentrations. These processes contributed to an inverse relationship between CO2 and O2 concentrations within the shallow aquifers studied.
Similar content being viewed by others
Introduction
In hydrogeological studies, natural groundwater chemical characteristics vary both temporally and spatially; with the sources of these variances being quite numerous. Among many cited, the proportionality between increased water mineralization and prolonged water–rock interactions, whether upstream to downstream, is a common source of hydrochemical composition change (Langbein and Dawdy 1964; Meybeck 1976; Peters 1984; Kilham 1990). Aerated water conditions and watershed lithological heterogeneity also produce spatial variations in aquifer hydrochemical characteristics, as well (Peters 1984; O’Connor 1967; Phillips 1971; Cook et al. 1989).
The study of shallow aquifer vulnerability in arid regions with respect to high evaporation rates, and increased contamination from agricultural, domestic or industrial sources has been the subject of several recently published research (Amharref et al. 2007; Omosuyi and Osehale 2012; Mohamed et al. 2015). It is currently accepted that the combination of the three aforementioned effects (i.e., water–rock interactions, aerated water conditions and lithological heterogeneity within the watershed), coupled with low precipitation, lead to increased water mineralization and eventual salt paragenesis within the endorheic region beneath the stream bed in the unsaturated zone of a shallow groundwater system. Resulting groundwater vulnerability is dependent on the thickness of the capillary zone above the shallow aquifer water table, and zone proximity to the soil-atmosphere interface. Groundwater vulnerability, however, is not exclusively limited to shallow water table depths. In certain regions of the world, deep groundwater may also become vulnerable under high surface temperatures, as fluctuations may cause changes in hydraulic conductivity, viscosity and groundwater flow velocity within that deep reservoir system (Anderson 2005).
The aim of this work is to study both mechanisms responsible for water quality change, and processes affecting observed hydrogeochemical characteristic variations in shallow Algerian Saharan aquifers. A literature search was initially performed to identify recent studies along three lines of interest: (1) the impact of carbonate mineral equilibria, (2) mineral dissolution or precipitation with resulting ion exchange, and finally (3) the effect of CO2 dissolution on groundwater chemistry within saline aquifer systems. First, two sequential alluvial aquifer geochemical studies were reviewed. Research studies conducted highlighted the relationship and behavior exhibited at different saturation indexes (SI) of carbonate minerals in coastal aquifers located in Pondicherry, India (Chidambaram et al. 2011, 2012). These SIs showed that increasing values of log pCO2 during water–rock interaction directly influenced mineral dissolution processes. Secondly, a descriptive statistics and geochemical modeling study was reviewed to better understand hydrogeochemical processes involving silicate weathering or cation exchange, and the origins of groundwater chemical components in the coastal part of a similarly arid region in Calabar, Nigeria (Edet et al. 2011). Finally, a numerical model of a CO2 leakage scenario occurring during geological storage and sequestration within the Jurassic Dogger Limestone formation in the Paris Basin, Paris, France, was reviewed to determine the various impacts CO2 might have on groundwater quality and hydrochemical characteristics. This aforementioned geologic situation was of interest to our purposes, because of calculations regarding both the CO2 rate of migration, and the amount of released CO2 gas from pressurized solutions. The analysis of said sequestered CO2 gas, stored in close proximity to overlying groundwater, showed the subsequent dissolution of CO2 in the adjacent aquifer; resulting in an overall drop in pH within that underground system (Humez et al. 2011).
Study area
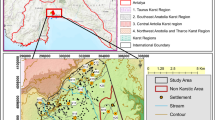
The study area is located in the city of Ouargla, in the northeastern part of the Algerian lower-Sahara (Fig. 1). This area is 800 km away from the capital city of Algiers and bounded by the following wilayas or provinces: El Oued, Biskra and Djelfa to the North, Ghardaia in the West, Illizi and Tamanrasset to the South, and the Tunisian border in the East.

Location map of the study area
The Ouargla region is characterized by a hyperarid desert climate. Data from the National Meteorological Office “NMO” (Ouargla station) in the period between 1996 and 2010 have shown an average annual temperature of 23.4 °C, with a minimum mean of 4.8 °C in January, and a maximum mean of 43 °C in July. Rainfall is very sparse and irregular with an average value of around 39.1 mm/year. These intermittent rains are marked by low or varying quantities, such as 117.8 mm in 2004 and 5.9 mm in 2001. Evaporation is very high that is materialized by an average of 3085 mm/year. It represents an evaporation rate of about 9 mm/day, when it gets its maximum during July 16 mm/day (Idder et al. 2011).
The Ouargla basin is a large geologic depression with an area of approximately 750 km2. It is located in the bed of the low fossil valley of the M’ya wadi which extends from the Tademaït plateau in the In-Salah region, Tamanrasset in the south; to Sebkha Safioune located 40 km north of the city of Ouargla. The Oued Mya converges with Oued Igharghar from a source south of the Ahaggar mountains in the Oued Righ valley; finally draining into Chott Melrhir (Ballais 2010).
In the Ouargla region, the width of the Oued M’ya varies between 4 and 30 km. The average valley gradient is very low at 1‰, gently sloping northward from Ouargla to Sebkha Safioune.
The western boundary is marked by Cenozoic sandstone plateau cliffs covered with gravel and pebbles, and associated reddish sands overlooking the valley at about 220 m in elevation (Aumassip et al. 1972). The eastern boundary is much lower in elevation 160 m, and intensively eroded. It is made of sandstone overlain by a huge Aeolian dune. Where, the basin is mainly dominated by alluvial deposits.
Wetlands appear as flooded depressions. Chotts and Sebkhas, as they are called, are endorheic structures composed of gypsum saline soils. These wetland areas encompass about 25,000 ha, whereas wetlands occupied by palm groves does not exceed 5,000 ha (Idder et al. 2011).
During the Neolithic age, these Chotts and Sebkhas were fed naturally by groundwater contributions (Aumassip et al. 1972). Currently, they receive waters of anthropogenic origin via infiltration of water surpluses from palm plantations and wastewater, causing a marked water table rise in the shallow aquifer. During hot and dry periods, areas flooded in Sebkhas during winter months dry up as a result of evaporation; forming gypsum and other salt-bearing crusts.
Materials and methods
Experimental protocol: sampling locations and water analysis
Groundwater samples were collected during two contrasting temperature periods in both June (Summer) and February (Winter). Surface water piezometers were utilized and situated in various locations differentiated by natural geogenic and anthropogenic conditions. Electrical conductivity (EC), temperature (°C), pH and dissolved oxygen (DO) were measured in situ immediately before filtration. Initially, 18 groundwater samples, taken from the surface, were collected in February during a seasonally high groundwater dilution period. Five months later, 27 additional samples were collected in June during a seasonally high groundwater evaporation period. All groundwater samples were stored in the dark for less than 24 h at low temperatures (< 4 °C) in polyethylene bottles. Ca2+, Mg2+, Cl− ions and alkalinity (HCO3−) were determined by volumetric method (Rodier et al. 1996) at Sonatrach Haoudh Berkaoui Physico-chemical Analysis Laboratory located in Ouargla, Algeria. Na+, K+ ions were analyzed by IC9200 flame photometer, and SO42− ion was analyzed by HACH-DR/2000 spectrophotometer. All measured groundwater physico-chemical data may be found on Table 1 for Summer and Winter.
The majority of the piezometers are shown in (Fig. 2), where static level depths of the groundwater in shallow aquifer range from 0.2 up to 10 m. Most of these water gauge points were localized at urban agglomerations, chotts, sebkhas and palm grove elevations.

Shallow aquifer static level depths
Experimental protocol: activity calculations and concentration factors
Calculation of activities and equilibrium diagram
Solution ion activities were calculated from total concentrations using the "Aqua" Ion Pair software program, and an association model from “GYPSOL” (Valles 1987; Valles et al. 1992, 1997) based on the Debye–Hückel law and extended by the Scatchard Deviation function (Scatchard 1936).
The equilibration level for the partial pressure of CO2 was estimated from pH measurements and carbonate alkalinity (Bourrié 1976; Keller et al. 1987).
Estimate of the concentration factor (CF)
From the study of solution chemistry, followed by the concentration process, it is understood that any variance in exhibited chemical behavior is dependent upon the major elements or ions present (Valles et al. 1997; Al-Droubi 1976; Fritz 1981; Gac 1980). In the studied high concentration range, chloride ion is ubiquitous and conservative (Feth 1981). Therefore, it can be considered an inert solute tracer against the porous unsaturated zone sediment matrix above the shallow aquifer. Chloride ion may also serve as a criterion for concentration factor (CF) estimations and is equilibrated to the ratio between the chloride content of a shallow groundwater sample as compared to that of dilute groundwater.
Results and discussion
Water balance with calcite
Chemical activity calculations for aqueous species permitted the generation of an equilibrium diagram between groundwater solutions and the mineral calcite [CaCO3(s)] (Fig. 3).

Water-calcite equilibrium diagram
Groundwater samples collected and analyzed are well within thermodynamic equilibrium range with calcite. This is consistent with the general presence of that particular carbonate mineral throughout the study area.
The partial pressure of CO2 in the samples becomes variable at 10–3.5 or 0.000316 bars. Comparatively, atmospheric CO2 can reach up to 10–1 or 0.1 bars: reflecting a high level of CO2(g). Inverse relationship is observed between pCO2 in equilibrated water and dissolved oxygen (Fig. 4). This behavior may be explained by dissolved oxygen consumption and CO2 production mechanisms from biological activity within the shallow groundwater system.

Relationship between dissolved oxygen (O2) and log(pCO2)
Another important relationship considered was that between pressure and temperature. Generally, thermodynamic equilibrium with dissolved gases in groundwater relies on both pressure and temperature regimes in subjected aquifer system. Considering pressure alone, CO2 solubility in water is a function of its partial pressure in an aquifer; with groundwater fluid pressure being proportional to aquifer depth. With a stated increase in pCO2, groundwater pH would tend to decrease; and in an unbuffered aqueous system, carbonate mineral dissolution would eventually result. Conversely, pCO2 (as well as dissolved oxygen) levels in water are inversely proportional to temperature. As a shallow aquifer would be more directly influenced by surface temperatures in an arid or hyperarid climate, higher temperatures affecting the capillary zone may cause lower pCO2 (and dissolved oxygen) levels in those groundwaters. Therefore, the determination of the pCO2 in consideration of these two important parameters is rather complex at best.
Equilibrium with sulfated and chlorinated minerals
Overall, analyzed groundwater samples mostly remained at equilibrium with gypsum [CaSO4 • 2H2O(s)] (Fig. 5a). However, some samples reached equilibrium with mirabilite [Na2SO4 • 10H2O(s)], as well. This trend was found to be prevalent during the seasonally high groundwater dilution period in February, due to reduced solubility of this particular mineral at low temperatures (Fig. 5b). More saturated solutions reaching equilibrium with halite [NaCl(s)] were only found in samples P160 and P165 collected at sebkha Oum Raneb. Halite saturation was due to very shallow water table depths measured at 0.30 and 0.88 m in June (Summer), and 0.40 and 1.00 m in February (Winter); as well as water quality being affected by mineral dissolution from surrounding saline soils (Fig. 5c).

Relationship between ion activity [log (ax)] for sulfated and chlorinated minerals
Diagrams of concentration (molality vs. CF)
As equilibrium with the mineral halite was achieved with a small number of highly saturated samples, chloride ion (Cl−) was considered the most conservative major element constituent present. This ion was subsequently used to estimate a concentration factor (CF) in the form of a ratio between chloride concentration measured in a sample solution compared to the minimum chloride ion concentration observed in a sample series (Valles 1987). The concentration factor calculated varied from 1 to 513 for surface waters. This CF variation signifies that water mineralization depends mainly on measured mineral constituent concentration in evaporating waters within an arid or hyperarid climate. Therefore, evaporation directly affected groundwaters located at the surface of the shallow water table.
In dilute waters, the molality of sodium ion (Na+) increased generally at a slower rate than chloride ion (Cl−) due to preferential ion adsorption onto clay particles. However, in this study, the line progression of both chloride and sodium ion concentrations eventually became identical (Fig. 6), due to high concentrations in water.

Ion concentration diagram (Molality vs. CF)
Figure 6 shows that water sample solution alkalinity was found to be lower in moles of dissolved calcium. Calcite residual alkalinity was therefore negative when sample water solutions were saturated. Alkalinity remained stable, however, with calcium content increase (Gac et al. 1979; Ribolzi et al. 1993) due to calcium equilibrium with fluoride ion (Barbiero et al. 1995; Nezli et al. 2009).
With gypsum precipitation, calcite + gypsum residual alkalinity was positive, where SO42− + alkalinity > Ca2+ loads in moles. Sulfate content increased while calcium content stabilized, and the chemical activity of calcium decreased with an increase in ionic strength. Successive precipitation of calcite and gypsum led to an increase in the Mg2+/Ca2+ ratio when water salinity increased.
During the winter season, when lower temperatures prevail and sodium sulfate mineral is less soluble, the precipitation of mirabilite, magnesium sulfates (hexahydride) and sodium + magnesium sulfates, such as blœdite [Na2Mg(SO4)2 • 4H2O(s)] takes place. Halite mineral precipitation is observed in the most concentrated or supersaturated saline waters, and these mineral precipitation processes have been described at length by Valles (Valles 1987). However, within the expected general variation by dissolved element constituents, some divergence does appear within said trends.
H+ ion activity was highly variable around the trend curve (Fig. 7). This variability was less remarkable with alkalinity, but remained somewhat important for calcium and other major elements measured. These results may be attributed to pCO2 heterogeneity; as shown by chemical equilibrium calculations, and the layout of the sample water-calcite equilibrium diagram (Fig. 3).

H + ion concentration diagram
H+ ion activity distribution was observed to be high when pCO2 fluctuated strongly. This result may be attributed to high aerated conditions of groudwater, and spatial variability of biological activity producing CO2 concentrations within the shallow aquifer environment.
Conclusions
In this Ouargla region shallow aquifer study, major element concentrations were controlled by a complex interaction among numerous factors and various mechanisms. Arid climate induced a high concentration of shallow water mineral precipitates, resulting in the formation of saturated saline solutions and salt paragenesis. Physico-chemical mechanisms observed were Na+/Ca2+ ion exchange, and the successive precipitation of calcite, gypsum, mirabilite or blœdite and halite. Biogeochemical processes favored by anthropic effects (wastewater) were evidenced by CO2 concentration variability and biological processes were proposed as responsible for the inverse relationship observed between measured CO2 and O2 concentrations in shallow groundwater samples. Other hydrogeochemical processes (e.g., mineral precipitation and dissolution) were also active within the endorheic zone of each shallow system, as represented by measured data variation from water samples collected from seasonally high evaporation and dilution periods in February (Winter) and June (Summer), respectively.
Mechanisms responsible for measured chemical composition variability in analyzed shallow groundwater samples were multiple, naturally occurring and highly variable in intensity. In this work, mechanisms associated with carbonate equilibrium, partial pressure of CO2 and dissolved oxygen produced from biogeochemical reactions, aquifer temperature and depth; ion exchange and successive salt paragenesis were considered. As our goal was to identify mechanisms that caused variability, it was soon apparent that mechanism interdependence and complexities made it difficult to differentiate between different chemical composition sources within our measured groundwater samples independently. Although a challenging undertaking, it is suggested that in the future, an attempt should be made to establish a hierarchical organization of mechanism intensities, in spite of the many interactions that might affect the carbonate equilibria studied. This is beyond the scope of this paper, but it will be the basis for subsequent research issuing from this same study site.
References
Al-Droubi A (1976) Geochemistry of salts and concentrated solutions by evaporation. Thermodynamic model simulation; application to saline soils of Chad. Geological Sciences Memoire, 46. Strasbsourg: Louis-Pasteur University. Institute of Geology Library
Amharref M, Aassine S, Bernoussi AS et al (2007) Cartography of groundwater pollution vulnerability: application at Gharb Plain (Morocco). Water Sci Rev des Sciences de l’Eau 20(2):185–199. https://doi.org/10.7202/015812ar
Anderson MP (2005) Heat as a groundwater tracer. Groundwater 43(6):951–968. https://doi.org/10.1111/j.1745-6584.2005.00052.x
Aumassip G, Dagorne A, Estorges P et al (1972) Brief Review on the evolution of a quaternary landscape and the population of the Ouargla region. Libyca XX 1:205–257
Ballais JL (2010) From mythical wadis to artificial rivers, the hydrography of the Lower Algerian Sahara. Physio-Géo Revues 4:107–127. https://doi.org/10.4000/physio-geo.1173
Barbiero L, Valles V, Regearg A (1995) Precipitation of fluorite and geochemical control of calcium in alkaline soils of Niger. Consequences for a quantitative estimate of the evolution of geochemical soil. Comptes Rendus de l’Académie des Sciences Paris 321:1147–1154
Bourrié G (1976) Relationship between pH, alkalinity, buffering and CO2 equilibrium in natural waters. Soil Sci du Sol 3:141–159
Chidambaram S, Prasanna MV, Karmegam U et al (2011) Significance of pCO2 values in determining carbonate chemistry in groundwater of Pondicherry Region, India. Front Earth Sci 5(2):197–206. https://doi.org/10.1007/s11707-011-0170-5
Chidambaram S, Prasanna MV, Singaraja C et al (2012) Study on the saturation index of the carbonates in the groundwater using WATEQ4F in layered coastal aquifers of Pondicherry. J Geol Soc India 80(6):813–824. https://doi.org/10.1007/s12594-012-0210-0
Cook PG, Walker GR, Jolly ID (1989) Spatial variability of groundwater recharge in a semiarid region. J Hydrol 111(1–4):195–212. https://doi.org/10.1016/0022-1694(89)90260-6
Edet AE, Preston MR, Worden RH et al (2011) Hydrogeochemical processes in a shallow coastal plain sand aquifer and tidal river systems (Calabar, Southeastern Nigeria): tracking wastewater and seawater pollution in ground and river waters. Environ Earth Sci 65(7):1933–1953. https://doi.org/10.1007/s12665-011-1175-9
Feth JH (1981) Chloride in Natural Continental Water. A Review, U. S. Geologic Survey Water Supply Paper 2176, U. S. Government Printing Office, Washington, D. C. http://pubs.usgs.gov/wsp/2176/report.pdf
Fritz B (1981) Thermodynamic study and modelling of hydrothermal and diagenetic reactions. Geological Sciences Memoire, 65: Strasbsourg: Louis-Pasteur University. Institute of Geology Library
Gac JY (1980) Geochemistry of the drainage basin of Lake Chad. Documents and Works of the O. R.S.T.O.M., 123. http://horizon.documentation.ird.fr/exl-doc/pleins_textes/pleins_textes_6/Tra_d_cm/00039.pdf
Gac JY, Al-Droubi A, Paquet H et al (1979) Chemical model for origin and distribution of elements in salts and brines during evaporation of waters. Application to some saline lakes of Tibesti, Chad. Phys Chem Earth 11:149–158. https://doi.org/10.1016/0079-1946(79)90018-1
Humez P, Audigane P, Lions J et al (2011) Modeling of CO2 leakage up through an abandoned well from deep saline aquifer to shallow fresh groundwaters. Transp Porous Media 90(1):153–181. https://doi.org/10.1007/s11242-011-9801-2
Idder, T, Idder A et Mensous M (2011) Ecological consequences of a non-rational agricultural water management of in Algerian oases of the Sahara (Case of an Oasis in Ouargla)/Les conséquences écologiques d’une gestion non raisonnée des eaux agricoles dans les oasis du Sahara algérien (Cas de l’oasis de Ouargla). International Symposium on ecological, economic and social uses of agricultural water in Mediterranean, University of Provence, Marseille, 20–21 January 2011, 12 p
Keller C, Bourrie G, Vedy JC (1987) Alkalinity forms in infiltration waters Influence of heavy metals contained in composts. Soil Sci du Sol 25(1):17–29
Kilham P (1990) Mechanisms controlling the chemical composition of lakes and rivers: Data from Africa. Limnol Oceanogr 35(1):80–83. https://doi.org/10.4319/lo.1990.35.1.0080/pdf
Langbein WB, Dawdy DR (1964) Occurrence of dissolved solids in surface waters of the United States; U. S. Geological Survey Professional Paper 501-D; U. S. Government Printing Office, Washington, D. C.: D115–D117
Meybeck M (1976) Total mineral dissolved transport by world major rivers/transport en sels dissous des plus grands fleuves mondiaux. Hydrol Sci Bull 21(2):265–284. https://doi.org/10.1080/02626667609491631
Mohamed EA, El-Kammar AM, Yehia MM et al (2015) Hydrochemical evolution of inland lakes’ water: a study of major element geochemistry in the Wadi El Raiyan depression, Egypt. J Adv Res. https://doi.org/10.1016/j.jare.2014.12.008
Nezli IE, Achour S, Djidel M et al (2009) Presence and origin of fluoride in the complex terminal water of Ouargla Basin (Northern Sahara of Algeria). Am J Appl Sci 6(5):876–881. https://doi.org/10.3844/ajassp.2009.876.881
O’Connor DJ (1967) The temporal and spatial distribution of dissolved oxygen in streams. Water Resour Res 3(1):65–79. https://doi.org/10.1029/WR003i001p00065
Omosuyi GO, Osehale A (2012) Groundwater vulnerability assessment in shallow aquifers using geoelectric and hydrogeologic parameters in Odigbo, Southwestern Nigeria. Am J Sci Ind Res 3(6):501–512. https://doi.org/10.5251/ajsir.2012.3.6.501.512
Peters NE (1984) Evaluation of environmental factors affecting yields of major dissolved ions of streams in the United States. U. S. Geological Survey Water Supply Paper 2228; U. S. Government Printing Office, Alexandria, VA. URL: http://pubs.usgs.gov/wsp/2228/report.pdf
Phillips KN, Van Denburgh AS (1971) Hydrology and geochemistry of abert, summer and goose lakes, and other closed-basin lakes in South-Central Oregon; U. S. Geological Survey Professional Paper 502-B; U. S. Government Printing Office, Washington D. C. URL: http://pubs.usgs.gov/pp/0502b/report.pdf
Ribolzi O, Valles V, Barbiero L (1993) Geochemical control of water via the formation of calcite in a mediterranean and tropical environment: equilibrium and water budget arguments. Soil Sci du Sol 31(2):77–95
Rodier J, Bazin C, Broutin J-P et al (1996) The analysis of water—natural waters, residual waters, seawater, 8th edn. Dunod, Paris
Scatchard G (1936) Concentrated solutions of strong electrolytes. Chem Rev 19(3):309–327. https://doi.org/10.1021/cr60064a008
Valles V (1987) Water and salt transfer modelling in clay soils: Application to irrigation assay calculations. Geological Sciences Memoire, 79. Strasbourg: Louis-Pasteur University, Institute of Geology Library
Valles V, Cockborne A M (1992) Development of an applied geochemistry software program for water quality studies. In: Alteration and Restoration of Continental Water Quality, Seminary Meeting at Port-Leucate, October 1 -2: 27–30
Valles V, Rezagui M, Auque L et al (1997) Geochemistry of saline soils in two arid zones of the Mediterranean Basin. 1. Geochemistry of the chott melghir-mehrouane watershed in Algeria. Arid Soil Res Rehabil 11(1):71–84. https://doi.org/10.1080/15324989709381460
Acknowledgements
The authors are pleased to acknowledge to C. Mei-Ling HAMILTON for her helps in the language performance.
Funding
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Medjani, F., Djidel, M., Labar, S. et al. Groundwater physico-chemical properties and water quality changes in shallow aquifers in arid saline wetlands, Ouargla, Algeria. Appl Water Sci 11, 82 (2021). https://doi.org/10.1007/s13201-021-01415-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13201-021-01415-3