
Abstract
Self-organization in π-conjugated polymers gives rise to a highly ordered lamellar structure, in which inter-chain stacking spontaneously forms two-dimensional conjugated sheets. This multi-layer stacked nature of semicrystalline polymers allows the inclusion of various functional molecules. In particular, redox-triggered ion-intercalation is an ideal system for molecular doping, for which extremely high charge carrier density has been achieved. Here, we conducted a detailed structural analysis and electron density simulation to pinpoint exactly where the guest dopants are located periodically in the void space in a polymer’s lamellae. Our findings are indicative of an intercalation compound of layered polymers and a guest intercalant. We show that a homogeneous cocrystal structure can be realized throughout the host polymer medium, which is proved by the observation of coherent carrier transport. The intercalation cocrystal nature gives the best achievable doping level in semicrystalline polymers and excellent environmental stability. These findings should open up possibilities for tuning the collective dynamics of functional molecules through intercalation phenomena.
Similar content being viewed by others
Introduction
Doping of semiconductor materials is an essential technology common to any electronic devices, by which the charge carrier density can be tuned by several orders of magnitude, allowing precise control of the Fermi energy in electronic systems1,2. For the inorganic semiconductor Si, for example, impurity doping is achieved by substituting Si atoms in a periodic crystal with an electron-accepting or electron-donating element. The original, periodic crystal structure of the Si can be maintained even after the implantation of impurity dopants2,3. In organic semiconductors (OSCs), on the other hand, the fundamental doping process can be very different. This is because the doping agents for OSCs are not atoms, but are molecules having a unique size and conformation. Molecular doping via redox reactions between the host OSC and a guest dopant has been studied extensively in material sciences, chemistry, and physical engineering4,5,6,7,8,9,10,11,12. It remains a challenge to achieve extremely high doping levels, though heavily doped polymers have been produced via self-compensation by covalently bounded counter-ions13,14,15 and photo-doping16.
Recently, relatively high doping levels (nearly one charge per monomer unit of a π-conjugated polymer, i.e., a half-filled state) have been achieved via solid state diffusion17,18,19,20 or anion exchange21,22, where molecular dopants are introduced sequentially in the pre-deposited thin film of semicrystalline polymers. To achieve extremely high doping levels via molecular engineering, the molecular dopant must be inserted selectively into void spaces in semicrystalline π-conjugated polymers17,18,19,20,23,24,25, where alkyl side-chains would normally be located, i.e., between the polymer’s lamellae. Hence, the inclusion of dopant molecules is severely limited, not only by the original crystal structure of OSCs, but also by the structural admixture between the host OSC and guest dopants7,8,9,10. Although this may impose an apparent limitation on the maximum achievable doping level in OSCs, it is controversial how a “soft lattice” in semicrystalline polymers can be susceptible to dopant molecules, and to what extent the delicate balance in the structural admixture can be maintained.
In this study, we consider redox active radical salts as an acceptor dopant. A standard polythiophene-based, semicrystalline polymer can be doped efficiently with radical salts, resulting in remarkably high doping levels. Importantly, we perform detailed structural analysis and electron density simulations and show that the position of a guest anion inserted into a polymer’s lamellae is confined spatially in the restricted void in each lamella; in other words, layered π-conjugated polymers and intercalant anions can cooperatively build a supramolecular cocrystal. We demonstrate the best possible doping level for the closest packing structure, which is estimated to be exactly one-electron per monomer unit. This finding will help us understand coherent carrier transport in this system, and enable the design of interaction phenomena between the host and guest through molecular engineering.
Results and discussion
Chemical doping with radical salts
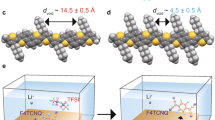
We focus on donor–acceptor systems composed of a p-type polymeric semiconductor, poly[2,5-bis(3-tetradecylthiophen-2-yl)thieno[3,2-b]thiophene] (PBTTT), and functional dopants. PBTTT is a widely studied, semicrystalline polymer, which spontaneously develops a two-dimensional lamellar structure from a solution process (Fig. 1a)26,27,28,29. To accomplish p-type molecular doping, a solid-state thin film of PBTTT is immersed in a dopant-dissolved solution (see detailed doping methods in Supplementary Note 1). Here, four types of acceptor dopants are investigated: 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ), molybdenum tris(1-(trifluoroacetyl)-2-(trifluoromethyl)ethane-1,2-dithiolene) (Mo(tfd-COCF3)3)30, Li-bis(trifluoromethylsulfonyl)imide (Li-TFSI via anion exchange route), and tris(4-bromophenyl)ammoniumyl bis(trifluoromethylsulfonyl)imide (TBPA-TFSI). Molecular structures and electrostatic potential maps are shown in Fig. 1b, and the energy alignment of the redox potentials is shown in Fig. 1c. TBPA•+ is a family of nitrogen centered radical cations, which has been investigated as strong oxidants that can externally generate radical cations31,32. Among them, tris(4-bromophenyl)ammoniumyl hexachloridoantimonate (TBPA-SbCl6), termed “magic blue”, is recognized by chemists for having a strong oxidation ability and simple electron transfer reactions. In general, the oxidation ability and stability of TBPA radical salts can be readily tuned by aryl substituents and counter-anions32. We selected the TFSI anion for its size and hydrophobicity, which brings high stability, and for the good electrical performance of the doped polymer, which was revealed in our previous anion exchange doping study21.

a Illustration of chemical doping of a PBTTT thin film. b Chemical structures of employed dopants and (c) their reported redox potentials accompanied with the approximate valence band top of PBTTT. d UV-Vis-NIR spectra, e conductivities, and (f) photoelectron yield spectroscopy measurements for pristine (black), F4TCNQ (blue), Mo(tfd-COCF3)3 (green), Li-TFSI via anion-exchange (orange), and TBPA-TFSI (red) doped PBTTT thin films. The error bars in the conductivities represent the uncertainty in the thickness of the PBTTT thin films and represent one standard deviation.
Ultraviolet-visible near-infrared (UV-vis-NIR) spectroscopy confirms that a neutral absorption of PBTTT centered at 553 nm is bleached and a broad absorption in NIR region emerges (Fig. 1d), which is consistent with p-type doping of the polymer via redox reaction with acceptor dopants17,18,19. By comparing the conventional dopants F4TCNQ and Mo(tfd-COCF3)3, bleaching of neutral absorption and an increase in the broad absorption in the NIR region occur for anion-exchange doping and TBPA-TFSI doping, suggesting that a higher doping level can be realized for anion-exchange and TBPA-TFSI doping. Particularly, in the doping process with a TBPA radical salt, the TBPA radical cation is responsible for an initial one-electron transfer, mainly due to the half-cell reaction: TBPA•+ + e− → TBPA. This converts the TBPA radical cation to a neutral state, while leaving the TFSI− as a counter-anion to guarantee charge neutrality with respect to the positively charged polymer. Note that the residual, neutral TBPA, is not present on the surface of PBTTT thin films because it dissolves well in acetonitrile.
The doping level of PBTTT thin films was further evaluated by electrical conductivity measurements and photoelectron yield spectroscopy (PYS) (see detailed information in Supplementary Note 2). The conductivity of a PBTTT thin film doped by anion exchange and with TBPA-TFSI increased significantly by a factor of ~3 compared with doping solely with F4TCNQ or Mo(tfd-COCF3)3 (Fig. 1e). In addition, PYS measurements allow us to qualitatively monitor the degree of doping; the ionization potential that corresponds to the work function (Φeff) was evaluated from the energy threshold of the photoemission yield, γ (Fig. 1f, and Supplementary Note 2). The Φeff for a pristine PBTTT thin film was measured to be 4.8 eV, which is close to the literature value of the highest occupied molecular orbital (HOMO)26,27,28,29. A positive shift in the Φeff for the doped films is clearly observed as the doping level increases, and the largest shift is observed for PBTTT doped with TBPA-TFSI. These shifts agree with p-type doping of PBTTT, where electrons are removed from the HOMO band21. The Φeff values are evaluated to be 5.2 eV for F4TCNQ doping, 5.4 eV for Mo(tfd-COCF3)3 doping, 5.4 eV for anion-exchange doping, and 5.6 eV for TBPA-TFSI doping, which roughly agree with the estimated redox potentials of the dopants (Fig. 1c)10,30. We emphasize that a Φeff of 5.6 eV is even larger than that achieved for anion exchange doping21. Although changes in conductivity values likely correlate well with the shift of Φeff, this correlation is not quantitatively understood. Assuming the rigid band approximation, i.e., the band calculation based on the neutral state of PBTTT is applicable to the highly-doped state, the calculated Fermi level shift from anion-exchanged sample to TBPA-doped sample is estimated to be 0.13 eV, which is inconsistent with the experimental result (0.2 eV shift). We do not speculate, but merely note that the discrepancy may be due to the violation of rigid band approximation. In this work, changes in doping levels were discussed qualitatively from PYS and optical absorption measurements. In addition, we emphasize that there is ambiguity in determining the carrier density and mobility from the Hall effect measurements when carriers are partially localized18,19. The overall results suggest that the donor–acceptor system tries to minimize the Gibbs free energy via a charge transfer interaction; electrons within the HOMO band of the polymer are transferred to the level of the redox potential (electron affinity) of the dopants, such that Φeff is close to the redox potential of the dopant, and the resulting donor–acceptor formation minimizes the Gibbs free energy at equilibrium such that no further charge transfer occurs. Clearly, the strong oxidation capability of the TBPA radical cation is advantageous for improving the doping levels of the polymer. We expect that any other salts based on radical cations with highly anodic redox potentials can serve as an efficient p-type dopant.
Structural analysis
This observation of remarkably high doping levels indicates that TFSI anions are incorporated into the polymer, such that they are in counter-balance to the positively charged PBTTT, which raises the question regarding where such high-density anions, equivalent to the amount of PBTTT monomers, can reside within the semicrystalline polymer network. To assess this, we performed X-ray diffraction (XRD) measurements and an electron density analysis. Out-of-plane and in-plane XRD profiles for pristine (black), F4TCNQ-doped (blue), and TBPA-TFSI doped PBTTT (red) thin films are shown in Fig. 2a, b. Typically, several orders of (h00) diffraction peaks, according to the lamellar spacing, are observed in the out-of-plane direction along the scattering vector qz. The diffraction peaks for doped PBTTT are found to shift to shorter qz, which corresponds to an expansion of the lamellar spacing17,33,34,35,36. Surprisingly, a 25% d-spacing expansion is obtained for the TBPA-TFSI doped PBTTT (dh00 shift in top panel of Fig. 2c). On the other hand, the in-plane diffraction along qxy after doping shows different behavior; the diffraction peaks assigned to (003), corresponding to the periodicity of the monomer repeat unit, is insensitive to doping, and those assigned to (010), corresponding to the π-stacking direction, shift to a larger scattering vector direction as the doping level increases. These XRD results suggest that dopants reside selectively in the alkyl side chain regions (resulting in lamellar expansion), and do not distribute into planar polymer planes (π–π spacing). The decrease in d0k0 may be attributed to suppression of torsion in the PBTTT backbone upon doping21 or to ionic interactions between positively charged PBTTT and counter-anions37.

a Out-of-plane XRD measurements of chemically doped PBTTT thin films. The peak marked with an asterisk is attributed to aggregated F4TCNQ on the surface of the thin film. b In-plane XRD measurements of chemically doped PBTTT thin films. The peaks with the square markers are mainly attributed to the periodicity of the PBTTT backbone in the main chain direction, while the peaks with the triangle markers arise from the periodicity of π stacking of the PBTTT. c d-spacings extracted from XRD measurements, where dh00 corresponds to stacking of PBTTT sheet structures in the out-of-plane direction, d00l corresponds to the length of the repeating unit of the PBTTT backbone, and d0k0 corresponds to the π stacking distance in the in-plane direction. The error bars for the d-spacings were determined from uncertainties in the fitting and represent one standard deviation.
Although spatial selectivity of dopant inclusion in the present semicrystalline polymer and dopant system has been observed17,21, a critical difference is found in PBTTT highly doped with TBPA-TFSI. Figure 2a shows the out-of-plane diffraction peaks (h00) for pristine and F4TCNQ doped PBTTT, exhibiting a monotonic decrease with increasing the Miller index (h00). In striking contrast, the (300) diffraction peak for TBPA-TFSI doped PBTTT vanishes, with its intensity being comparable to the noise level of the measurements, while higher order diffraction peaks (400) and (500) are clearly observed. Typically in XRD measurements of semicrystalline polymers, higher order diffraction peaks diminish rapidly as the Miller index increases because the degree of structural disorder for a polymer can be much larger than that for a single crystal27,38,39,40,41,42. Thereby, the missing (300) diffraction peak for the TBPA-TFSI doped PBTTT might not be explained by established diffraction theory for paracrystalline materials used for disordered polymers.
Loss of diffraction intensity commonly occurs in single-crystal XRD due to selection rules. This is a consequence of destructive interference of scattered X-rays43. The intensity of diffraction in a periodic system is proportional to the square of the absolute value of the structure factor, which can be derived from a Fourier transform and a convolution of the electron density of a repeating motif. Thus, the electron density of a repeating motif determines whether the interference is constructive or destructive. In turn, analyzing higher-order diffraction intensities allows us to determine the periodic electronic structures of layered compounds33,44. In order to examine the loss of the (300) peak and obtain a detailed electron density profile in doped PBTTT thin films, an XRD intensity simulation along the out-of-plane direction was conducted for pristine, TBPA-TFSI, and TBPA-SbCl6 doped PBTTT. The degree of (300) loss is found to be different between SbCl6 and TFSI anions, although both achieve similar doping levels.
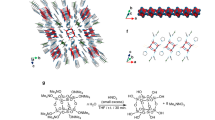
In our simulation, the electron density profile for isolated, monomer PBTTT was initially obtained from density functional theory (DFT) calculations with the Gaussian 09 package, projected in the out-of-plane direction (z-axis). This was then used to produce the electron density profile of a z-axis periodic PBTTT lattice using the experimentally determined dh00 = 21.4 Å. Figure 3a shows the one-dimensional electron density N(z) along the z-axis, where the electron density profile for the repeating motifs is shown separately for PBTTT backbones (orange) and alkyl chains (gray), and the total electron density profile, namely the sum of each motif, is shown as a solid curve. A Fourier transformation was applied to the electron density profile of the PBTTT lattice to obtain the structural factor. For doped PBTTT, the electron density profile for anions was added to that for the pristine PBTTT, where the periodic electron densities were reconstructed using the experimentally determined dh00 = 26.7 Å for TFSI− and 24.9 Å for SbCl6−. Figure 3b, c shows the electron density profile with TFSI (red) and SbCl6 (purple). We introduce four variable parameters: zanion, the weighted-center position of an anion relative to that of the polymer, ϕalkyl, the tilting angle of the alkyl chains relative to the out-of-plane direction, nanion, the amount of anions per monomer of PBTTT, and σ, the degree of Gaussian disorder. These four variable parameters were estimated by the least squares method so that all the simulated intensities of the (h00) diffraction peaks match the experimental intensities. The detailed method is described in Supplementary Note 3.

Models of (a) pristine PBTTT, (b) PBTTT doped with TFSI anion, and (c) PBTTT doped with SbCl6 anion, obtained from our simulations. The models are shown as side views, where the left axis represents the position in the out-of-plane direction. Left panels: Electron density profiles, N(z), for the repeating motifs for the PBTTT backbone (orange), alkyl chains (gray), TFSI anions (red), and SbCl6 anions (purple). Middle panels: Electron density profiles, N(z), for the repeating motifs integrated for all the components. Right panels: Side views of the modeled PBTTT and chemically doped PBTTT obtained from our simulations. d Comparison of experimentally obtained and simulated intensities of \(\left(h00\right)\) diffraction peaks for pristine and chemically doped PBTTT thin films. The error bars in the experimentally obtained peak intensities were determined from uncertainties in the Gaussian fitting of spectra and represent one standard deviation.
Figure 3a–c shows simulation results for electron density profiles that reproduce the (h00)-dependent XRD intensities. The agreement for the (h00)-dependent XRD intensities between experiments and simulation is shown in Fig. 3d. By optimizing the four variable parameters to achieve a global minimum of the fitting (see Table 1), a monotonic decay in the XRD intensities with respect to the Miller index due to structural disorder, and a characteristic decrease of (300) intensities are reproduced. We do not detail the mechanism of the disappearance of the (300) peaks (a more detailed explanation is shown in Supplementary Note 3), but we emphasize that a unique XRD profile such as in the present host–guest system, particularly for the observed destructive interference of XRD, can occur only when the intercalant is precisely located within the host lattice with the molecular precision preserved. Although there are four variable parameters in our simulation, the vital parameters determining the outcome of the experiments are found to be the position of the anions zanion and the number of anions nanion. These two parameters are determined explicitly to be zanion ~ 5 Å and nanion = 0.9–1.1, meaning that the weighted-center position of the anions is exactly 5 Å away from the backbone of the PBTTT, and there is almost one anion paired with each PBTTT monomer unit. This agrees with the experimentally observed carrier concentration, as discussed later. To verify this, we performed X-ray photoemission spectroscopy (XPS) analysis. The elemental analysis for the elemental composition ratio of carbon to fluorine for TFSI-doped PBTTT shows that the TFSI− content relative to PBTTT is 0.86 (see Supplementary Note 4), which is consistent with the XRD simulation results.
The heterogeneous, layered system of semicrystalline conjugated polymer and guest molecule is analogous with an intercalation compound. Intercalation is a chemical process in which guest agents, atoms, molecules, or clusters are incorporated into the interlayer gallery space of layered hosts, eventually forming hybrid heterostrucured materials. The host materials can be inorganic or organic, and their applications include batteries and electric and ionic conductors45,46,47. In ionic intercalation compounds, a guest ion intercalates into an oppositely charged sheet of the host two-dimensional material, resulting in an expansion of the van der Waals gap. A charge transfer interaction, i.e., a redox interaction, facilitates stabilization of the host and guest sheets. Such a controlled structure of the host and guest offers a platform for designing a system with desirable functionalities. Intercalation compounds based on graphite and the transition metal dichalcogenide have been widely studied45,46,47, but intercalation phenomena are also found in organic materials such as small molecular conductors48 and lipids49. However, there have been no previous reports on realizing intercalation compounds with a layered, semicrystalline polymer as the host platform, accommodating extremely high-density guest agents periodically.
Coherent carrier transport
In the hybrid host–guest system demonstrated here, the spatially confined guest anions may give rise to unique electronic functionalities because these anions inserted periodically into the polymer medium are in counter-balance to the positively charged polymer. Measuring the Hall effect is a powerful way to determine the charge density and the Hall mobility, and to distinguish the nature of the carrier localization. The Hall voltage, that is the transverse electromotive force generated in a conductor when placed in a magnetic field perpendicular to the current, is measurable only when the coherent wave packet of a delocalized electron that can be described with a semiclassical Boltzmann transport framework experiences the magnetic field17,21,22. In other words, charge carriers that undergo hopping transport never generate an ideal Hall voltage. Figure 4a shows the representative time domain profile for the transverse voltage at 240 K for TBPA-TFSI doped PBTTT thin films. A clear Hall voltage responding to the applied magnetic field B is observed. From the standard expression of the Hall effect, the Hall mobility μH and the Hall carrier density nH are determined and shown in Fig. 4b. A reasonably high nH of ~1.5 × 1021 cm−3 is achieved, concomitant with a high μH of 2 cm2 V−1 s−1 (Fig. 4b). This carrier density is identical to one hole per monomer unit or slightly greater. Importantly, the values for the charge carrier density derived separately from the Hall effect (Fig. 4b), electron density simulation (Table 1) and XPS elemental analysis (Supplementary Note 4) show almost perfect agreement, which strongly suggests that the proposed intercalation system with spatially confined guest anions is realized predominantly for the entire thin film. Note that the similar supramolecular cocrystal has been observed for the PBTTT/fullerene derivative system33, where the amount of intercalant was found to be limited by unavoidable aggregation and phase separation. A critical factor that differentiates these supramolecular cocrystals is that the creation of doping-induced cocrystals can be initiated by redox-triggered ion-intercalation, which enables an ideal production of homogeneous cocrystals without phase separation or aggregation.

a Hall voltages and the applied magnetic field B in the out-of-plane direction and (b) Hall carrier density nH derived from the inverse Hall coefficient \({n}_{{\rm{H}}}={(e{R}_{{\rm{H}}})}^{-1}\) and Hall mobility μH estimated from μH = RHσ for PBTTT thin films doped with F4TCNQ (blue), Mo(tfd-COCF3)3 (green), anion-exchange (orange), and TBPA-TFSI (red) at 240 K. The error bars in nH and μH represent uncertainties in extracting the Hall voltage from the fitting and the thickness of the polymer thin films and represent one standard deviation. c Out-of-plane XRD measurements of a PBTTT film with a thickness of 5 μm before and after TBPA-TFSI doping. d Miller index dependence of FWHM for pristine and TBPA-TFSI doped PBTTT thick film.
The spatial homogeneity of TBPA-TFSI doping presented in this work is further assessed using a thick polymer film. A 5-μm-thick PBTTT film was deposited via a modified drop-casting method, and was immersed in a TBPA-TFSI solution (see Supplementary Note 6). Figure 4c plots the out-of-plane XRD patterns for pristine and TBPA-TFSI doped PBTTT thick films. Even for the 5-μm-thick film, the (h00) diffraction peaks are found to shift to shorter qz with no splitting or broadening of the full width at half maximum (FWHM) (Fig. 4d). In addition, reduction of the (300) diffraction intensity for doped films was observed, similar to the doped thin films. The FWHM for the diffraction peaks decreases after doping, which demonstrates that the degree of cumulative disorder decreases by doping (Fig. 4d). Note that the X-ray penetration depth used here was estimated to be approximately 10 μm, and is larger than the film thickness, ensuring that the entire film was evaluated. These results suggest that TFSI anions are incorporated into the entire bulk of the 5-μm-thick PBTTT film, and the proposed intercalation supramolecular structure can be established. Details of the dopant dynamics still remain unknown. Considering that the chemical doping is mainly driven by the redox potential difference, which is on the order of sub-electronvolts, and is far higher than the thermal energy, spontaneous bulk doping can be a consequence of the host–guest system minimizing the Gibbs free energy. We note that various factors50, such as size, solvation of the dopant anion, softness of the polymer lattice, and void space in the polymer film, play important roles, and will be assessed in the future.
Ambient stability
The inclusion of functional anions into layered media is known to change the physicochemical properties34,36,45,46,47. Surprisingly, we found that the environmental durability of the doping effect depends strongly on the incorporated functional anions. We investigated the changes in the doping effect for TBPA-X (X = PF6, SbCl6, and TFSI) doped thin films by optical absorption and conductivity measurements. The effect of doping for TBPA-PF6 and TBPA-SbCl6 doped PBTTT was found to decrease rapidly after 88 h storage in ambient conditions of 22 ∘C and 40% relative humidity. The bleached optical absorption of the doped films returned to the neutral state (Fig. 5a, b) and the conductivity decreased by several orders of magnitude (Fig. 5c). For TBPA-TFSI doped PBTTT, changes in the optical absorption spectra seem to be largely suppressed and the degradation of the conductivity is much slower. It is reasonable to envisage that the doping effect of anions can be quenched by unintentional H2O adsorption by exposure to ambient air. We therefore speculate that the H2O molecule adsorption rate may correlate with the intercalation structure, and depend on the incorporated anions. For TBPA-TFSI doped PBTTT, the number of H2O molecules that diffuse through the polymer void and are chemisorbed at the surface of TFSI anions may decrease significantly. Figure 5d–f illustrates DFT simulation results for the surface electrostatic potential and their space filling in the present systems with one anion per monomer unit (see more details in Supplementary Note 7). For the TFSI anions, a strong, albeit local, surface electrostatic potential was obtained, particularly at the single nitrogen and four oxygen sites, which is comparably strong as the PF6 anions. In general, a large electrostatic potential on a molecular surface is likely to produce strong electrostatic interactions and water adsorption51; thus, intuitively the best stability will be found for TBPA-SbCl6 doped PBTTT. However, TBPA-SbCl6 doped PBTTT shows poor stability, as comparable as TBPA-PF6 doped PBTTT. Instead, TFSI-doped PBTTT shows the best stability. We speculate that this non-intuitive result originates from the unique conformation of TFSI anions relative to the polymer backbone; the nitrogen and two of the four oxygen sites in the TFSI anions that should interact strongly with the H2O molecules are sterically protected by the polymer backbone, and become inaccessible to H2O molecules (Fig. 5e). Furthermore, the intercalation compound TFSI-doped PBTTT may approach a closest-packed structure, for which the filling of the void space between each lamellae may help to reduce the accessible surface for H2O molecules. The effects of void filling reducing the accessible surface area has been investigated for amorphous polymers, for which a molecular additive in the void in amorphous polymers and optimization of dopant size have been found to improve device stability significantly52,53. Therefore, supramolecular engineering to form intercalation compounds that can potentially confine the position and conformation of reactive sites of functional anions may lead to further improvements of the physicochemical properties.

UV-Vis-NIR spectra for a pristine (gray), TBPA-PF6 (light blue), TBPA-SbCl6 (purple), and TBPA-TFSI (red) doped PBTTT thin films. Data were acquired (a) as doped and (b) after 88 h air exposure. c Ambient stability of conductivities of TBPA-PF6 (light blue), TBPA-SbCl6 (purple), and TBPA-TFSI (red) doped PBTTT thin films. Electrostatic potential map on molecular surfaces for ion pairs of (d). PBTTT/PF6, (e) PBTTT/SbCl6, and (f) PBTTT/TFSI, together with their space-filling, van der Waals-based representation. The calculations of these potentials were conducted with Spartan'16 software with the M06-2X functional and 6-31++G(d,p) basis set, where a pseudopotential is applied to the Sb atom.
Conclusion
In this study, we have demonstrated doping of polymeric semiconductors with radical salts, where radical cations with strong redox activity are responsible for electron transfer. Our observations show that at the extremely high doping levels achieved by the present doping method, a soft lattice of semicrystalline polymers maintains its molecular precision by the periodic inclusion of dopant anions, for which the position and conformation of the guest dopant can be confined spatially in the restricted void space in the polymer’s lamellae. Realizing closest-packed intercalation supramolecules leads to the best attainable doping level in semicrystalline conjugated polymers and excellent environmental stability due to the limited accessible surface. These findings should therefore shed light on possibilities for the storage, transport, and conversion of functional molecules and ions through intercalation phenomena, and the construction of host–guest systems of crystalline doped polymers to achieve desirable optoelectronic properties.
Methods
Chemical doping
PBTTT thin films were fabricated on low-impurity glass substrates, except for those used in the photoemission yield spectroscopy (PYS) analysis. The PYS data were obtained on indium-tin-oxide (ITO) substrates. PBTTT thin films were deposited via spin-coating. The resulting films were annealed on a hotplate at 180 ∘C for 1 h, then slowly cooled to room temperature. The film thicknesses were determined to be 42 ± 2 nm. The PBTTT thin films were doped by immersing solid-state thin films into dopant solutions for 10 min in N2-purged vials. After cooling the vial to room temperature, the film was removed from the vial and the residual solvent was blown off under a flow of N2. More detailed film preparations and doping procedures are presented in Supplementary Note 1 and 2.
XRD analysis
Out-of-plane and in-plane XRD data were acquired using a RIGAKU SmartLab with a MicroMax-007HF X-ray generator, employing Cu Kα radiation (λ = 0.15418 nm) in conjunction with a Si substrate with a naturally formed oxide layer. In our simulation, density functional theory (DFT) calculations using Gaussian 09 were initially employed to obtain electron densities for each component of pristine and doped PBTTT thin films; electron densities for a monomer of PBTTT backbone without alkyl chains, an alkyl chain (all-trans conformation), and anions were separately calculated. The B3LYP functional and the 6-31+G(d) basis set were employed, except for the Sb atom, for which the Lanl2DZ basis set was employed. These electron densities were projected into the z-axis to give one-dimensional electron density profiles along the out-of-plane direction (see more details in Supplementary Note 3).
Transport measurements
Hall effect measurements were performed using a Hall bar geometry, where the doped PBTTT thin film was patterned by dry etching in order to perform precise measurements of local potentials of probes along a channel. The Hall bar devices were fabricated on a 0.7-mm-thick glass substrate. The PBTTT thin film was deposited in a similar manner and then patterned into the Hall bar geometry by laser etching using an yttrium aluminum garnet (YAG) laser (see more details in Supplementary Note 5).
Data availability
The data that support the plots within this paper and the other findings of this study are available from the corresponding author (Shun Watanabe, swatanabe@edu.k.u-tokyo.ac.jp) upon request.
References
Sze, S. M. & Ng, K. K. Physics of semiconductor devices (John Wiley & Sons, 2006).
Bean, K. E. & Runyan, W. Semiconductor integrated circuit processing technology (Addison-Wesley, 1990).
Levy, R. Microelectronic materials and processes, vol. 164 (Springer Science & Business Media, 1989).
Shirakawa, H., Louis, E. J., MacDiarmid, A. G., Chiang, C. K. & Heeger, A. J. Synthesis of electrically conducting organic polymers: halogen derivatives of polyacetylene, (CH)x. J. Chem. Soc. Chem. Commun. 578–580 (1977).
Heeger, A. J., Kivelson, S., Schrieffer, J. & Su, W.-P. Solitons in conducting polymers. Rev. Mod. Phys. 60, 781 (1988).
Prosa, T., Winokur, M., Moulton, J., Smith, P. & Heeger, A. X-ray-diffraction studies of the three-dimensional structure within iodine-intercalated poly(3-octylthiophene). Phys. Rev. B 51, 159 (1995).
Kar, P. Doping in conjugated polymers (John Wiley & Sons, 2013).
Lüssem, B., Riede, M. & Leo, K. Doping of organic semiconductors. Phys Status Solidi A 210, 9–43 (2013).
Salzmann, I., Heimel, G., Oehzelt, M., Winkler, S. & Koch, N. Molecular electrical doping of organic semiconductors: fundamental mechanisms and emerging dopant design rules. Acc. Chem. Res. 49, 370–378 (2016).
Jacobs, I. E. & Moulé, A. J. Controlling molecular doping in organic semiconductors. Adv. Mater. 29, 1703063 (2017).
Tietze, M. L. et al. Elementary steps in electrical doping of organic semiconductors. Nat. Commun. 9, 1–9 (2018).
Gaul, C. et al. Insight into doping efficiency of organic semiconductors from the analysis of the density of states in n-doped C60 and ZnPc. Nat. Mater. 17, 439–444 (2018).
Tang, C. G. et al. Doped polymer semiconductors with ultrahigh and ultralow work functions for ohmic contacts. Nature 539, 536 (2016).
Png, R.-Q. et al. Madelung and Hubbard interactions in polaron band model of doped organic semiconductors. Nat. Commun. 7, 1–9 (2016).
Tan, J.-K., Png, R.-Q., Zhao, C. & Ho, P. K. Ohmic transition at contacts key to maximizing fill factor and performance of organic solar cells. Nat. Commun. 9, 3269 (2018).
Lin, X. et al. Beating the thermodynamic limit with photo-activation of n-doping in organic semiconductors. Nat. Mater. 16, 1209–1215 (2017).
Kang, K. et al. 2D coherent charge transport in highly ordered conducting polymers doped by solid state diffusion. Nat. Mater. 15, 896 (2016).
Fujimoto, R. et al. Control of molecular doping in conjugated polymers by thermal annealing. Organic Electr. 47, 139–146 (2017).
Fujimoto, R. et al. Molecular doping in organic semiconductors: fully solution-processed, vacuum-free doping with metal–organic complexes in an orthogonal solvent. J. Mater. Chem. C 5, 12023–12030 (2017).
Kim, Y. et al. Enhanced charge injection properties of organic field-effect transistor by molecular implantation doping. Adv. Mater. 31, 1806697 (2019).
Yamashita, Y. et al. Efficient molecular doping of polymeric semiconductors driven by anion exchange. Nature 572, 634–638 (2019).
Watanabe, S. et al. Validity of the Mott formula and the origin of thermopower in π-conjugated semicrystalline polymers. Phys. Rev. B 100, 241201 (2019).
Hamidi-Sakr, A. et al. A versatile method to fabricate highly in-plane aligned conducting polymer films with anisotropic charge transport and thermoelectric properties: the key role of alkyl side chain layers on the doping mechanism. Adv. Funct. Mater. 27, 1700173 (2017).
Ueji, K., Ohno, M., Takeya, J. & Watanabe, S. Correlation between coherent charge transport and crystallinity in doped π-conjugated polymers. Appl. Phys. Expr. 12, 011004 (2018).
Aubry, T. J. et al. Tunable dopants with intrinsic counterion separation reveal the effects of electron affinity on dopant intercalation and free carrier production in sequentially doped conjugated polymer films. Adv. Funct. Mater. 2001800 (2020).
McCulloch, I. et al. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat. Mater. 5, 328 (2006).
Chabinyc, M. L., Toney, M. F., Kline, R. J., McCulloch, I. & Heeney, M. X-ray scattering study of thin films of poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene). J. Am. Chem. Soc. 129, 3226–3237 (2007).
McCulloch, I. et al. Semiconducting thienothiophene copolymers: design, synthesis, morphology, and performance in thin-film organic transistors. Adv. Mater. 21, 1091–1109 (2009).
Cho, E. et al. Three-dimensional packing structure and electronic properties of biaxially oriented poly(2,5-bis(3-alkylthiophene-2-yl)thieno[3,2-b]thiophene) films. J. Am. Chem. Soc. 134, 6177–6190 (2012).
Paniagua, S. A. et al. Production of heavily n-and p-doped CVD graphene with solution-processed redox-active metal–organic species. Mater. Horizons 1, 111–115 (2014).
Dapperheld, S., Steckhan, E., Brinkhaus, K.-H. G. & Esch, T. Organic electron transfer systems, II substituted triarylamine cation-radical redox systems–synthesis, electrochemical and spectroscopic properties, hammet behavior, and suitability as redox catalysts. Chemische Berichte 124, 2557–2567 (1991).
Connelly, N. G. & Geiger, W. E. Chemical redox agents for organometallic chemistry. Chem. Rev. 96, 877–910 (1996).
Miller, N. C. et al. Use of X-ray diffraction, molecular simulations, and spectroscopy to determine the molecular packing in a polymer-fullerene bimolecular crystal. Adv. Mater. 24, 6071–6079 (2012).
Patel, S. N. et al. Morphology controls the thermoelectric power factor of a doped semiconducting polymer. Sci. Adv. 3, e1700434 (2017).
Guardado, J. O. & Salleo, A. Structural effects of gating poly(3-hexylthiophene) through an ionic liquid. Adv. Funct. Mater. 27, 1701791 (2017).
Jacobs, I. E. et al. Polymorphism controls the degree of charge transfer in a molecularly doped semiconducting polymer. Mater. Horizons 5, 655–660 (2018).
Garcia-Yoldi, I., Miller, J. S. & Novoa, J. J. Theoretical study of the electronic structure of [TCNQ]22− (TCNQ = 7,7,8,8-tetracyano-p-quinodimethane) dimers and their intradimer, long, multicenter bond in solution and the solid state. J. Phys. Chem. A 113, 7124–7132 (2009).
Kline, R. J., McGehee, M. D. & Toney, M. F. Highly oriented crystals at the buried interface in polythiophene thin-film transistors. Nat. Mater. 5, 222–228 (2006).
Chabinyc, M. L. X-ray scattering from films of semiconducting polymers. Polymer Rev. 48, 463–492 (2008).
Salleo, A., Kline, R. J., DeLongchamp, D. M. & Chabinyc, M. L. Microstructural characterization and charge transport in thin films of conjugated polymers. Adv. Mater. 22, 3812–3838 (2010).
Rivnay, J., Noriega, R., Kline, R. J., Salleo, A. & Toney, M. F. Quantitative analysis of lattice disorder and crystallite size in organic semiconductor thin films. Phys. Rev. B 84, 045203 (2011).
Brinkmann, M. Structure and morphology control in thin films of regioregular poly(3-hexylthiophene). J. Polymer Sci. Part B: Polymer Phys. 49, 1218–1233 (2011).
Warren, B. E. X-ray Diffraction (Courier Corporation, 1990).
Pruvost, S., Hérold, C., Hérold, A. & Lagrange, P. Structural study of novel graphite-lithium-calcium intercalation compounds. Eur. J. Inorganic Chem. 2004, 1661–1667 (2004).
He, P., Yu, H. & Zhou, H. et al. Layered lithium transition metal oxide cathodes towards high energy lithium-ion batteries. J. Mater. Chem. 22, 3680–3695 (2012).
Wan, C. et al. Flexible n-type thermoelectric materials by organic intercalation of layered transition metal dichalcogenide TiS2. Nat. Mater. 14, 622 (2015).
Jache, B. & Adelhelm, P. Use of graphite as a highly reversible electrode with superior cycle life for sodium-ion batteries by making use of co-intercalation phenomena. Angew. Chem. Int. Ed. 53, 10169–10173 (2014).
Saito, G. & Yoshida, Y. Development of conductive organic molecular assemblies: organic metals, superconductors, and exotic functional materials. Bull. Chem. Soc. Japan 80, 1–137 (2007).
Ziblat, R., Leiserowitz, L. & Addadi, L. Crystalline lipid domains: characterization by X-ray diffraction and their relation to biology. Angew. Chem. Int. Edi. 50, 3620–3629 (2011).
Kroon, R. et al. Bulk doping of millimeter-thick conjugated polymer foams for plastic thermoelectrics. Adv. Funct. Mater. 27, 1704183 (2017).
Hagelin, H., Murray, J. S., Politzer, P., Brinck, T. & Berthelot, M. Family-independent relationships between computed molecular surface quantities and solute hydrogen bond acidity/basicity and solute-induced methanol O–H infrared frequency shifts. Can. J. Chem. 73, 483–488 (1995).
Nikolka, M. et al. High operational and environmental stability of high-mobility conjugated polymer field-effect transistors through the use of molecular additives. Nat. Mater. 16, 356 (2017).
Thomas, E. M. et al. Effects of counter-ion size on delocalization of carriers and stability of doped semiconducting polymers. Adv. Electr. Mater. 2000595 (2020).
Acknowledgements
S.W. acknowledges support from PRESTO-JST through the project “Hyper-nanospace Design Toward Innovative Functionality” (Grant no. JPMJPR151E), and from the Leading Initiative for Excellent Young Researchers of JSPS. T.O. acknowledges support from PRESTO-JST through the project “Scientific Innovation for Energy Harvesting Technology” (Grant no. JPMJPR17R2). This work was also supported in part by JSPS KAKENHI grants (nos. JP17H06123, JP20K15358, JP20H00387, JP20K20562, and JP20H05868).
Author information
Authors and Affiliations
Contributions
T.O., Y.Y., and S. W. conceived the proof-of-concept of the doping method. Y.Y., K.U., J. Tsurumi, Y.T., S. Kohno, and S.W., designed and performed the experiments and analyzed the data. T.K., H.K., and S. Kumagai, synthesized the series of TBPA dopants. S.W., Y.Y., and T.K., wrote the manuscript. S.W. and J. Takeya, supervised the work. All authors discussed the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary handling editor: John Plummer
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamashita, Y., Tsurumi, J., Kurosawa, T. et al. Supramolecular cocrystals built through redox-triggered ion intercalation in π-conjugated polymers. Commun Mater 2, 45 (2021). https://doi.org/10.1038/s43246-021-00148-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-021-00148-9
This article is cited by
-
Band transport evidence in PEDOT:PSS films using broadband optical spectroscopy from terahertz to ultraviolet region
Communications Materials (2024)
-
Doping of molecular semiconductors through proton-coupled electron transfer
Nature (2023)
-
Doped semiconducting polymer nanoantennas for tunable organic plasmonics
Communications Materials (2022)
