

Abstract
In this research, we aimed to establish a guideline for designing electron mediators suitable for biofuel cells. A redox potential simulator was fabricated by combining density functional theory calculation and experiment, allowing us to select molecules with appropriate redox potentials efficiently. Previously, mediators have been developed depending on the trials and errors; thus, our strategy will speed up the development of biofuel cells with outstanding performances.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
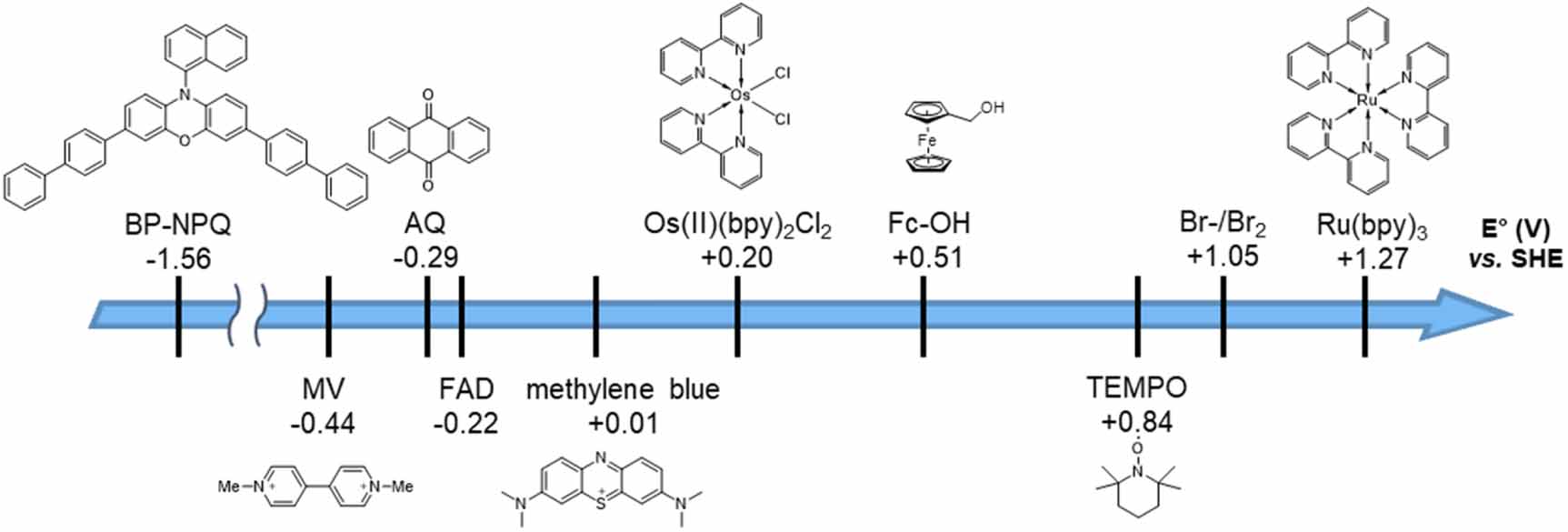
The demand for alternative resources of fossil fuels has triggered us to develop new energy systems. Biofuel cells (BFCs) are among the most attractive power sources that utilize enzymatic reactions to generate electricity from sugars, lactic acid, and other biogenic molecules. BFCs are biocompatible and operative at mild temperatures (20 °C–40 °C) and a nearly neutral pH; therefore, applications in a wide range of electronic devices, including wireless wearable smart sensors and implantable biomedical devices, have been actively investigated [1–3]. Despite these advantages, BFCs have not participated in the practical application due to the limited power density. Modified electrodes for BFCs have been proposed to address this issue; a high power density of 2.67 mW g−1 was achieved by employing carbon nanotubes [4], and stable BFC was fabricated by immobilizing enzymes on electrodes using a sol-gel matrix or polymer [5, 6]. As another approach, mediated electron transfer (MET) through redox-active molecules, so-called electron mediators (MEDs), have been employed. The MEDs exhibit redox activity and act as an electron shuttle that mediates electron transfer between materials such as enzymes and electrodes. Their electronic properties are used in a wide range of fields such as biological reactions [7, 8], photoredox catalysts [9], and Li–O2 batteries [10, 11]. Examples of MEDs reported to date are shown in figure 1. In BFCs, MEDs are used to improve the efficiency of structurally limited electron transfer of biocatalysts [12, 13]. Metal complexes, such as osmium and iron complexes [14, 15], and redox-active organic compounds, such as naphthoquinones, are potential candidates of MEDs in BFCs.

Figure 1. Redox potentials of possible electron MEDs (vs. SHE) at pH 7.
Download figure:
Standard image High-resolution imageThe selection of a MED with a high redox potential can increase the rate of electron transfer from the enzyme or coenzyme to the electrode [16, 17]. In contrast, the redox-mediated process reduces the resulting cell overvoltage (ΔEcell) (figure 2) [18]. Therefore, MEDs with suitable redox potential for the enzymes are required on both electrodes to maximize the performance of BFCs. Since the redox potential correlates with the electronic state of the molecule, it can be adjusted by changing the core structure, the number of redox-active sites, and the type of functional groups [19].

Figure 2. MET system. ΔEcell is smaller than the ideal value of 1.2 V (by glucose oxidation and oxygen reduction) and 0.8 V (by direct electron transfer between enzymes) at pH 7.
Download figure:
Standard image High-resolution imageIn this study, we adopted a density functional theory (DFT) calculations to simulate redox potential. A redox potential simulator was developed by calibrating the calculated and measured reduction potentials, which enabled the simulation of redox potentials of unexplored molecules. The simulator suggested the phenazine can be a core structure for the MEDs. Phenazine is more stable than traditional MEDs, such as quinones, due to the polycondensed aromatic structure, but its derivatives have not been investigated as MEDs for BFCs. Based on the simulation, we propose several candidate structures of phenazine-based MEDs.
2. Methods
2.1. Calculation of reduction potential
For the simulation of redox potentials, we focused on the calculation of the reduction potentials. The reduction potential (E0) can be calculated by the following equation: E0 = −ΔG0(t)/(−nF), where ΔG0(t) is Gibbs free energy change, n is the number of electrons participating in the reaction, and F is Faraday constant.
Here, ΔG(t) is the energy change during the reduction process of MED, where an electron is added to the MED. Following Coote's calculation of the reduction potential in acetonitrile [20], we simulate the redox potential by calculating ΔG(t) by the DFT method. BFCs employ enzymatic reactions which are operated in water; therefore, the influence of water is necessary to consider for the ΔG(t) calculation. The free energy change of isolated molecule in the gas state, ΔG(gas), can be calculated at a standard B3LYP6-31G level. Besides, the solvation energies, ΔG(solv., MED) and ΔG(solv., MED.−), can be calculated using the conductor-like polarizable continuum (CPCM) model. In other words, ΔG(t) in water where BFCs operate can be expressed by the following equation: ΔG(t) = ΔG(gas) + ΔG(solv., MED.−) − ΔG(solv., MED). The summary of the calculation method is shown in figure 3.

Figure 3. Scheme for the calculation of ΔG(t).
Download figure:
Standard image High-resolution imageThe Gibbs free energies for each state are defined as follows: the vacuum state MED is G(gas, MED), after adding one electron to the vacuum state MED is G(gas, MED.−), the hydrated MED is G(solv., MED), and hydrated MED after adding one electron is G(solv., M.−). Gauss View software was used to create input files corresponding to these four states. The charge and number of spins for MED were set to 0 and 1, and those for MED.− were set to −1 and 2. In the case of MED in the cationic state, the charge and the spin numbers of the original structure were 1 and 1, and those after reduction were 0 and 2, respectively. In the case of a solvated state, 'scrf = (cpcm, solvent = water)' was added to the calculation condition. Structural optimization was performed using these input files, and the Gibbs free energy (G') and its thermal correction (GibbsCorr) of the optimized structure were obtained from the output data. The corrected free energy in each state, G(gas) = G'(gas) + GibbsCorr, was calculated from the output data. Next, ΔG was calculated by the following formula: ΔG(gas) = G(gas, MED.−)−G(gas, MED)—5/2 RT. Similarly, ΔG(solv., MED) and ΔG(solv., MED.−) were calculated, and finally, E0 was obtained from the following two equations

2.2. Experimental measurement of redox potential by cyclic voltammetry
The redox potentials of the prepared phenazine derivatives were measured by cyclic voltammetry (CV) in a standard three-electrode system using a SI1287 potentiostat (Solartron Analytical, Hampshire, UK). In the three-electrode system, following electrodes were used; a glassy carbon electrode polished with alumina slurry (0.05 μm) as a working electrode, Pt wire as a counter electrode, and Ag/AgCl as a reference electrode. The CVs were measured in an aqueous electrolyte solution composed of a 10 μl MED solution (0.1 M solution in DMSO) and 10 ml phosphate buffer solution (0.1 M, pH 7) containing NaCl (137 mM) and KCl (2.7 mM) at a scan rate of 10 mV s−1. Since phenazines are not well soluble in water (ca. 10 μg ml−1), assistance with DMSO is necessary.
2.3. Synthesis of phenazine-type MEDs
2.3.1. Typical procedure for the AcOH-catalyzed synthesis of phenazine
Following a reported procedure [21], a round-bottom flask equipped with a magnetic stir bar was charged with 1,2-dihydroxybenzene (132 mg, 1.2 mmol, 1.0 equiv.) and dichloromethane (11 ml). The reaction vessel was cooled in an ice-water bath for 10 min. To the solution, a solution of sodium periodate (279 mg, 1.3 mmol, 1.1 equiv.) and distilled water (5 ml) was added dropwise by syringe over 5 min with cooling. After 10 min, the mixture was extracted with dichloromethane. To this solution, including crude 1,2-benzoquinone in dichloromethane, was mixed with acetic acid (0.8 ml) and o-phenylenediamine (1.2 mmol, 1.0 equiv.) at room temperature. After 30 min, the reaction mixture was warmed to 50 °C and stirred for 5 h. After quenching with saturated aq. NaHCO3, the mixture was extracted with ethyl acetate. The organic layers were dried over MgSO4, filtered, and the solvent was removed in vacuo. The crude residue was purified by silica gel column chromatography (hexane/ethyl acetate = 1/5–1/2) to give 6a. For the synthesis of 6b, 4-methyl-1,2-phenlylenediamine was used instead of o-phenylenediamine.
2.3.2. Typical procedure for the Nd(OTf)3-catalyzed synthesis of phenazine
Following a reported procedure [22], a round-bottom flask equipped with a magnetic stir bar was charged with 1,2-dihydroxybenzene (220 mg, 2.0 mmol, 1.0 equiv.), manganese dioxide (869 mg, 10 mmol, 5.0 equiv.) and dichloromethane (20 ml) at room temperature. After 3 h, the mixture was filtered using a celite pad. To the filtrate, o-phenylenediamine (2.0 mmol, 1.0 equiv.) and neodymium(III) triflate (237 mg, 0.4 mmol, 20 mol%) were added at room temperature, and stirred overnight. After quenching with saturated aq. NaHCO3, the mixture was extracted with chloroform. The organic layers were dried over MgSO4, filtered, and the solvent was removed in vacuo. The crude residue was purified by silica gel column chromatography (hexane/ethyl acetate = 1/5–1/2) to give 6a. For the synthesis of 6c and 6d, 4-chloro-1,2-phenlylenediamine and 4-cyano-1,2-phenlylenediamine were respectively used instead of o-phenylenediamine.
2.3.3. Typical procedure for the synthesis of benzophenazine
For the synthesis of 6f, 1,2-naphtoquinone (95.3 mg, 0.6 mmol) was reacted with o-phenylenediamine (65.5 mg, 0.6 mmol) in AcOH (0.4 ml) at room temperature for 30 min, then heated to 50 °C for 5.5 h. The reaction mixture was added saturated aq. NaHCO3, extracted with AcOEt, then purified by column chromatography with silica gel to obtain yellow powder.
2.3.4. Typical procedure for the cationic derivatization of phenazine
A round-bottom flask equipped with a magnetic stir bar was charged with phenazine (180 mg, 1.0 mmol, 1.0 equiv.) and nitrobenzene (0.9 ml) at 120 °C. To the mixture, dimethyl sulfate (0.25 ml, 2.6 mmol, 2.6 equiv.) was added, and the solution was stirred vigorously for 10 min. The precipitates formed were collected by filtration. Thorough washing with diethyl ether gave the pure product.
3. Results and discussion
First, we surveyed the core structure of commercially available MEDs. The redox potentials of various molecules, such as 1,4-naphthoquinone (1), 1,2-naphthoquinone (2), phenanthrenequinone (3), benzoquinone (4), and anthraquinone (5), were measured or collected from the database. As a result, phenazine (6), which has not been employed as MED in BFCs so far, whereas used in organic batteries [23], was found as a candidate core structure with a desirable redox potential of −0.35 V.
Next, to construct a redox potential simulator, we searched for a simulation method that shows a good correlation between the measurements and calculations. The affinity of a molecule with an electron is correlated to its lowest unoccupied molecular orbital (LUMO) [16]. DFT calculations were performed at the B3LYP/6-31G+(d) level using Gaussian 09 software. The LUMO energy values were calculated and plotted as a function of the redox potentials for molecules 1–6. However, a linear relationship between LUMO energy and redox potential was not observed among these molecules. One of the reasons for the unsuccess would be the LUMO calculations were performed on isolated molecules in the gaseous state. Therefore, we took into account the effect of solvent for the calculation; in BFCs MEDs are solvated with water. Furthermore, we decided not to rely on the LUMO level, but on the energy change between a molecule and its reduced form to accurately simulate the redox potential. The detailed calculation method is shown in section 2.1. The calculated redox potentials and measured redox potentials were plotted; as a consequence, a linear correlation was observed (figure 4). The linear regression of the data in figure 4 indicates the correlation coefficient (R2) of 1.00, which is very linear because of the non-functionalization of the core structures. But, when any functional group is introduced, a core structure and a functional group contribute to the redox reaction; thus, it cannot lie on the line.

Figure 4. The relationship between measured redox potentials and calculated reduction potentials for 1–6.
Download figure:
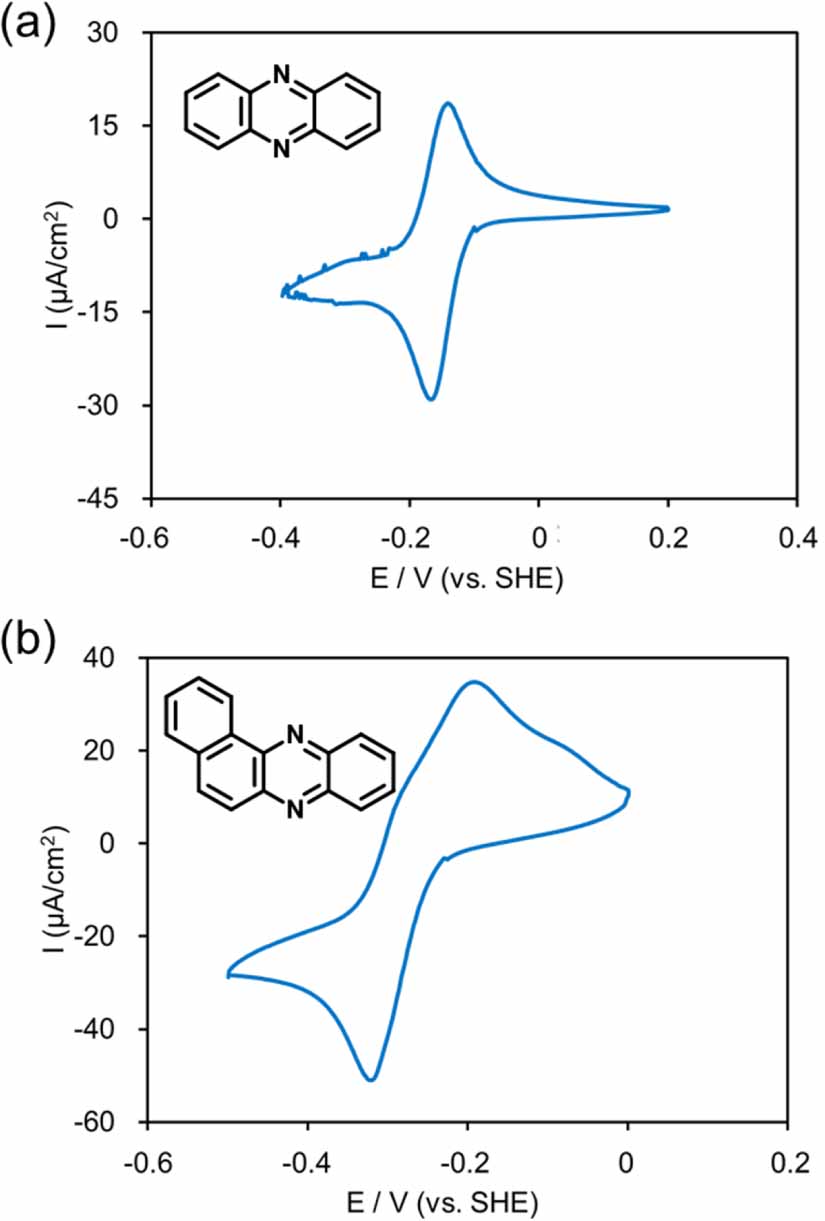
Standard image High-resolution imageThe electrochemical properties of the phenazine and their derivatives were investigated by CV in a three-electrode cell. Selected CVs are shown in figures 5(a) and (b), each displaying one reversible redox wave at E1/2 of −0.261 and −0.158 V (vs. SHE), corresponding to the reduction of phenazine to dihydrophenazine and its re-oxidation, respectively [24]. The redox potentials of several types of phenazines determined by CV curves were depicted in table 1. The redox potentials of phenazine were shifted to the positive potential side by the introduction of electron-withdrawing groups such as chloro (6c) and cyano (6d) groups from −0.158 to −0.054 and −0.112 V (vs. SHE). The methylation of nitrogen by dimethyl sulfate (6e) increased the redox potential. When a more expanded conjugate system (6f) was employed, the redox potential shifted to the negative side.

Figure 5. Selected cyclic voltammograms of (a) phenazine (6a) and (b) benzo[a]phenazine (6f).
Download figure:
Standard image High-resolution imageTable 1. Redox potentials of various phenazines.
| Entry | Chemical name | E1/2 (V) (vs. SHE) |
|---|---|---|
| 1 | Phenazine (6a) | −0.158 |
| 2 | 2-Methylphenazine (6b) | −0.143 |
| 3 | 2-Chlorophenazine (6c) | −0.112 |
| 4 | 2-Cyanophenazine (6d) | −0.054 |
| 5 | Phenazine methosulfate (6e) | 0.085 |
| 6 | Benzo[a]phenazine (6f) | −0.261 |
Using readily available phenazine derivatives (6a–6e), the calculated reduction potentials and measured redox potentials were plotted, showing an excellent linear correlation (R2 = 0.9827) (figure 6). This indicates that we have successfully fabricated a simulator to precisely predict the redox potential in phenazines with various functional groups. However, benzophenadine (6f) was slightly out of this correlation may be due to the extended π system. When 6f is included and the data were fitted by linear regression, R2 value was 0.89. In contrast, when 6f is excluded, R2 became 0.9827. Such intentional control of data is not recommended from statistical point of view. However, when the number of samples is small because of synthesis technique, it is not the main point to have a significant focus on statistics. We consider it suffices to know whether the redox potential becomes higher or lower when a particular functional group is introduced. We want to collect data on the redox potentials of vast numbers of redox-active compounds and analyze statistically in the future for more detailed studies.

Figure 6. Correlation between measured redox potentials and calculated reduction potentials for phenazine derivatives 6. The calibration line was prepared excluding 6f.
Download figure:
Standard image High-resolution imageThe MED performance of phenazines was evaluated by CV measurements. The measurement conditions were the same as those of redox potential measurements. The results obtained by adding 1 μM FAD-GDH to a PBS solution prepared at a glucose concentration of 0.1 M are shown in figure 7. Phenazine derivative 6e, known as MED for a conventional BFC, showed catalytic currents, while other phenazines did not work as MED. These results indicate that a structure with a redox potential higher than 0 V (vs. SHE) or a structure ionized by methylation is required to function as a MED.

Figure 7. Evaluation of MED performance using CV in the presence or absence of enzyme using PBA/glucose electrolyte. (a) phenazine methosulfate (6e), (b) 2-cyanophenazine (6d).
Download figure:
Standard image High-resolution image4. Conclusion
In this research, the redox potential simulator was fabricated by combining DFT calculation and experiment. As MEDs on the anode side of BFCs, control the redox potential higher than ca. −0.3 V is desired. This is because the BFCs for power generation require higher cell voltage by using MED with a redox potential close to that of the enzyme. In contrast, for sensor applications, a high response related to the current density is required; thus, MEDs with higher redox potentials would be suitable. Using the simulator, we have predicted the redox potentials of unexplored molecules. As a result of the simulation, we propose new phenazine derivatives 6g–6j with redox potentials from −0.3 to 0 V (figure 8). For the choice of candidate phenazine derivatives, we randomly attached various functional groups, such as electron-withdrawing F, Cl, CF3, COOH, CONH2, NO2, and electron-donating Me, OMe, NMe2 groups, on phenazine. Some of these molecules are difficult to synthesize in the current synthetic chemistry techniques; therefore, future development in the field of organic synthesis is keenly desired.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Candidates of MEDs with desirable redox potentials. Simulated redox potentials are shown.
Download figure:
Standard image High-resolution image{kind=link}
Acknowledgments
This work was financially supported by JST SICORP, ANR (ANR-15-JTIC-0002-01), and JSPS Kakenhi (20H05224, 17K18992).