
Abstract
Coronavirus disease 2019, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is still a pandemic around the world. Currently, specific antiviral drugs to control the epidemic remain deficient. Understanding the details of SARS-CoV-2 structural biology is extremely important for development of antiviral agents that will enable regulation of its life cycle. This review focuses on the structural biology and medicinal chemistry of various key proteins (Spike, ACE2, TMPRSS2, RdRp and Mpro) in the life cycle of SARS-CoV-2, as well as their inhibitors/drug candidates. Representative broad-spectrum antiviral drugs, especially those against the homologous virus SARS-CoV, are summarized with the expectation they will drive the development of effective, broad-spectrum inhibitors against coronaviruses. We are hopeful that this review will be a useful aid for discovery of novel, potent anti-SARS-CoV-2 drugs with excellent therapeutic results in the near future.
Similar content being viewed by others

1 Introduction
COVID-19, caused by the novel coronavirus SARS-CoV-2, has developed into a serious public health event around the world. To date, more than 108 million individuals have been infected by SARS-CoV-2 in at least 210 countries/territories [1]. Like MERS-CoV and SARS-CoV, SARS-CoV-2 belongs to the beta-coronavirus family that is thought to spread mainly through person-to-person contact and air. Their positive, single-strand RNA genomes are a typical family characteristic, exhibiting a spherical protein envelope and diameter of about 65–125 nm [2].
Liu and co-workers reported an electron microscopy image of isolated SARS-CoV-2 [3]. The envelope consists of Spike protein (S), RNA and nucleocapsid-protein (N), membrane glycoprotein (M) and envelope glycoprotein (E) (Fig. 1). The S protein (also named S glycoprotein), embedded in the virus envelope, is a transmembrane protein with molecular weight of about 150 kDa. In the process of cell entry (Fig. 1), the S protein acts in a pivotal role during invasion of SARS-CoV-2 into host cells. It enters via interaction with angiotensin-converting enzyme 2 (ACE2) receptors located on the cell surface of type II pneumocytes [4]. Enzymatic hydrolysis of the complex between SARS-CoV-2 and host cells is processed by the type II transmembrane serine protease TMPRSS2. After cleavage of ACE-2 and activation of S protein, SARS-CoV-2 fuses to the host cell membrane [5]. Blocking the S protein/ACE2 interaction, or deactivating TMPRSS2, have been identified as promising strategies to prevent viral cell entry. However, once fusion occurs after uncoating of the SARS-CoV-2 envelope, the positive single-strand RNA is released in the cytoplasm and moves to the cell nucleus whereupon it becomes translated to produce nonstructural proteins and viral polymerases. After advancing through the sequence of translation-proteolysis-RNA synthesis, viral structural proteins are synthesized from subgenomic mRNA, and these are further packaged into new virion assemblies [6]. Importantly, a variety of nonstructural proteins exhibit multiple functions that affect the survival and toxicity of coronaviruses. For example, the main protease (Mpro) promotes cytokine expression and cleavage of viral polyproteins. The RNA-dependent RNA polymerase (RdRp) takes a central role in the replication of coronavirus (Fig. 1) [7, 8]. As Mpro and RdRp act a pivotal part in replication and transcription of coronavirus, extensive effort has been devoted to the development of Mpro and RdRp inhibitors to control diseases caused by coronavirus.

The life cycle of SARS-CoV-2, including: membrane fusion, endocytosis, uncoating, translation of RNA, proteolysis, assembly and exocytosis
In light of the infectious route outlined above, and with a view to uncover new inhibitors/drug candidates that interrupt this paradigm, our perspective focuses on understanding the structural biology and medicinal chemistry of various important proteins (Spike, ACE2, TMPRSS2, RdRp and Mpro) in the life cycle of SARS-CoV-2. The representative broad-spectrum antiviral drugs, especially those against homologous virus SARS-CoV, are also summarized with the expectation that may aid in driving the development of anti-SARS-CoV-2 drugs.
2 S protein, ACE2 and TMPRSS2, the Key Proteins for Cell Entry of SARS-CoV-2
Numerous studies have demonstrated that virus infection begins with the recognition and binding of the virus to the receptors on the host cell surface [9]. For coronaviruses, entry into host cells occurs through receptor-mediated recognition by the transmembrane S protein followed by membrane fusion [4, 10, 11]. Since SARS emerged in 2002, the structural biology-based mechanisms of infection by SARS-CoV have been widely studied. The human angiotensin I-converting enzyme 2 (hACE2) was proven to be the main entry receptor. Once it is recognized by the SARS-CoV S protein, the cellular serine protease TMPRSS2 is recruited to cleave the viral S protein, an obligatory action that must occur before membrane fusion [4, 12,13,14]. Recent studies indicate that SARS-CoV-2 also takes advantage of the host proteins TMPRSS2 and ACE2 for cell entry. Recent evidence shows that the affinity of SARS-CoV-2 S protein to ACE2 is higher than that of SARS-CoV. This fact may be the reason for higher rates of transmission of SARS-CoV-2 in humans [15].
2.1 The Structure of SARS-CoV-2 S Protein
The S protein is a trimeric, class I fusion protein with homotrimers protruding from the viral surface and two functional subunits located near the amino (S1) and carboxy (S2) termini [15,16,17]. The virus uses the S1 subunit for binding to the receptors of the host cell. The S2 subunit is employed for the virus-host membrane fusion [18]. Like other class I fusion glycoproteins, membrane fusion activity depends on the proteolytic cleavage of S protein into S1 and S2 subunits. Cell entry of SARS-CoV-2 is accomplished by the engagement between a receptor-binding domain (RBD) in the homotrimeric S protein and ACE2 receptor followed by virus-host membrane fusion [19, 20]. Destroying the key role of S protein in the process of infection is the main purpose of neutralizing antibodies and the interest of vaccine design as well as therapeutic interventions [21]. Wrapp et al. first used cryo-electron microscopy (cryo-EM) to investigate the SARS-CoV-2 S protein, and reported the structure in the pre-fusion conformation (Fig. 2a) [15]. They obtained the crystal structure of an asymmetrical trimer at 3.5-Å resolution. As shown in Fig. 2a, the single RBD presents an up conformation that corresponds to the receptor-accessible state. They also demonstrated that the structures of different domains, including RBDs, N-terminal domains (NTDs), SD1/2s and S1/2s in SARS-CoV-2, have a high degree of homology with corresponding domains in SARS-CoV. The root mean square difference (RMSD) values range from 2.0 to 3.0 Å. [15]. As shown in Fig. 2b, the noteworthy difference between these two S proteins from SARS-CoV-2 and SARS-CoV is the position of the RBDs. The SARS-CoV RBD highlighted by the white color packs tightly against the NTD of the adjacent protomer, while the angle of SARS-CoV-2 RBD is closer to the core cavity of the trimer [15].

a Cryo-EM structure of the SARS-CoV-2 S protein in the prefusion conformation (PDB ID 6VSB) [15]; b an overlay of RBDs from SARS-CoV-2 and SARS-CoV based on the position of their respective adjacent NTD: SARS-CoV-2 RBD (green), SARS-CoV RBD (white), SARS-CoV-2 NTD (purple), SARS-CoV NTD (white) [15]
In the novel coronavirus of most recent interest, an “RRAR” furin recognition site was observed at the boundary near the S1/S2 protease cleavage site that is often found to be related to high virulence [4, 16, 22, 23]. Walls et al. examined cryo-EM structures of the SARS-CoV-2 S ecto-domain trimer, and made a detailed comparison with that of SARS-CoV [4]. They observed that the SARS-CoV-2 S protein exists in a cross-domain conformation that appears to be a 160-Å-long trimer, which closely resembles the structure of the S protein of SARS-CoV. Within the trimer, the S1 subunit presents a V-shaped structure, while the S1 subunit is in an extended conformation. Walls et al. also identified that in the SARS-CoV-2 RBD, a structural opening is essential to initiate a conformational change for recognition of ACE2 that then leads to membrane fusion and cell entry. They also found a 75% sequence homology in the RBD and 88% sequence homology in the S2 subunit between SARS-CoV and SARS-CoV-2. Overall, the structure to emerge from the SARS-CoV-2 S protein is similar to that of all reported coronaviruses, especially to that of SARS-CoV.
To investigate the evolutionary origin of SARS-CoV-2 and gain better insight into the emergence of the COVID-19 pandemic, Wrobel et al. [24] characterized the structures of S protein of SARS-CoV-2 virus (Fig. 3). In the closed conformation (Fig. 3a), the surface of the RBD is wrapped in the interior of the trimer, which makes it unable to interact with the ACE2 receptor. Furin cleavage sites associated with increased pathogenicity were observed between S1 and S2 subunits. In the intermediate conformation (Fig. 3b), one of the three RBDs is disordered, while the other two keep a similar conformation with the closed form. Moreover, the centroids of the two NTDs that are in closest contact with the disordered RBD are shifted by 2.5–2.9 Å compared to that of closed form. The disordered RBD in the intermediate form leads to a decrease in the stability of the S protein, which ultimately enables the formation of an open form. In the open conformation (Fig. 3c), one of the RBDs is completely exposed to the ACE2-interacting surface after rotating ~ 60°, and the other two RBDs are consistent with that of closed and intermediate forms. This rotation is facilitated by the movement of NTD in the adjacent and same chain.

The three structures of S protein of SARS-CoV-2 are calculated from micrographs of furin-cleaved material: a closed (PDB ID 6ZGI); b intermediate (PDB ID 6ZGH); c open (PDB ID 6ZGG) forms. The three monomers are colored orange, light blue and pale green [24]
2.2 The Structural Basis of hACE2 Recognizing S Protein of SARS-CoV-2
Commonly expressed in lungs, intestine, kidneys and heart, ACE2 is a type I membrane protein [25,26,27]. Full-length hACE2 consists of a carboxy terminal collectrin-like domain (CLD) and an amino terminal peptidase domain [26, 28]. However, the monoclonal antibodies that once were effective against SARS-CoV RBD do not neutralize SARS-CoV-2 and block virus infection because of some structural difference between two RBDs from SARS-CoV-2 and SARS-CoV [15, 29]. B0AT1 as the gene mutated in Hartnup disorder is a neutral amino acid (NAA) transporter that regulates the transport of NAAs into intestinal cells in a Na+-dependent manner. Reported evidence shows that B0AT1 employs ACE2 as a chaperone protein to achieve membrane trafficking [30]. However, it is unclear how B0AT1 interacts with ACE2. Recently, on the basis of the assumption that the structure of full-length ACE2 may be disclosed in the presence of B0AT1, Yan et al. reported the 3D structure of the hACE2-B0AT1 complex and the RBD-ACE2-B0AT1 ternary complex that provides the structural basis for understanding SARS-CoV-2 recognition and infection by hACE2 [31]. To reveal these differences and solve this problem, Wang, Li and co-workers determined the co-crystal structures of SARS-CoV-2 RBD in complex with ACE2 [32, 33]. As shown in Fig. 4a, b, the entire binding region of SARS-CoV-2 RBD with ACE2 is nearly congruent with that of SARS-CoV RBD and ACE2. The detailed structural analysis of SARS-CoV-2 RBD reveals that the majority of residues involved in binding either RBD are highly conserved, and the functionalities involved in side chain attachments in SARS-CoV-2 are nearly coincident with those in the SARS-CoV RBD [32]. Extensive studies show that SARS-CoV RBD contains a receptor-binding motif (RBM) and a core [13]. The contact interface between SARS-CoV-2 RBM and ACE2 is broader and tighter than that between SARS-CoV RBM and ACE2 [33].

a The co-crystal structures of SARS-CoV-2 RBD in complex with ACE2. b Superposition of the ridge in SARS-CoV RBM (yellow) and SARS-CoV-2 RBM (cyan). c Superposition of the ridge from another visual angle (PDB ID 6VW1) [33]
Shang et al. confirmed that the significant structural change between RBMs from SARS-CoV-2 and SARS-CoV is the conformation of the loops in the binding model of RBM with ACE2 receptor (Fig. 4b, c) [33]. Specifically, SARS-CoV RBM contains a P-P-A-residue motif in this loop; SARS-CoV-2 RBM, by contrast, contains a G-V-E-G-residue motif; the two kinds of residue motifs allow the loop to adopt a distinct conformation (Fig. 4c). Additionally, there is a hydrogen bond (H bond) between Ala475 and Asn487 in the main chain of SARS-CoV-2 RBM, and hydrophobic interaction was formed by the Phe486 of SARS-CoV-2 RBM with residues Tyr83, Leu79 and Met82 of hACE2. In summary, these structural traits of SARS-CoV-2 RBM are characterized by a higher binding affinity to ACE2 than seen for SARS-CoV.
The results of previous studies indicate that the activation of the envelope glycoproteins of various viruses, including Ebola virus, MERS-CoV, influenza virus and SARS-CoV, is mediated by the host cell protease [34, 35]. Hoffmann and colleagues demonstrated that the cleavage and priming of the S protein of SARS-CoV-2 that is employed for virus-host membrane fusion and cell entry is mediated by TMPRSS2 [18]. Cells, such as sustentacular cells of the olfactory epithelium, susceptible to SARS-CoV-2 virus infection have high ACE2 and TMPRSS2 expression [36]. Theoretically, the prevention of SARS-CoV-2 virus cell entry could be achieved by inhibiting TMPRSS2.
2.3 Potential Drugs for Blocking the SARS-CoV-2 Cell Entry
ACE2 and TMPRSS2 receptors are recruited by the SARS-CoV-2 S protein to enable virus entry into cells. Inhibitors that target these key proteins as well as S protein sites and block SARS-CoV-2 cell entry need to be found and tested for their effectiveness as therapeutic agents [18].
Arbidol (1, an indole-based small-molecule inhibitor), as shown in Fig. 5, is a broad-spectrum antiviral agent developed by the former Soviet Union Medical Chemistry Research Center. It inhibits cell entry of enveloped viruses by blocking virus-host cell membrane fusion [37,38,39]. Li's group reported that Arbidol inhibits SARS-CoV-2 cell entry in in vitro experiments. Molecular docking studies showed that it may do so by targeting S protein [16].

Reported compounds that block the SARS-CoV-2 cell entry
The serine protease inhibitor camostat (2, Fig. 5) is approved in Japan (1985) for treatment of chronic viral infections [34, 40], oral squamous cell carcinoma [41] and chronic pancreatitis [42]; it functions by targeting the serine protease TMPRSS2 that SARS-CoV-2 is reported to use for S protein priming [18]. It was found that camostat mesylate inhibits viral entry and reduces SARS-CoV-2 infection of Calu-3 lung cells [18]. Nafamostat (3) is another TMPRSS2 inhibitor approved in Japan for the treatment of acute pancreatitis, diffuse intravascular coagulation and anticoagulation in cardiopulmonary bypass [43, 44]. Nafamostat has been confirmed to block the membrane fusion of the virus with the lung Calu-3 host cell expressing TMPRSS2 by inhibiting the TMPRSS2 protease activity [45]. In view of the highly conserved amino acid sequence in S proteins of MERS-CoV and SARS-CoV-2, Xiao and co-workers [46] demonstrated that nafamostat exhibited good inhibitory activity against SARS-CoV-2 infection with EC50 of 22.50 μM, indicating that nafamostat as a TMPRSS2 inhibitor can prevent SARS-CoV-2 infection.
Chloroquine (4, Fig. 5), initially developed for the treatment of malaria and inflammatory (e.g., rheumatoid) arthritis [47], was reported to be highly effective in the management of SARS-CoV-2 infection in vitro by interfering with ACE2 [48]. The latest clinical trials carried out in China in COVID-19 patients show that chloroquine effectively shortens the time course of the disease and reduces deterioration of lungs due to pneumonia with acceptable safety constraints [49]. Although the in vitro inhibitory effect of chloroquine on several viruses including SARS-CoV-2 and its multiple mechanisms of action have been elucidated [50], its clinical pharmacology is still unclear [51]. The death of a man in Arizona reported by the New York Times [52] and the serious adverse reactions [53] induced a scientific debate engaged by several scientists on the effective use of chloroquine in the prevention and treatment of COVID-19. At present, there are no studies to evaluate the use of chloroquine for prophylaxis, and there is no convincing evidence to prove its significant clinical efficacy [51]. Accordingly, large, randomized, double-blind clinical trials with proper monitoring are required to determine whether chloroquine can be used as a powerful weapon to prevent and treat COVID-19 and has acceptable safety.
3 RNA-Dependent RNA Polymerase (RdRp)
RNA-dependent RNA polymerase [RdRp, also referred to as nonstructural protein 12 (nsp12)] plays an important role in the synthesis of viral RNA from RNA templates, and especially in the replication and transcription of coronaviruses [54]. Therefore, RdRp has been the preferred target for the treatment of viral infection, such as SARS, MERS and chronic hepatitis viruses [55]. Recent sequence alignment studies have revealed that the SARS-CoV-2 RdRp shares highly homologous sequences with SARS-CoV RdRp [54]. Because blocking RdRp activity in SARS-CoV has been historically successful, it has also rapidly emerged as a promising therapeutic approach to the treatment of COVID-19.
3.1 Structure of RdRp
To facilitate the structure-based drug design, Rao et al. report the 3D structure of the SARS-CoV-2 full-length nsp12-nsp7-nsp8 complex at 2.9-Å resolution using cryo-EM (Fig. 6) [56]. This complex exhibited RNA polymerization activity in the presence of adenosine triphosphate (ATP) [57]. They also identified a newly discovered β hairpin domain at the amino termini of RdRp, in addition to the polymerase core that is conserved throughout the viral polymerase family. Overall, the architecture of the SARS-CoV-2 nsp12-nsp7-nsp8 complex is nearly identical with that of SARS-CoV [56]. The main difference between the RdRps from SARS-CoV-2 and SARS-CoV is the residue cluster Ala4-Arg118 and the residue cluster Asn215-Asp218, both of which are highly ordered in the SARS-CoV-2 nsp12 but less ordered in SARS-CoV nsp12 [56]. Qi Peng et al. reported the reduced polymerase activity and thermostability of the SARS-CoV-2 nsp12-nsp7-nsp8 core polymerase complex compared to that of SARS-CoV through 3D structure comparison, and this feature might contribute to the fitness of SARS-CoV-2 to human hosts [58]. Hillen et al. presented a 3D structure of the SARS-CoV-2 RdRp in an active form that mimics the replicating enzyme. The complex contains the viral proteins nsp12, nsp8 and nsp7, and more than two turns of RNA template–product duplex, while the two copies of nsp8 are mainly responsible for the binding with the opposite sides of the cleft and position of the second turn of RNA. The long helical extensions in nsp8 protrude along the exiting RNA, forming positively charged “sliding poles”, which could account for the known processivity of RdRp that is required for replicating the long genome of coronaviruses [59]. Liming Yan et al. determined the SARS-CoV-2 replication and transcription complex (RTC) which consisted of a template-primer RNA, nsp7 and nsp8, and two helicase molecules (nsp13-1 and nsp13-2) and revealed that the nsp13-1 motif is essential for the enhancement of the helicase activity of mini RTC [60].

Cryo-EM structure of the nsp12-nsp7-nsp8 complex of SARS-CoV-2 (PDB ID 6M71) [56]
Structures of a template-primer RNA bound to SARS-CoV-2 RdRp and in complex with the antiviral drug remdesivir (5, GS-5734, Fig. 8) were determined using cryo-EM structural studies by Yin and co-workers [57]. Remdesivir is a prodrug that needs to be converted to the active triphosphate form [remdesivir triphosphate (RTP)] within the cells. It then covalently combines with the active center at the catalytic site of RdRp (the primer located at the +1 position in Fig. 7a) in the form of a nucleoside triphosphate active metabolite and indirectly inhibits viral RdRp transcription activity through strand termination at arbitrary positions of the template sequence. The backbones of RNA phosphate-ribose are involved in most protein-RNA interactions that directly involve 2′-OH groups (Fig. 7a). The RNA polymerization activity was completely inhibited by 1 mM RTP despite the exposure to physiological concentrations of ATP (10 mM) [57].

a Remdesivir monophosphate is covalently combined with the primer; b X-ray co-crystal structure of RNA bound RdRp in complex with remdesivir (PDB ID 7BV2) [57]
3.2 Drug Repurposing for Treatment of COVID-19 by Inhibiting RdRp
The ongoing SARS-CoV-2 infection highlights the significance and urgency of antiviral agents [55]. Among several antiviral drugs, remdesivir, reported as an anti-Ebola virus inhibitor, is one of the most promising and hopeful antiviral agents used to control SARS-CoV-2 replication [61,62,63]. Remdesivir is a nucleoside analogue with broad-spectrum antiviral efficacy in HAE cells. The half-maximal effective concentration (EC50 value) for SARS-CoV and MERS-CoV are 69 nM and 74 nM, respectively [46, 63]. Unexpectedly, remdesivir less efficiently inhibits SARS-CoV-2 replication with an EC50 value of 0.77 μM. The co-crystal structure of RNA bound to RdRp in complex with remdesivir illustrates that the adenosine analogue of remdesivir monophosphate forms two H bonds with a uridine base from the template strand and additional base-stacking interactions with upstream adenosine bases from the primer strand (Fig. 7b) [57].
As shown in Fig. 8, like remdesivir, other RdRp-targeted inhibitors, such as ribavirin, EIDD-2081, galidesivir, favipiravir, sofosbuvir and IDX-184, are all nucleotide analogues. They have been shown to be efficient in inhibiting viral RdRp in their triphosphate active forms [57, 64]. Figure 9 shows the molecular docking calculations performed by our group, which indicates that their binding configurations are nearly the same as remdesivir; i.e., they all covalently bind to RdRp in a ribosyl monophosphate form at the +1 position of the primer strand. Ribavirin is a broad-spectrum antiviral drug prescribed for the treatment of diseases caused by viral infections, such as hepatitis C and some viral hemorrhagic fevers. The antiviral activity against SARS-CoV was estimated to be at a concentration of 50 mg/ML in vitro [61, 65,66,67,68]. EIDD-2801 bearing good oral bioavailability is a prodrug of EIDD-1931 that has wide-spectrum antiviral activity against influenza virus and multiple coronaviruses, such as MERS-CoV, SARS-CoV and SARS-CoV-2. Specifically, the potency of EIDD-2801 in preventing SARS-CoV-2 replication is 3–10 times stronger than that of remdesivir [57, 69, 70]. This may be due to the two extra H bonds formed by the 4-hydroxyimino group of the cytidine ring with Lys545 and the cytidine base with the guanine base (Fig. 9b) [57]. Galidesivir (BCX4430) is an adenine analogue originally developed to treat hepatitis C virus. It is currently undergoing safety testing in early clinical studies and evaluation of its effectiveness in treating yellow fever. In preclinical studies, it has shown activity against a variety of RNA viruses, including SARS and MERS [71].

The structures of representative RdRp inhibitors

The proposed binding modes of inhibitors [ribavirin (a), EIDD-2081 (b), galidesivir (c), favipiravir (d), sofosbuvir (e) and IDX-184 (f)] with RdRp (PDB ID 7BV2)
Of the pyrimidine analogues, favipiravir (T-705) is also a novel RdRp-targeted inhibitor that is initially phosphoribosylated by enzymes and then converted to favipiravir-ribofuranosyl-5′-triphosphate (an active conformation) that exhibits influenza RdRp inhibitory activity with a half-maximal inhibitory concentration (IC50) of 341 nM. Recently, preliminary clinical trials have shown favipiravir has encouraging anti-polymerase activity for use in treatment of patients infected by SARS-CoV-2 [57, 71,72,73].
Sofosbuvir (PSI-7977) and IDX-184 are HCV RNA replication inhibitors. Using docking data, Elfiky et al. showed that IDX-184 and sofosbuvir tightly bind to the SARS-CoV-2 RdRp. They suggested that further optimization of these two compounds could result in a more potent compound that may be effective against SARS-CoV-2 [74,75,76].
4 The Main Protease (Mpro, Also Named a Chymotrypsin-Like Cysteine Protease, 3CLpro)
According to recent research [77, 78], two polyproteins (pp1a and pp1ab) in SARS-CoV-2 are coded in the two open reading frames, ORF1a and ORF1b, that consist of approximately two-thirds of the entire genome length. Subsequently, the two polyproteins are processed into various nonstructural proteins by employing the two viral-borne proteases, Mpro and PLpro (papain-like protease). That the role of Mpro is vital in adjusting coronavirus replication and transcription of the viral life cycle has been demonstrated in two major studies [79, 80]. The homologous protein of Mpro does not exist in humans. Therefore, the possibility of targeting Mpro by various therapeutic reagents has been viewed as a promising strategy for the treatment of coronavirus diseases. Efforts to target Mpro have been given considerable attention from numerous researchers [81, 82].
At present, the detailed catalytic mechanism of SARS-CoV-2 Mpro action is still unknown. Because it belongs to the β-coronavirus family, SARS-CoV-2 Mpro may share similar catalytic mechanisms with SARS-CoV Mpro and MERS-CoV Mpro. As previously reported, SARS-CoV Mpro and MERS-CoV Mpro cleave the polyproteins that generally favor Leu-Gln↓(Ser, Ala, Gly) sequence sites in the manner of a nucleophile [83,84,85,86,87]. Recently, Shang et al. [88] showed that MERS-CoV Mpro and SARS-CoV Mpro exploit the existence of a partial negative charge cluster (Arg-Tyr-Asp) and a conserved water molecule to accelerate catalysis. They inferred the existence of a partial negative charge cluster (PNCC) at the active interface and suggested it become a potential target for interposing a blocking agent that cancels the dipole moment of PNCC in the prospect that it inhibits proteolytic activity. Following the widening COVID-19 pandemic, an increasing number of groups from all over the globe are testing drug candidates that target SARS-CoV-2 Mpro.
4.1 The Structure of SARS-CoV-2 Mpro
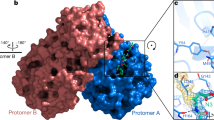
The first crystal structure of SARS-CoV-2 Mpro was examined at a 2.1-Å resolution after co-crystallization with the inhibitor N3 (Fig. 10a, b) [82]. Subsequently, structures of its inhibitor-free form, as well as an α-ketoamide inhibitor-co-crystalized form, were also made available (Fig. 10c) [89]. The Mpro of SARS-CoV-2 exists as a homodimer, which is consistent with the composition of most known coronaviruses [82]. As shown in Fig. 10a, each monomer is composed of three domains (domains I–III). Domains I and II involve residues 8–101 and residues 102–184, respectively. They comprise six-stranded antiparallel β-barrels. Domain III is linked to domain II via a long loop region (residues 185–200) and consists of a globular cluster of five α-helices that are closely related to the dimerization of the Mpro. The protease catalytic cavity occupies the cleft between domains I and II. There are two deep-buried subsites (S1 and S1′) and three shallow subsites (S2-S4) in the pocket [90], and their roles in binding mode will be described in detail in the following. It is worth noting that an active dyad Cys145-His41 (His41 serves as a proton acceptor, and Cys145 serves as the nucleophile) located in the cavity is highly conserved in the Mpro from other coronaviruses (Fig. 10c) [91,92,93,94].

Protein sequence alignment of Mpro in SARS-CoV [95] and SARS-CoV-2 [89] indicates up to 96% amino acid sequence homology with only 12 different residues between the two proteases. The residue Cys145 that forms a C–S covalent bond with inhibitors is a key site in the substrate-binding pocket of Mpro [96]. The site is absolutely conserved in both proteases. In view of the above, repurposing of protease inhibitors that bind to Cys145 of SARS-CoV Mpro may be a promising approach for treatment of COVID-19 to meet the great challenge to discover effective and specific antiviral drugs. It is worth mentioning that the Ser46 residue at the active site of SARS-CoV-2 Mpro may influence the behavior of the substrate-binding pocket and enable novel blocking interactions with protease inhibitors.
By superposition of their crystal structures, Jin et al. demonstrated that the substrate-binding pockets for the Mpro proteases for the 12 reported coronaviruses are highly conserved. This suggests that the previously tested drugs that target this pocket may be useful as broad-spectrum antiviral inhibitors [82, 97]. Thus, the repurposing of already-approved antiviral agents or antiviral drug candidates in various stages of clinical trials may become a feasible approach to speed discovery of effective drugs to control the spread of COVID-19. Previous investments in time and money may help to compensate for the time-consuming and high costs of conventional drug development.
4.2 Inhibitors Targeting SARS-CoV-2 Mpro and the Binding Modes Between Them
Although there is a dearth of specific antiviral drugs to mitigate the COVID-19 epidemic, several drug candidates having encouraging clinical potential and have been described by numerous investigators. Zhang and co-workers discovered a peptidomimetic α-ketoamide analogue 12 (see Fig. 11a) with picomolar level inhibitory activity (EC50 of 400 pM) against MERS-CoV and low-micromolar inhibition (EC50 value in μM) against SARS-CoV in cell-based assays [98]. Given the highly conserved substrate-recognition pocket in two Mpros from SARS-CoV-2 and SARS-CoV, the same authors treat 12 as a reasonable starting point for exploration of novel SARS-CoV-2 Mpro inhibitors. Impeded by the poor metabolic stability (T1/2 = 0.3 h) of 12, they initially developed structural modifications on analogue 12 to improve its half-life in plasma [89]. Replacing the styryl group in the P4 moiety with a tert-butyloxycarbonyl protecting group (Boc-NH) that protects the amide bond in the P3 moiety with a pyridone, and converting the benzyl group in the P1′ moiety to a cyclopropyl group, these changes generated significant improvement (~ three fold) in the plasma half-life in mice of compound 13 (T1/2 = 1 h), relative to compound 12. However, they also resulted in approximately 13.3-fold loss of inhibitory activity (IC50 value reduced from 0.18 to 2.39 μM). To restore the antiviral activity of 13, a smaller cyclopropyl group was introduced in the P2 moiety to replace the cyclohexyl functionality that appears extensively in a number of broad-spectrum inhibitors of Mpro. In addition, the newly substituted cyclopropyl group in the P1′ moiety was returned to a benzyl group. The resulting compound 14 was able to inhibit SARS-CoV-2 Mpro at sub-micromolar concentrations (IC50 = 0.67 μM). In addition, it possessed longer half-life when administrated systemically in mice (T1/2 = 1.8 h). Importantly, compound 14 showed favorable antiviral efficacy in SARS-CoV-2-infected Calu3 cells in an in vitro model with an EC50 value of 4–5 μM. However, upon deprotecting the amine function in moiety P4, compound 15 was formed and was almost completely inactive. In addition, compound 15 showed favorable absorption, distribution, metabolism and excretion (ADME) properties and lung tropism, stimulating the development of the pyridone-containing inhibitors for anti-COVID-19 drugs.

a Structures of α-ketoamide inhibitors 12, 13, 14 and 15 (the modification process is highlighted with background color); b co-crystal structure of inhibitor 14 in the active site of SARS-CoV-2 Mpro (PDB: 6Y2F) [89]
As shown in Fig. 11b, compound 14 locates in the substrate-recognition pocket of SARS-CoV-2 Mpro in an extended conformation. The S1 subsite, composed of His163, Phe140, His172, Cys145 and Glu166 residues, securely anchors the P1 γ-lactam moiety via three H bonds linking between the lactam and Phe140, Glu166 and His163. In addition, the α-ketoamide moiety in 14 was expected to target residue Cys145 to form a stable C–S covalent bond. The benzyl group in the P1′ moiety extends into the bulky S1′ subsite that consists of Cys145, Gly143, His41 and Thr25 residues to interact with Gly143, His41 and Thr25 by van der Waals interaction. The hydrophobic S2 subsite, bearing a stereoscopic volume of 28 Å3 occupied by Met165, Met49 and His41 residues, suitably accommodates the cyclopropylmethyl group in P2. It is worth mentioning that the S2 pocket (with a stereoscopic volume of 252 Å3) in SARS-CoV Mpro is larger than the corresponding S2 pocket of SARS-CoV-2 Mpro, enabling it to adapt to the larger bulky cyclohexylmethyl group in P2 of compound 12 [98]. The pyridone ring in P3 of 14 occupies the S3 subsite formed by the side chains of Glu166 and Gln189 that is stabilized with a H bond between the carbonyl oxygen of pyridine and the main-chain amide of the Glu166 residue. The protecting Boc-NH group in P4 faces towards Pro168, but does not occupy the authorized S4 subsite of the protease.
Based on the substrate-recognition pocket of homologous virus SARS-CoV Mpro (Fig. 12), Dai et al. recently reported two novel peptide-mimetic-based lead compounds (15a and 15b) for SARS-CoV-2 Mpro [99]. On the basis of the core structure of the peptidomimetic covalent inhibitor, a number of privileged groups have been inserted into reported Mpro inhibitors, including:
-
1.
An aldehyde group expected as a warhead to form a C–S bond with the Cys145 residue;
-
2.
A cyclohexyl or 3-fluorophenyl group that is expected to insert deeply into the S2 subsite; and
-
3.
An indole group expected to form new H bonds with S4 subsite.

Medicinal chemistry information of novel peptidomimetic-based inhibitors 16a–b against SARS-CoV-2 Mpro from design to candidates
These were introduced into P1′ (pink mark), P2 (green mark) and P3 (red mark) moieties, respectively, when the fixed (S)-γ-lactam ring was substituted as the P1 moiety. Subsequently, extensive in vitro and in vivo studies led to the identification of the antiviral drug candidates 16a and 16b (in Fig. 12), which demonstrated excellent in vitro potency (IC50, SARS-CoV-2 Mpro of 53 nM and 40 nM, respectively) and good bioavailability (fraction of dose F achieving systemic circulation of 87.8% and 85.9%, respectively). Compounds 16a and 16b displayed excellent cell-based antiviral efficacy (EC50 = 0.53 μM, and 0.72 μM, respectively) with acceptable cytotoxicity (CC50 > 100 μM). In co-crystalized structures of 16a (PDB ID 6LZE) and 16b (PDB ID 6M0K) with SARS-CoV-2 Mpro, both inhibitors were securely anchored in the substrate-binding pocket through the C–S covalent bond with the Cys145 residue. They were further stabilized by an H bond between aldehyde groups and the Cys145 residue. In addition, the cyclohexyl or 3-fluorophenyl group is inserted deeply into the S2 subsite and held by π–π stacking and hydrophobic interactions. The indole group occupies the S4 subsite as expected.
In order to address unmet clinical need in COVID-19 therapy, Gao et al. recently described the biological activities of GC376 (17) and boceprevir (18) against Mpro and SARS-CoV-2 according to drug repurposing strategy (Fig. 13) [100]. As shown in Fig. 13, GC376, a broad-spectrum antiviral drug targeting cysteine protease, and boceprevir, an FDA-approved antiviral medication for the treatment of chronic hepatitis C, both can inhibit the SARS-CoV-2 Mpro activity well with IC50 values of 0.15 μM and 8.0 μM, respectively. Furthermore, GC376 and boceprevir displayed excellent anti-SARS-CoV-2 replication efficacy (EC50 = 0.70 μM, and 15.75 μM, respectively) with excellent selectivity over the bovine chymotrypsin and acceptable cytotoxicity. To interpret mechanisms of these two compounds against SARS-CoV-2 Mpro, they determined the co-crystal structures of GC376 (Fig. 13b) and boceprevir (Fig. 13d) [100]. Both of them were tightly anchored in the substrate-binding pocket through the C–S covalent bond with the Cys145 residue and further stabilized by several H bonds and hydrophobic interactions between ligands and residues.

The structures of GC376 a and boceprevir c; Co-crystal structures of GC376 b (PDB ID: 7C6S) and boceprevir d (PDB ID: 7C6U) in the active site of SARS-CoV-2 Mpro
Rao and colleagues identified that carmofur can be used as a promising lead compound to develop new anti-SARS-COV-2 drugs for treatment of COVID-19 (Fig. 14) [101]. Carmofur effectively inhibits SARS-CoV-2 replication in Vero cells (EC50 = 24.30 μM) with acceptable cytotoxicity (CC50 > 134 μM). The X-ray crystal structure of the carmofur/Mpro complex disclosed the elimination of pyrimidinedione (block B) and covalent modification of Cys145, which is caused by the sulfhydryl group of Cys145 attacking the carbonyl group in block A of carmofur. In addition, they observed that the residues Ser144 and Gly143 formed two hydrogen-bonding interactions with a carbonyl group in block A and a water bridge between oxygen atom of carbonyl in block A and Thr26. the fatty carbon chain tail is inserted deeply into the S2 subsite and held by hydrophobic interactions.

a The chemical structure of carmofur; b co-crystal structures of carmofur in the active site of SARS-CoV-2 Mpro (PDB ID: 7BUY)
4.3 Inhibitors Targeting SARS-CoV Mpro
The development of homologous virus SARS-CoV Mpro inhibitors acts an important role in driving rapid discovery of SARS-CoV-2 Mpro inhibitors, given the great many highly conserved active sites of Mpro in all known coronaviruses, especially in SARS-CoV and SARS-CoV-2. Currently, the reported peptide-mimetic-based covalent inhibitors targeting SARS-CoV Mpro are typically designed with a chemical “warhead” [81, 102]. The current research status involving peptidomimetic-based SARS-CoV Mpro inhibitors can be summarized based on specifying different types of warheads. They include Michael acceptors, aldehydes, substituted ketones, keto-glutamines, epoxides and boronic acid.
As shown in Fig. 15, AG7088 (20, rupintrivir), a potent ethoxy-containing Michael acceptor-based peptidomimetic inhibitor, has already been clinically studied for use against the common cold rhinovirus 3C protease (HRV2 3Cpr°) [103]. AG7088 has been proposed as a starting point in the drug design of new SARS-CoV Mpro inhibitors [79, 104,105,106]. In 2005, Yang and co-workers identified a benzyloxy-containing compound 21 (N3, an analogue of AG7088, Fig. 15) with a Michael acceptor as a broad-spectrum anti-coronavirus inhibitor (e.g., SARS-CoV Mpro and MERS-CoV Mpro) [82, 97].

The structures of representative peptidomimetic inhibitors 20–28 incorporating various warheads (identified in pink clouds)
Yang and colleagues disclosed a novel, potent aldehyde-based SARS-CoV Mpro inhibitor 22 (TG-02005221, Fig. 15) [107]. Compound 22 displayed robust antiviral activity with a Ki value of 53 nM as well as favorable pharmacokinetic profiles in rodents. The X-ray crystal structure of SARS-CoV Mpro in complex with 22 showed that compound 22 was tightly anchored on the active site of Mpro via a covalent C–S bond, together with 10 H bonds and an extensive assemblage of hydrophobic interactions (PDB ID 2GX4).
Cai and co-workers reported a series of new SARS-CoV Mpro inhibitors that employed a fluoromethyl ketone group as a new warhead, and also replaced the widely studied (S)-γ-lactam ring in the P1 moiety with a dimethylamino oxoethyl group (corresponding to the ring opening of the lactam) [108]. The most potent compound 23 (Fig. 15) showed good cell-based antiviral efficacy (EC50 = 2.5 μM) with low toxicity in mice.
Inspired by the electron-withdrawing aspects of the fluoromethyl ketone group in 23, the effects of other types of electron-withdrawing groups (thiazolyl and benzothiazolyl) on anti-SARS-CoV Mpro replication activity were investigated by Hayashi and co-workers. They identified two lead compounds, 24 and 25 (Fig. 15), with potent antiviral efficacy [109,110,111,112]. Compound 24 brandishes a thiazolyl ketone warhead and (S)-γ-lactam at P1, showing favorable inhibitory activity against SARS-CoV Mpro (Ki = 2.2 μM, IC50 = 9.5 μM) [109]. Compound 25, bearing a benzothiazolyl ketone warhead, exhibited excellent inhibitory activity with a Ki value of 3.1 nM [110]. The co-crystal structure of SARS-CoV Mpro in complex with 24 reveals that the ketone group of 24 is converted to an oxyanion after attack by the thiol of the Cys145 residue. As a result, the arch-reason for ketone as an acceptable warhead is the formation of multiple H bonds between the generated oxyanion and three Mpro residues: Cys145, Gly143 and Ser144. In addition, the nitrogen in the thiazole plays an important role in stabilizing the binding model of 24 with SARS-CoV Mpro by forming an H bond with the His41 residue.
In 2004, as the promising effects of glutamine analogues against hepatitis A virus (HAV) became evident [113, 114], Vederas and co-workers reported a series of phthalhydrazide-based keto-glutamine analogues as novel SARS-CoV Mpro inhibitors [115]. Compound 26 (Fig. 15) was identified as the most potent anti-SARS-CoV Mpro inhibitor with an IC50 of 0.6 μM [115]. Molecular modeling studies of 26 indicated that the bulky phthalhydrazide group was accommodated at the S1′ subsite of the enzyme and formed a key H bond between Asn142 and the nitro group attached to the phthalhydrazide. In light of the novel phthalhydrazide-containing ketone-based warhead of 26, Zhang and co-workers proposed the introduction of an α-ketoamide warhead into the P1′ moiety based on de-cyclization of the phthalhydrazide group in 26 [98]. Extensive biological evaluation led to the broad-spectrum inhibitor 12 (mentioned in Fig. 11 above) that demonstrated inhibition of the activity of SARS-CoV Mpro with a low-micromolar IC50 value (0.71 μM). Moreover, further crystallographic study of 12 (PDB ID: 2BX4) demonstrated that the α-keto-carbon formed a hemithioacetal by a condensation reaction with the mercapto group of Cys145.
James et al. reported that a novel substrate-like aza-peptide inhibitor in compound 27 (Fig. 15) contained an epoxy structure with prominent activity [Kinact/Ki (SARS-CoV Mpro) = 1900 ± 400 M−1S−1] [116, 117]. The co-crystal structures of SARS-CoV Mpro in complex with 27 demonstrated the formation of a C–S covalent bond between the epoxide C-3 and sulfur atom in Cys145 (PDB ID 2A5K) [117]. Interestingly, a symmetric peptide-like inhibitor 28 (Fig. 15) [118] with a Ki value of 73 nM for SARS-CoV Mpro was disclosed based on in silico optimization of a noncovalent HIV protease inhibitor by Wong and co-workers [119].
Besides the peptidomimetic inhibitors mentioned above, small-molecule inhibitors targeting SARS-CoV Mpro have also been extensively explored. Wong and co-workers produced a series of nitroaniline derivatives, among which compound 29 (Fig. 16) was discovered and functions as a competitive, noncovalent inhibitor targeting SARS-CoV Mpro; it displays potent inhibitory efficacy (Ki and IC50 values of 0.03 and 0.06 μM, respectively) [120]. Currently, the increasing exploitation of metal-conjugated compounds as broad-spectrum antiviral agents has been widely investigated [121,122,123,124]. In one example, Hsu and co-workers discovered a mercury-conjugated inhibitor 30 (Fig. 16) that effectively inhibits the activity of SARS-CoV Mpro (Ki = 0.3 μM) by utilizing the affinity of the thiol group in Cys145 to Hg2+ ions [125]. Another example is the zinc-conjugated inhibitor 31 (Fig. 16) reported by Lee and co-workers [126]. Compound 31 displayed good antiviral activity with a Ki value of 0.05 μM by benefiting from the binding of Zn2+ ions to the His41-Cys145 catalytic dyad.

The structures of small-molecule inhibitors 29–32 against SARS-CoV Mpro (warheads are identified in pink clouds)
Bacha and co-workers [127] discovered an attractive subsite at the cluster of residues Ser147-Ser139-Ser144 that are close by the catalytic residues in SARS-CoV Mpro. As the cluster of Ser residues in all reported coronavirus Mpro is highly conserved, this cluster may provide a common target site of broad-spectrum anti-coronavirus inhibitors [128]. Since the efficacy of aryl boronic acid derivatives against serine protease was demonstrated in the 1970s, a great number of boronic acid compounds have been investigated for use as protease inhibitors that feature both site selectivity and host safety [129, 130]. The recognized mechanism of inhibition is the production of a boronate-based tetra-coordinate complex by a nucleophilic coordination process between the –OH group in serine and boronic acid derivatives [128]. In view of the above recognized mechanism, a series of bifunctional boronic acid-conjugated SARS-CoV Mpro inhibitors were evaluated by Bacha et al., ultimately leading to the potent antiviral inhibitor 32 (Fig. 16) with a Ki value of 0.04 μM [127].
Non-peptide small molecules, which usually noncovalently or reversibly interact with protein, are an important class of inhibitors against SARS-CoV Mpro. These small molecules were discovered by a fragment-screening campaign or high-throughput screening or reasonable molecular design. Generally, compared with covalent inhibitors, these inhibitors have fewer side effects and lower toxicity. Recently, Jian and colleagues [131] gave a great description of non-peptide SARS-CoV Mpro inhibitors from 2010 to 2020, involving the structural characteristics, binding modes and SARs. However, difference in binding mode of between peptide and non-peptide inhibitors that plays a key role in discovery of new Mpro inhibitors were not illustrated in detail by them. Herein, we conduct a contrastive analysis of the superposition mode of co-crystal complex to understand this difference (Fig. 17). Akaji et al. [132] described that a novel decahydroisoquinolin scaffold in compound 33 contained an aldehyde group with prominent activity against SARS-CoV Mpro (IC50 of 108 μM). According to the superimposition of N3 (a representative peptide inhibitor, Fig. 10b) and compound 33 bound to SARS-CoV Mpro, the hydrophobic interaction in the large S2 pocket and the H bonds in the S1 pocket are consistent. The aldehyde group in 33 was expected to target residue Cys145 to form a stable C–S covalent bond, which is consistent with the Michael acceptor moiety in N3. The Ala-isoxazole moiety in N3 occupied the S4 pocket and forms a hydrogen bond interaction with residue Thr190, which is not observed between 33 and the S4 pocket. Jacobs et al. [133] reported the identification of a representative non-peptidic inhibitor 34 with excellent activity against SARS-CoV Mpro (IC50 of 4.8 μM) through virtual screening approaches. As shown in Fig. 17c, the superimposition of N3 and 34 bound to SARS-CoV Mpro have confirmed that the 3-pyridyl group, the tert-butyl anilido group and tert-butyl amide group of 34 occupied the S1, S2 and S3 pockets, respectively, similar to the results obtained with the P1, P1′, P2 and P3 of peptide inhibitor N3. Compound 34 was a noncovalent SARS-CoV Mpro inhibitor different from most reported peptide inhibitors that act via covalent modification of the enzyme. Turlington et al. [134] reported the biological activity of a series of non-covalent benzotriazole derivatives from the NIH Molecular Library Probe Production Center Network (MLPCN). Compound 35 (Fig. 17a) was identified as the most potent anti-SARS-CoV Mpro inhibitor with an IC50 of 6.2 μM. As shown in Fig. 17d, for compound 35, the key H bonds between the benzotriazole moiety and S1 pocket, the hydrophobic interaction between the 4-acetamidoaniline moiety and S2 pocket, and the two H bonds formed with residue Glu166 were observed, which was also observed for compound N3. Overall, peptide inhibitors aim to achieve irreversible inhibition effects by the formation of a C–S bond between electrophilic warhead groups and residue Cys145. Non-peptide small molecules usually utilize a noncovalent mechanism of action to accomplish efficient inhibition effects involving H bonds, hydrophobic interaction and van der Waals forces.

a The chemical structures of representative non-peptide SARS-CoV Mpro inhibitors; b superposition of co-crystal structures of N3 (yellow, PDB ID: 2HOB) and compound 33 (violet, PDB ID: 4TWY) with SARS-CoV Mpro; c superposition of co-crystal structures of N3 (yellow) and compound 34 (cyan, PDB ID: 3V3M) with SARS-CoV Mpro; d superposition of co-crystal structures of N3 (yellow) and compound 35 (pale green, PDB ID: 4MDS) with SARS-CoV Mpro. The H bonds are colored in wheat or green
5 Conclusion
The COVID-19 pandemic has dealt a painful blow to the safety of human life and the economic development of the world. A novel coronavirus, SARS-CoV-2, has been identified as the causative agent of the disease. Although extensive investigations are being carried out to fight SARS-CoV-2 infection, there are no specific antiviral agents available to manage this epidemic at present. Consequently, drug development against SARS-CoV-2 is an extremely urgent and challenging goal for which the understanding of the structural biology of SARS-CoV-2 and intimate knowledge of molecular structures involved in implementing infection and replication mechanisms are of crucial importance. It has been confirmed that the virus takes advantage of the host proteins TMPRSS2 and ACE2 to invade the human body, and RdRp and Mpro are vital in adjusting coronavirus replication and transcription of the viral life cycle. In this perspective, we mainly focused on the structural biology and medicinal chemistry of key proteins in the life cycle of SARS-CoV-2. Some of the successful and candidate inhibitors that have raised interest because of their effectiveness on homologous viruses in the coronavirus family are also summarized in this report. The protracted time needed for new drug development makes it highly desirable to repurpose existing antiviral agents that have been approved for clinical use against homologous viruses whenever they evidence effectiveness against SARS-CoV-2. Because many antiviral targets show highly conserved sequences among viruses in the coronavirus family, this underscores the reasonable possibility of rapid identification of both existing drugs and modifications to existing drugs that will enable effective control over the spread of COVID-19. Evaluation of these begins with an understanding of the modes of action of some representative broad-spectrum antiviral drugs, especially those against the homologous virus SARS-CoV. These are summarized in this report with the hope of driving the development of broad-spectrum inhibitors against SARS-CoV-2. In conclusion, we believe that this review will be useful to aid the research community in the discovery of novel and potent anti-SARS-CoV-2 inhibitors for distribution to clinics in the near future.
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- COVID-19:
-
Coronavirus disease 2019
- CLD:
-
Collectrin-like domain
- EC50 :
-
Half-maximal effective concentration
- IC50 :
-
Half-maximal inhibitory concentration
- Mpro :
-
Main protease
- NAA:
-
Neutral amino acid
- NTD:
-
N-terminal domain
- PLpro :
-
Papain-like protease
- PNCC:
-
Partial negative charge cluster
- RBD:
-
Receptor-binding domain
- RBM:
-
Receptor-binding motif
- RdRp:
-
RNA-dependent RNA polymerase
- RMSD:
-
Root mean square difference
- RTP:
-
Remdesivir triphosphate
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- TMPRSS2:
-
Transmembrane protease serines 2
References
https://www.who.int/emergencies/diseases/novel-coronavirus-2019
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506
Liu C, Yang Y, Gao YZ, Shen CG, Ju B, Liu CC, Tang X, Wei JL, Ma XM, Liu WL, Xu SM, Liu YX, Yuan J, Wu J, Liu Z, Zhang Z, Wang PY, Liu L (2020) Viral architecture of SARS-CoV-2 with post-fusion spike revealed by cryo-EM. BioRxiv. https://doi.org/10.1101/2020.1103.1102.972927
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292
Rabi FA, Al Zoubi MS, Kasasbeh GA, Salameh DM, Al-Nasser AD (2020) SARS-CoV-2 and coronavirus disease 2019: what we know so far. Pathogens 9:231
Oberfeld B, Achanta A, Carpenter K, Chen P, Gilette NM, Langat P, Said JT, Schiff AE, Zhou AS, Barczak AK, Pillai S (2020) SnapShot: COVID-19. Cell 181:954–954
Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66:193–292
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423
Wang QH, Zhang YF, Wu LL, Niu S, Song CL, Zhang ZY, Lu GW, Qiao CP, Hu Y, Yuen KY, Wang QS, Zhou H, Yan JH, Qi JX (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181:894–904
Tortorici MA, Veesler D (2019) Structural insights into coronavirus entry. Adv Virus Res 105:93–116
Tortorici MA, Walls AC, Lang Y, Wang C, Li Z, Koerhuis D, Boons GJ, Bosch BJ, Rey FA, de Groot RJ, Veesler D (2019) Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct Mol Biol 26:481–489
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454
Li F, Li WH, Farzan M, Harrison SC (2005) Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309:1864–1868
Li WH, Zhang CS, Sui JH, Kuhn JH, Moore MJ, Luo SW, Wong SK, Huang IC, Xu KM, Vasilieva N, Murakami A, He YQ, Marasco WA, Guan Y, Choe HY, Farzan M (2005) Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24:1634–1643
Wrapp D, Wang NS, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260–1263
Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811
Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3:237–261
Hoffmann M, Kleine-Weber H, Schroeder S, Krueger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Mueller MA, Drosten C, Poehlmann S (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280
Lv Z, Deng YQ, Ye Q, Cao L, Sun CY, Fan C, Huang W, Sun S, Sun Y, Zhu L, Chen Q, Wang N, Nie J, Cui Z, Zhu D, Shaw N, Li XF, Li Q, Xie L, Wang Y, Rao Z, Qin CF, Wang X (2020) Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science 369:1505–1509
Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, Qi F, Bao L, Du L, Liu S, Qin C, Sun F, Shi Z, Zhu Y, Jiang S, Lu L (2020) Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 30:343–355
Gao Q, Bao L, Mao H, Wang L, Xu K, Yang M, Li Y, Zhu L, Wang N, Lv Z, Gao H, Ge X, Kan B, Hu Y, Liu J, Cai F, Jiang D, Yin Y, Qin C, Li J, Gong X, Lou X, Shi W, Wu D, Zhang H, Zhu L, Deng W, Li Y, Lu J, Li C, Wang X, Yin W, Zhang Y, Qin C (2020) Development of an inactivated vaccine candidate for SARS-CoV-2. Science 369:77–81
Bosch BJ, Bartelink W, Rottier PJM (2008) Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol 82:8887–8890
Belouzard S, Chu VC, Whittaker GR (2009) Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 106:5871–5876
Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ, Gamblin SJ (2020) SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol 27:763–767
Wu C, Zheng S, Chen Y, Zheng M (2020) Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCoV, in the nasal tissue. medRxiv. https://doi.org/10.1101/2020.1102.1111.20022228
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87:E1–E9
Zhang H, Kang ZJ, Gong HY, Xu D, Wang J, Li ZF, Cui XG, Xiao JR, Meng T, Zhou W, Liu JM, Xu HJ (2020) The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes. bioRxiv. https://doi.org/10.1101/2020.1101.1130.927806
Zhang H, Wada J, Hida K, Tsuchiyama Y, Hiragushi K, Shikata K, Wang H, Lin S, Kanwar YS, Makino H (2001) Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem 276:17132–17139
Tian XL, Li C, Huang AL, Xia S, Lu SC, Shi ZL, Lu L, Jiang SB, Yang ZL, Wu YL, Ying TL (2020) Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microb Infect 9:382–385
Kowalczuk S, Bröer A, Tietze N, Vanslambrouck JM, Rasko JEJ, Bröer S (2008) A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J 22:2880–2887
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367:1444–1448
Lan J, Ge JW, Yu JF, Shan SS, Zhou H, Fan SL, Zhang Q, Shi XL, Wang QS, Zhang LQ, Wang XQ (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581:221–224
Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R Jr, Nunneley JW, Barnard D, Poehlmann S, McKerrow JH, Renslo AR, Simmons G (2015) Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 116:76–84
Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Poehlmann S, Soilleux EJ (2012) Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 7:e38567
Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R (2020) Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci 11:1555–1562
Shi L, Xiong H, He J, Deng H, Li Q, Zhong Q, Hou W, Cheng L, Xiao H, Yang Z (2007) Antiviral activity of arbidol against influenza A virus, respiratory syncytial virus, rhinovirus, coxsackie virus and adenovirus in vitro and in vivo. Arch Virol 152:1447–1455
Zhong Q, Yang Z, Liu Y, Deng H, Xiao H, Shi L, He J (2009) Antiviral activity of Arbidol against Coxsackie virus B5 in vitro and in vivo. Arch Virol 154:601–607
Teissier E, Zandomeneghi G, Loquet A, Lavillette D, Lavergne JP, Montserret R, Cosset FL, Boeckmann A, Meier BH, Penin F, Pecheur EI (2011) Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS ONE 6:e15874
Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S (2012) Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol 86:6537–6545
Ohkoshi M, Oka T (1984) Clinical experience with a protease inhibitor N,N-dimethylcarbamoylmethyl 4-(4-guanidinobenzoyloxy)-phenylacetate methanesulfate for prevention of recurrence of carcinoma of the mouth and in treatment of terminal carcinoma. J Maxillofac Surg 12:148–152
Sai JK, Suyama M, Kubokawa Y, Matsumura Y, Inami K, Watanabe S (2010) Efficacy of camostat mesilate against dyspepsia associated with non-alcoholic mild pancreatic disease. J Gastroenterol 45:335–341
Hiraishi M, Yamazaki Z, Ichikawa K, Kanai F, Idezuki Y, Onishi K, Takahama T, Inoue N (1988) Plasma collection using nafamostat mesilate and dipyridamole as an anticoagulant. Int J Artif Organs 11:212–216
Hirota M, Shimosegawa T, Kitamura K, Takeda K, Takeyama Y, Mayumi T, Ito T, Takenaka M, Iwasaki E, Sawano H, Ishida E, Miura S, Masamune A, Nakai Y, Mitoro A, Maguchi H, Kimura K, Sanuki T, Ito T, Haradome H, Kozaka K, Gabata T, Kataoka K, Hirota M, Isaji S, Nakamura R, Yamagiwa K, Kayaba C, Ikeda K (2020) Continuous regional arterial infusion versus intravenous administration of the protease inhibitor nafamostat mesilate for predicted severe acute pancreatitis: a multicenter, randomized, open-label, phase 2 trial. J Gastroenterol 55:342–352
Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue J-i, Matsuda Z (2016) Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus s protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 60:6532–6539
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271
Said A, Bock S, Lajqi T, Mueller G, Weindl G (2014) Chloroquine promotes IL-17 production by CD4(+) T cells via p38-dependent IL-23 release by monocyte-derived Langerhans-like cells. J Immunol 193:6135–6143
Colson P, Rolain JM, Lagier JC, Brouqui P, Raoult D (2020) Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int J Antimicrob Agents 55:105932
Gao J, Tian Z, Yang X (2020) Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends 14:72–73
Principi N, Esposito S (2020) Chloroquine or hydroxychloroquine for prophylaxis of COVID-19. Lancet Infect Dis 20:1118–1118
White NJ, Watson JA, Hoglund RM, Chan XHS, Cheah PY, Tarning J (2020) COVID-19 prevention and treatment: a critical analysis of chloroquine and hydroxychloroquine clinical pharmacology. PLoS Med 17:e1003252
DeJong C, Wachter RM (2020) The risks of prescribing hydroxychloroquine for treatment of COVID-19—first, do no harm. JAMA Intern Med 180:1118
Gerard A, Romani S, Fresse A, Viard D, Parassol N, Granvuillemin A, Chouchana L, Rocher F, Drici M-D, French Network Pharmacovigilance C (2020) “Off-label” use of hydroxychloroquine, azithromycin, lopinavir-ritonavir and chloroquine in COVID-19: a survey of cardiac adverse drug reactions by the French Network of Pharmacovigilance Centers. Therapie 75:371–379
Lung J, Lin YS, Yang YH, Chou YL, Shu LH, Cheng YC, Liu HT, Wu CY (2020) The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J Med Virol 92:693–697
Shannon A, Le NTT, Selisko B, Eydoux C, Alvarez K, Guillemot JC, Decroly E, Peersen O, Ferron F, Canard B (2020) Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 exonuclease active-sites. Antiviral Res 178:104793–104793
Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Hu T, Hua T, Zhang B, Yang X, Li J, Yang H, Liu Z, Xu W, Guddat LW, Wang Q, Lou Z, Rao Z (2020) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368:779–782
Yin WC, Mao CY, Luan XD, Shen DD, Shen QY, Su HX, Wang XX, Zhou FL, Zhao WF, Gao MQ, Chang SH, Xie YC, Tian GH, Jiang HW, Tao SC, Shen JS, Jiang Y, Jiang HL, Xu YC, Zhang SY, Zhang Y, Xu HE (2020) Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368:1499–1504
Peng Q, Peng R, Yuan B, Zhao J, Wang M, Wang X, Wang Q, Sun Y, Fan Z, Qi J, Gao GF, Shi Y (2020) Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep 31:107774
Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P (2020) Structure of replicating SARS-CoV-2 polymerase. Nature 584:154–156
Yan L, Zhang Y, Ge J, Zheng L, Gao Y, Wang T, Jia Z, Wang H, Huang Y, Li M, Wang Q, Rao Z, Lou Z (2020) Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat Commun 11:5874
Jean SS, Lee PI, Hsueh PR (2020) Treatment options for COVID-19: the reality and challenges. J Microbiol Immunol Infect 53:436–443
Li G, De Clercq E (2020) Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 19:149–150
Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, Smith EC, Case JB, Feng JY, Jordan R (2018) Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 9:e00221-e218
Warren TK, Wells J, Panchal RG, Stuthman KS, Garza NL, Van Tongeren SA, Dong L, Retterer CJ, Eaton BP, Pegoraro G, Honnold S, Bantia S, Kotian P, Chen X, Taubenheim BR, Welch LS, Minning DM, Babu YS, Sheridan WP, Bavari S (2014) Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508:402–405
Savic D, Stojiljkovic M, Lavrnja I, Parabucki A, Bjelobaba I, Nedeljkovic N, Herdegen T, Pekovic S (2014) Ribavirin shows immunomodulatory effects on activated microglia. Immunopharmacol Immunotoxicol 36:433–441
Chan JFW, Yao Y, Yeung ML, Deng W, Bao L, Jia L, Li F, Xiao C, Gao H, Yu P, Cai JP, Chu H, Zhou J, Chen H, Qin C, Yuen KY (2015) Treatment with lopinavir/ritonavir or interferon-beta 1b improves outcome of MERS-CoV infection in a nonhuman primate model of common marmoset. J Infect Dis 212:1904–1913
Refaat B, El-Shemi AG, Ashshi AM (2015) The effects of pegylated interferon-α and ribavirin on liver and serum concentrations of activin-A and follistatin in normal Wistar rat: a preliminary report. BMC Res Notes 8:265
Abdel-Hamid NM, Wahid A, Nazmy MH, Eisa MAM (2016) Synergistic effects of Jerusalem artichoke in combination with pegylated interferon Alfa-2a and ribavirin against hepatic fibrosis in rats. Asian Pac J Cancer Prev 17:1979–1985
Toots M, Yoon JJ, Cox RM, Hart M, Sticher ZM, Makhsous N, Plesker R, Barrena AH, Reddy PG, Mitchell DG, Shean RC, Bluemling GR, Kolykhalov AA, Greninger AL, Natchus MG, Painter GR, Plemper RK (2019) Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci Transl Med 11:eaax5866
Sheahan TP, Sims AC, Zhou S, Graham RL, Pruijssers AJ, Agostini ML, Leist SR, Schafer A, Dinnon KH III, Stevens LJ, Chappell JD, Lu X, Hughes TM, George AS, Hill CS, Montgomery SA, Brown AJ, Bluemling GR, Natchus MG, Saindane M, Kolykhalov AA, Painter G, Harcourt J, Tamin A, Thornburg NJ, Swanstrom R, Denison MR, Baric RS (2020) An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med 12:5883
Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL (2013) Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100:446–454
Rocha-Pereira J, Jochmans D, Dallmeier K, Leyssen P, Nascimento MSJ, Neyts J (2012) Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem Biophys Res Commun 424:777–780
Murakami E, Tolstykh T, Bao H, Niu C, Steuer HMM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA (2010) Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem 285:34337–34347
Xu HT, Colby-Germinario SP, Hassounah SA, Fogarty C, Osman N, Palanisamy N, Han Y, Oliveira M, Quan Y, Wainberg MA (2017) Evaluation of sofosbuvir (beta-D-2’-deoxy-2’-alpha-fluoro-2’-beta-C-methyluridine) as an inhibitor of Dengue virus replication. Sci Rep 7:6345
Elfiky AA (2020) Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci 253:117592–117592
Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, Meng J, Zhu Z, Zhang Z, Wang J, Sheng J, Quan L, Xia Z, Tan W, Cheng G, Jiang T (2020) Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27:325–328
Song F, Shi N, Shan F, Zhang Z, Shen J, Lu H, Ling Y, Jiang Y, Shi Y (2020) Emerging 2019 novel coronavirus (2019-nCoV) pneumonia. Radiology 295:210–217
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (2003) Coronavirus main proteinase (3CL(pro)) structure: basis for design of anti-SARS drugs. Science 300:1763–1767
Hilgenfeld R (2014) From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J 281:4085–4096
Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH (2016) An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 59:6595–6628
Jin ZM, Du XY, Xu YC, Deng YQ, Liu MQ, Zhao Y, Zhang B, Li XF, Zhang LK, Peng C, Duan YK, Yu J, Wang L, Yang KL, Liu FJ, Jiang RD, Yang XL, You T, Liu X, Yang XN, Bai F, Liu H, Liu X, Guddat LW, Xu WQ, Xiao GF, Qin CF, Shi ZL, Jiang HL, Rao ZH, Yang HT (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293
Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD (2006) Severe acute respiratory syndrome coronavirus papain-like protease: tructure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA 103:5717–5722
Wojdyla JA, Manolaridis I, van Kasteren PB, Kikkert M, Snijder EJ, Gorbalenya AE, Tucker PA (2010) Papain-like protease 1 from transmissible gastroenteritis virus: crystal structure and enzymatic activity toward viral and cellular substrates. J Virol 84:10063–10073
Needle D, Lountos GT, Waugh DS (2015) Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr Sect D Struct Biol 71:1102–1111
St John SE, Tomar S, Stauffer SR, Mesecar AD (2015) Targeting zoonotic viruses: structure-based inhibition of the 3C-like protease from bat coronavirus HKU4-The likely reservoir host to the human coronavirus that causes Middle East respiratory syndrome (MERS). Biorg Med Chem 23:6036–6048
Tomar S, Johnston ML, John SES, Osswald HL, Nyalapatla PR, Paul LN, Ghosh AK, Denison MR, Mesecar AD (2015) Ligand-induced dimerization of Middle East respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals. J Biol Chem 290:19403–19422
Wang H, He S, Deng W, Zhang Y, Li G, Sun J, Zhao W, Guo Y, Yin Z, Li D, Shang L (2020) Comprehensive insights into the catalytic mechanism of Middle East respiratory syndrome 3C-like protease and severe acute respiratory syndrome 3C-like protease. ACS Catal 10:5871–5890
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368:409–412
Lu IL, Mahindroo N, Liang PH, Peng YH, Kuo CJ, Tsai KC, Hsieh HP, Chao YS, Wu SY (2006) Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J Med Chem 49:5154–5161
Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R (2002) Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J 21:3213–3224
Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci 100:13190–13195
Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z (2008) Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol 82:2515–2527
Wang F, Chen C, Tan W, Yang K, Yang H (2016) Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Sci Rep 6:22677
Tan J, Verschueren KHG, Anand K, Shen J, Yang M, Xu Y, Rao Z, Bigalke J, Heisen B, Mesters JR (2005) pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 354:40
Huang C, Wei P, Fan K, Liu Y, Lai L (2004) 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 43:4568–4574
Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Qi Z, Zhou Z, Pei D, Ziebuhr J, Hilgenfeld R, Ky Y, Wong L, Gao G, Chen S, Chen Z, Ma D, Bartlam M, Rao Z (2005) Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3:1742–1752
Zhang LL, Lin DZ, Kusov YR, Nian Y, Ma QJ, Wang J, von Brunn A, Leyssen P, Lanko K, Neyts J, de Wilde A, Snijder EJ, Liu H, Hilgenfeld R (2020) α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem 63:4562–4578
Dai WH, Zhang B, Su HX, Li J, Zhao Y, Xie X, Jin ZM, Liu FJ, Li CP, Li Y, Bai F, Wang HF, Cheng X, Cen XB, Hu SL, Yang XN, Wang J, Liu X, Xiao GF, Jiang HL, Rao ZH, Zhang LK, Xu YC, Yang HT, Liu H (2020) Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368:1331–1335
Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, Song H, Shi Y, Li X, Tan W, Qi J, Gao GF (2020) Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun 11:1
Jin Z, Zhao Y, Sun Y, Zhang B, Wang H, Wu Y, Zhu Y, Zhu C, Hu T, Du X, Duan Y, Yu J, Yang X, Yang X, Yang K, Liu X, Guddat LW, Xiao G, Zhang L, Yang H, Rao Z (2020) Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol 27:529–532
Ghosh AK, Xi K, Johnson ME, Baker SC, Mesecar AD (2006) Progress in anti-SARS coronavirus chemistry, biology and chemotherapy. Annu Rep Med Chem 41:183–196
Matthews DA, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, Hendrickson TF, Love RA, Prins TJ, Marakovits JT, Zhou R, Tikhe J, Ford CE, Meador JW, Ferre RA, Brown EL, Binford SL, Brothers MA, DeLisle DM, Worland ST (1999) Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci USA 96:11000–11007
Jenwitheesuk E, Samudrala R (2003) Identifying inhibitors of the SARS coronavirus proteinase. Bioorg Med Chem Lett 13:3989–3992
Ghosh AK, Xi K, Ratia K, Santarsiero BD, Fu WT, Harcourt BH, Rota PA, Baker SC, Johnson ME, Mesecar AD (2005) Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J Med Chem 48:6767–6771
Mukherjee P, Desai P, Ross L, White EL, Avery MA (2008) Structure-based virtual screening against SARS-3CLpro to identify novel non-peptidic hits. Biorg Med Chem 16:4138–4149
Yang S, Chen SJ, Hsu MF, Wu JD, Tseng CTK, Liu YF, Chen HC, Kuo CW, Wu CS, Chang LW, Chen WC, Liao SY, Chang TY, Hung HH, Shr HL, Liu CY, Huang YA, Chang LY, Hsu JC, Peters CJ, Wang AHJ, Hsu MC (2006) Synthesis, crystal structure, structure−activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J Med Chem 49:4971–4980
Zhang HZ, Zhang H, Kemnitzer W, Tseng B, Cinatl J, Michaelis M, Doerr HW, Cai SX (2006) Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronovirus (SARS-CoV) inhibitors. J Med Chem 49:1198–1201
Regnier T, Sarma D, Hidaka K, Bacha U, Freire E, Hayashi Y, Kiso Y (2009) New developments for the design, synthesis and biological evaluation of potent SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett 19:2722–2727
Konno S, Thanigaimalai P, Yamamoto T, Nakada K, Kakiuchi R, Takayama K, Yamazaki Y, Yakushiji F, Akaji K, Kiso Y, Kawasaki Y, Chen SE, Freire E, Hayashi Y (2013) Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Biorg Med Chem 21:412–424
Thanigaimalai P, Konno S, Yamamoto T, Koiwai Y, Taguchi A, Takayama K, Yakushiji F, Akaji K, Chen SE, Naser-Tavakolian A, Schön A, Freire E, Hayashi Y (2013) Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur J Med Chem 68:372–384
Thanigaimalai P, Konno S, Yamamoto T, Koiwai Y, Taguchi A, Takayama K, Yakushiji F, Akaji K, Kiso Y, Kawasaki Y, Chen SE, Naser-Tavakolian A, Schön A, Freire E, Hayashi Y (2013) Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: structure–activity relationship study. Eur J Med Chem 65:436–447
Ramtohul YK, James MNG, Vederas JC (2002) Synthesis and evaluation of keto-glutamine analogues as inhibitors of hepatitis A virus 3C proteinase. J Org Chem 67:3169–3178
Jain RP, Vederas JC (2004) Structural variations in keto-glutamines for improved inhibition against hepatitis A virus 3C proteinase. Bioorg Med Chem Lett 14:3655–3658
Jain RP, Pettersson HI, Zhang J, Aull KD, Fortin PD, Huitema C, Eltis LD, Parrish JC, James MNG, Wishart DS, Vederas JC (2004) Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J Med Chem 47:6113–6116
Asgian JL, James KE, Li ZZ, Carter W, Barrett AJ, Mikolajczyk J, Salvesen GS, Powers JC (2002) Aza-peptide epoxides: a new class of inhibitors selective for clan CD cysteine proteases. J Med Chem 45:4958–4960
Lee TW, Cherney MM, Huitema C, Liu J, James KE, Powers JC, Eltis LD, James MNG (2005) Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J Mol Biol 353:1137–1151
Shao YM, Yang WB, Peng HP, Hsu MF, Tsai KC, Kuo TH, Wang AHJ, Liang PH, Lin CH, Yang A-S, Wong CH (2007) Structure-based design and synthesis of highly potent SARS-CoV 3CL protease inhibitors. ChemBioChem 8:1654–1657
Lee T, Laco GS, Torbett BE, Fox HS, Lerner DL, Elder JH, Wong CH (1998) Analysis of the S3 and S3’ subsite specificities of feline immunodeficiency virus (FIV) protease: development of a broad-based protease inhibitor efficacious against FIV, SIV and HIV in vitro and ex vivo. Proc Natl Acad Sci U S A 95:939–944
Shie JJ, Fang JM, Kuo CJ, Kuo TH, Liang PH, Huang HJ, Yang WB, Lin CH, Chen JL, Wu YT, Wong CH (2005) Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J Med Chem 48:4469–4473
Tong L, Qian C, Massariol MJ, Bonneau PR, Cordingley MG, Lagace L (1996) A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature 383:272–275
Dhanak D, Burton G, Christmann LT, Darcy MG, Elrod KC, Kaura A, Keenan RM, Link JO, Peishoff CE, Shah DH (2000) Metal mediated protease inhibition: design and synthesis of inhibitors of the human cytomegalovirus (hCMV) protease. Bioorg Med Chem Lett 10:2279–2282
Yeung KS, Meanwell NA, Qiu Z, Hernandez D, Zhang S, McPhee F, Weinheimer S, Clark JM, Janc JW (2001) Structure-activity relationship studies of a bisbenzimidazole-based, Zn2+-dependent inhibitor of HCV NS3 serine protease. Bioorg Med Chem Lett 11:2355–2359
Han YS, Chang GG, Juo CG, Lee HJ, Yeh SH, Hsu JTA, Chen X (2005) Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry 44:10349–10359
Hsu JTA, Kuo CJ, Hsieh HP, Wang YC, Huang KK, Lin CPC, Huang PF, Chen X, Liang PH (2004) Evaluation of metal-conjugated compounds as inhibitors of 3CL protease of SARS-CoV. FEBS Lett 574:116–120
Lee CC, Kuo CJ, Hsu MF, Liang PH, Fang JM, Shie JJ, Wang AHJ (2007) Structural basis of mercury- and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett 581:5454–5458
Bacha U, Barrila J, Velazquez-Campoy A, Leavitt SA, Freire E (2004) Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 43:4906–4912
Smoum R, Rubinstein A, Dembitsky VM, Srebnik M (2012) Boron containing compounds as protease inhibitors. Chem Rev 112:4156–4220
Knott K, Fishovitz J, Thorpe SB, Lee I, Santos WL (2010) N-Terminal peptidic boronic acids selectively inhibit human ClpXP. Org Biomol Chem 8:3451–3456
Benderdour M, Bui-Van T, Dicko A, Belleville F (1998) In vivo and in vitro effects of boron and boronated compounds. J Trace Elem Med Biol 12:2–7
Liu Y, Liang C, Xin L, Ren X, Tian L, Ju X, Li H, Wang Y, Zhao Q, Liu H, Cao W, Xie X, Zhang D, Wang Y, Jian Y (2020) The development of coronavirus 3C-like protease (3CLpro) inhibitors from 2010 to 2020. Eur J Med Chem 206:112711
Shimamoto Y, Hattori Y, Kobayashi K, Teruya K, Sanjoh A, Nakagawa A, Yamashita E, Akaji K (2015) Fused-ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorg Med Chem 23:876–890
Jacobs J, Grum-Tokars V, Zhou Y, Turlington M, Saldanha SA, Chase P, Eggler A, Dawson ES, Baez-Santos YM, Tomar S, Mielech AM, Baker SC, Lindsley CW, Hodder P, Mesecar A, Stauffer SR (2013) Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J Med Chem 56:534–546
Turlington M, Chun A, Tomar S, Eggler A, Grum-Tokars V, Jacobs J, Daniels JS, Dawson E, Saldanha A, Chase P, Baez-Santos YM, Lindsley CW, Hodder P, Mesecar AD, Stauffer SR (2013) Discovery of N-(benzo 1,2,3 triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg Med Chem Lett 23:6172–6177
Acknowledgements
This research was supported in part by the National Key R&D Program (2017YFD0200501), the China Postdoctoral Science Foundation (2020M672390), the Key Research and Development Program of Hubei Province, China (2020BCB042), and the Science and Technology Program of Wuhan (no. 2019020701011460).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhuo, LS., Wang, MS., Yang, JF. et al. Insights into SARS-CoV-2: Medicinal Chemistry Approaches to Combat Its Structural and Functional Biology. Top Curr Chem (Z) 379, 23 (2021). https://doi.org/10.1007/s41061-021-00335-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-021-00335-9