
Abstract
Chinese plum (Prunus salicina L.), also known as Japanese plum, is gaining importance because of its extensive genetic diversity and nutritional attributes that are beneficial for human health. Single-nucleotide polymorphisms (SNPs) are the most abundant form of genomic polymorphisms and are widely used in population genetics research. In this study, we constructed high-quality SNPs through whole-genome resequencing of 67 Prunus accessions with a depth of ~20× to evaluate the genome-level diversity and population structure. Phylogenetic analysis, principal component analysis, and population structure profiling indicated that the 67 plum accessions could be classified into four groups corresponding to their origin location, the southern cultivar group (SCG), the northern cultivar group (NCG), the foreign cultivar group (FG), and the mixed cultivar group (MG). Some cultivars from South China clustered with the other three groups. The genetic diversity indices including private allele number, observed heterozygosity, expected heterozygosity, and the nucleotide diversity of the SCG were higher than those of the NCG. Gene flow from the SCG to FG was also detected. Based on the distribution of wild resources, we concluded that the domestication center of origin of the Chinese plum was southwestern China. This study also provided genetic variation features and the population structure of Chinese plum cultivars, laying a foundation for breeders to use diverse germplasm and allelic variants to improve Chinese plum varieties.
Similar content being viewed by others

Introduction
Plum is one of the most important fruit crops globally and, possesses extensive genetic diversity and high economic value (Topp et al. 2012). The largest plum producer is China, with an annual production of 6,801,187 metric tons in 2018, accounting for 53.9% of the world’s total (FAOSTAT 2018). Within the genus Prunus, the Chinese plum (Prunus salicina L., 2n = 2x = 16), also known as the Japanese plum, is widely grown for fresh market consumption and the canning industry, and includes both the pure Chinese plum and its hybrids with other diploid plum species, such as Prunus simonii Carr., Prunus cerasifera Ehrh., and Prunus americana Marsh. (Liu et al. 2007).
According to historical records of cultivation, the Chinese plum may have originated in the Yangtze River Basin, and there are abundant plum germplasm resources in China (Hartmann and Neumulle 2009). The Chinese plum has a long growing history and extensive geographical distribution, with more than 1000 indigenous plum cultivars in China, derived from Prunus salicina L. (Zhang 1990). Over 700 of these cultivars are currently preserved at the National Germplasm Repository for Plums and Apricots (NGRPA) located in Xiongyue, Liaoning Province, China (Wei et al. 2020). Yu et al. (2011) carried out comprehensive phenotyping of 405 Chinese plum cultivars and their hybrids from the NGRPA, and investigated a total of 32 morphological and agronomic characters. Coefficients of variation (CVs) ranged from 14.85 to 47.09%, suggesting that the genetic variability of Chinese plums is widely distributed.
Genetic variability is a prerequisite for any plant breeding program. Learning the extent and structure of genetic variation in germplasm collections is a crucial step for the efficient conservation and utilization of biodiversity in cultivated crops. For plum breeders, using diverse plum resources to broaden the genetic base of worldwide plum cultivars is a critical objective (Liu et al. 2007; Urrestarazu et al. 2018). Previous studies have conducted the genetic diversity of plums; for example, Zhang and Zhou (1998) took the lead in collecting plum germplasm resources and evaluating their genetic diversity based on morphological traits and isozyme polymorphisms. However, because morphological traits were highly susceptible to environmental factors, the estimates of genetic diversity were not precise. The isozymes had a low degree of polymorphism and hence were not efficient enough for the characterization of germplasm genetic diversity (Khush 2002). Current research applies DNA-based markers in plum genetic diversity analysis: markers include random amplified polymorphic DNA (RAPD) (Liu et al. 2006; Ben Tamarzizt et al. 2015); simple sequence repeats (SSRs) (Pop et al. 2018; Zhang et al. 2018; Acuña et al. 2019); and inter-simple sequence repeats (ISSRs) (Liu et al. 2007; Wu et al. 2019). DNA-based marker assessments showed that Chinese plums can generally be classified into two major groups: the southern cultivar group (SCG) and the northern cultivar group (NCG) (Liu 2005; Wei et al. 2019). A palynology study showed that the P. salicina system in South China was more primitive than that in North China, with the spread of P. salicina taking place from the south to the north (Guo 2006). A cultivar clustering approach, however, is inadequate for the integrated study of genetic diversity of the Chinese plum. Advances in next-generation sequencing (NGS) technologies, involving genotyping by sequencing (GBS), provide a great wealth of information that makes it possible to identify thousands of single nucleotide polymorphisms (SNPs). This information, after adequate filtering, allows to carry out detailed genetic diversity studies (Elshire et al. 2011; Salazar et al. 2019; Zhebentyayeva et al. 2019). Whole-genome resequencing can be used to obtain more SNP datasets for Prunus species. In the case of the peach, large-scale SNP data-based diversity analysis has boosted the deciphering of the evolution and domestication of peach germplasm resources (Cao et al. 2014; Li et al. 2019).
The present study was the first in which we used SNP markers generated from high depth whole-genome resequencing data (an average depth of ~20×) to elucidate the pattern of genetic diversity, population structure, and domestication of a diverse P. salicina collection. An in-depth understanding of such genetic relationships could benefit plum germplasm conservation and utilization and lead to new cultivar improvement.
Materials and methods
Plant materials
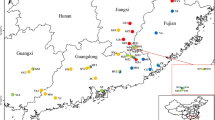
A diverse collection of 67 Prunus spp. accessions, including 65 P. salicina accessions and two P. simonii accessions, was selected for the whole-genome resequencing study (Table S1). These trees were accessed at the NGRPA located in Xiongyue county, Liaoning Province, China (40° 18′ N, 122° 16′ E) at a planting density of 3.0 × 4.0 m, trained to the open vase system with three or four main branches, and maintained under conventional management and pest control operations.
DNA extraction, library preparation, and sequencing
Genomic DNA was isolated from young, healthy leaf samples of 67 accessions using a modified cetyl trimethylammonium bromide (CTAB) protocol (Doyle and Doyle 1990). The quality and integrity of the DNA were examined using a NanoDrop® spectrophotometer (ND-1000, Thermo Fisher Scientific Inc., USA), followed by electrophoresis in 1% agarose gels. Quantification of the DNA samples was performed using Qubit™ (2.0 Fluorometer, Invitrogen, Carlsbad, CA, USA).
High-molecular-weight DNA aliquots with 230/260 and 260/280 ratios ranging between 1.8–2.0 and 1.8–2.2, respectively, were then sent to BGI (Shenzhen, China) for library construction and sequencing. The insert size of the libraries was 500 bp, and the length of the pair-end reads was 150 bp. All libraries were sequenced using the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA).
Read mapping, SNP calling, and SNP annotation
The qualified paired-end reads of each accession were aligned against the peach reference genome v2.0 (Verde et al. 2013) using BWA v0.7.12-r1039 with the parameters mem -t 4 -k 32 –M (Li and Durbin 2010), and SNPs were identified using SAMtools v1.4 (Li et al. 2009). Low-quality SNPs were filtered out by minimum minor allele frequency (mnMAF < 0.01) and missing data per site (MDpS > 10%), and finally converted into a variant call format file (VCF). Gene-based SNP annotation was performed using the ANNOVAR package v2018-04-16 (Wang et al. 2010). Based on the reference genome annotation, SNPs were categorized as occurring in exonic regions (overlapping with a coding exon); intronic regions (overlapping with an intron); upstream and downstream regions (within a 1 kb region upstream or downstream from the transcription start site); or intergenic regions. Additionally, heterozygosity was calculated using VCFtools v0.1.14 (Danecek et al. 2011).
Population structure analysis
TreeBeST v1.9.2 software was used to calculate the distance matrix (Vilella et al. 2009). RAxML v8.2.12 was used to construct the maximum likelihood (ML) phylogenetic tree, and 1000 bootstrap replicates were used (Stamatakis 2014). The resulting phylogenetic tree was visualized using MEGA v5.05 (Tamura et al. 2011).
The principal component analysis was performed using PLINK v1.07 software with default parameters (Purcell et al. 2007).
We used ADMIXTURE v1.23 to infer population structure (Alexander et al. 2009). To identify the best genetic clusters K, cross-validation error was tested for each K value from 2 to 10. The termination criterion was 1e−6 (stopping when the log-likelihood increased by less than 1e−6 between iterations).
Linkage disequilibrium (LD) analysis
Linkage disequilibrium (LD) was calculated using SNPs with MAF greater than 0.05, using PLINK v1.07 software with the following settings: --file --r2 --ld-window 99999 --ld-window-kb 200 --out. The LD decay was calculated based on the squared correlation coefficient (r2) values between the two SNPs and the physical distance between the two SNPs (Purcell et al. 2007).
Gene flow analysis
The TreeMix software v1.13 was used to evaluate the gene flow among different groups with the parameters -se-bootstrap-k 1000 -m, where the number (-m) varied from one to three (Pickrell and Pritchard 2012).
Genetic diversity and differentiation analysis
To evaluate the genetic diversity and differentiation, we used a 100-kb sliding window with a step size of 10 kb to calculate the number of private alleles (AP); observed heterozygosity (Ho); expected heterozygosity (He); nucleotide diversity (π); Tajima’s D value; Wright’s F-statistic (FIS); and population differentiation statistics (fixation index, FST) using VCFtools v0.1.14 (Danecek et al. 2011).
Results
Marker development based on whole-genome resequencing
The 67 plum accessions representing different geographic and morphological characteristics (Table S1) were sequenced using the Illumina HiSeq 2500 platform with a sequence depth higher than 20×; a total 462.6 Gb of clean data was retained with an average of 6.9 Gb for each accession after filtering out low-quality reads. The quality of the sequencing data was high with a Q30 > 85%, and the GC content fluctuating slightly around 38.0%. The mapping rate to the peach genome ranged from 85.73 to 88.57%, with an average of 87.49% (Table S1). This result indicated a high level of collinearity between the genomes of Chinese plum and peach.
We called SNPs with unique mapped reads, using SAMtools v1.4 software; a total of 16,600,033 SNPs and 2,107,664 indels were identified across the 67 accessions. After filtering out those of low quality, we obtained 14,549,234 SNPs that would ensure the accuracy and reliability of subsequent genetic diversity and population structure analyses (Fig. 1). Approximately 43.81% of the SNPs were located in intergenic regions, and 10.52% were in coding regions. The non-synonymous to synonymous substitution ratio (dN/dS) for the SNPs in the coding regions was 1.32. Transitions were found in 59.58% (10,315,764/16,600,033), with a transition/transversion ratio (Ts/Tv ratio) of 1.47 (Fig. 1, Table 1).

The statistics of markers generated from the whole-genome resequencing of the 67 plum accessions. a The chromosome-scale SNP distribution. b The distribution of transition/transversion ratio (Ts/Tv ratio). c The counts of different types of transitions and transversions. d The counts of genome-wide insertions and deletions
Phylogenetic and population structure
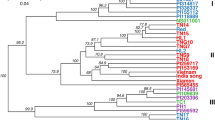
To explore the relationships among the Chinese plum accessions, a neighbor-joining phylogenetic tree of the 67 accessions was constructed using all SNPs (Fig. 2a). The phylogenetic tree classified the accessions into four main groups, which corresponded to their respective origin locations: (1) the southern cultivar group (SCG), comprising plum cultivars from Sichuan, Guizhou, Yunnan, Guangdong, Guangxi, Zhejiang, and Fujian Provinces; (2) the northern cultivar group (NCG), comprising plum cultivars mainly from Hebei, Henan, Shandong, Shaanxi Province, and the southern part of Liaoning Province; (3) the foreign cultivar group (FG), including plum cultivars from the USA and Japan; and (4) the mixed cultivar group (MG), comprising newly bred cultivars from the NGRPA and several cultivars originated SCG and FG. Notably, we found that “Saozouli” and “Shuili” cultivars, collected from South China, were clustered with the NCG; “Zaohuangli” and “Jinshali” with the FG; and “Wanshu huanai,” “Huahongli,” and “Abazhou meiguili” with the MG. These SCG samples showed some degree of correlation, suggesting a close relationship between the SCG and others groups. The results showed that the other groups potentially originated from the SCG.

The population structure of the 67 plum accessions. a Neighboring-joining phylogenetic tree constructed using SNPs at fourfold degenerate site. Each group was color coded. b Bayesian model-based clustering of the 67 plum accessions with the number of ancestry kinship (K) from 2 to 10. c Principle Component Analysis (PCA) of the 67 plum accessions. FG, the foreign cultivar group; MG, the mixed cultivar group; SCG, the southern cultivar group; NCG, the northern cultivar group
A population genetic structure analysis was performed, based on high-quality SNPs. We employed 5-fold cross-validation to infer the number of ancestral populations, K (Fig. 2b and Figure S1). When the K value was 2–4, the NCG exhibited a consistent genetic constitution with the SCG, suggesting that the NCG was derived from the SCG and, to some extent, had also undergone environmental/human selection. The population structure of the SCG was complex, especially when the K value was higher, suggesting a higher genetic diversity in the SCG, which was consistent with the π value analysis (Fig. 3b). For the FG, we found that the population structure was relatively independent, compared with the SCG and others. Thus, we postulated that the existence of intra-species hybrids in the FG group led to a high π value (Fig. 3b).

The genetic diversity of different groups. a The decay of linkage disequilibrium (LD) measured as the squared correlation coefficient (r2) by pairwise physical distance. b The nucleotide diversity (π) of different groups. c The genetic differentiation analysis between groups. The values between pairs indicate population divergence (FST). FG, the foreign cultivar group; MG, the mixed cultivar group; SCG, the southern cultivar group; NCG, the northern cultivar group
To further confirm the relationship among the cultivars, we performed principal component analysis (PCA) of the 67 plum accessions (Fig. 2c). As shown in the principal component plot for the first two principal components, the NCG exhibited a relatively close relationship with the SCG, which was consistent with the phylogenetic tree and population structure analysis. As shown in Table 1, a large number of rare alleles were lost in the NCG population. Therefore, we deduced that the NCG could be considered to be a more independent subgroup of the SCG and might have been derived from a specific ecotype.
Genetic diversity, differentiation, and inferred evolutionary path
As shown in Table 2, the expected heterozygosity (He) of the Prunus populations varied between 0.234 and 0.304; the observed heterozygosity (Ho) of the Prunus populations ranged between 0.328 and 0.429; Wright’s F-statistic (FIS) of the Prunus populations varied between −0.241 and −0.146; the number of private alleles (AP) in the Prunus populations varied between 546 and 31,285; and the nucleotide diversity (π) ranged between 0.00358 and 0.00467. The mean nucleotide variation of P. salicina was higher than that of other perennial crops, such as peach (π = 0.0015) (Verde et al. 2013), cassava (π = 0.0026) (Kawuki et al. 2009), and apricot (π = 0.0027) (Li et al. 2020), but was lower than that of date palms (π = 0.0092) (Hazzouri et al. 2015). The Tajima’s D values of the four groups all tested positive (1.002–1.497) and were significantly different from zero. Thus, the null hypothesis of neutral evolution was rejected. As shown in Fig. 3a, the LD decay rate was fastest in the SCG and slowest in the FG, which indicated that genetic recombination in the FG was difficult because of the narrow genetic origin of the artificial hybrid cultivars in the group.
The FST values of the four groups varied between 0.0485 and 0.1303 (Fig. 3). The FST values of the MG-FG, MG-SCG, and SCG-NCG pairs were relatively lower among all the group pairs analyzed, indicating that the genetic differences within populations were higher than those between populations and that there was possible genetic exchange between populations. The FST value between the NCG and FG groups was the highest of all the groups analyzed, possibly because of geographical isolation, low gene flow between populations, and significant genetic differences.
We analyzed the gene flow between the four geographic groups (Fig. 4b, c). Allowing one or two migration events (m = 1 or 2), we observed that gene flow occurred between the SCG and FG accessions, a likely reflection of their many shared genomic components due to hybridization in their domestication and breeding histories. Considering the geographical locations of the SCG and NCG, although these two ecological groups were close to each other, we found no gene flow between them, indicating a relatively independent domestication process.

The inferred evolutionary path (a) and gene flow analysis between populations as inferred by TreeMix using a model with one (b) and two (c) admixture events. Admixtures are colored according to their weight. FG, the foreign cultivar group; MG, the mixed cultivar group; SCG, the southern cultivar group; NCG, the northern cultivar group
Discussion
SNPs have become essential as markers for research in plant genetics because they occur in high frequencies, display a lower mutation rate compared to SSR-based markers, and are uniformly distributed across genomes (Carrasco et al. 2018). The NGS technologies allow discovery of large numbers of SNPs for extensive genetic studies at relatively low-cast. The technologies include within-species diversity analysis; linkage map construction; and genome-wide association studies (GWAS), which have led to significant advances in plant genetics and breeding (Cao et al. 2016; Li et al. 2019; Pinosio et al. 2020). The genus Prunus has shown conserved intraspecific and intragenic collinearity in the Rosaceae family, with the peach being considered a model species for the genus Prunus for multiple types of genetic research (Arús et al. 2012; Carrasco et al. 2018; Marti et al. 2018). Zhang et al. (2020) found that the correlation of eight chromosomes was greater than 0.8, indicating a high level of collinearity between the Japanese plum and peach genomes. A recently released plum genome has been published, and collinearity analysis has shown that the assembled genomes of the plum also exhibit a high level of genome synteny with the peach (Liu et al. 2020). For the first time, we obtained an average of 6.9 Gb of high-quality data for each acquisition based on whole-genome resequencing, where the mapping rate of 67 plum accessions with the peach genome sequence was above 85%. This result indicates that the peach genome sequence is highly effective as the reference genome of Chinese plum, and that the SNPs that were used ensure accurate and comprehensive population genetic analysis.
Based on the high-quality SNPs, the 67 plum accessions in this study were divided into four groups, the SCG, NCG, FG, and MG. The classification into these four groups was supported by the phylogenetic tree, population structure analysis, and PCA results, which partly aligned with the findings of a previous study which used RAPD, ISSR, and SSR markers (Liu et al. 2006; Liu 2005; Wei et al. 2019). We noticed that several cultivars from the southern part of China, present in the SCG, were distributed in the other three groups (Fig. 2a). We speculated that, for the SCG, the genetic background was relatively broader. Additionally, the fruit weight of the southern cultivars exhibited the highest genetic variation a previous analysis of the phenotypic variation of 405 plum accessions (Yu et al. 2011). The π, Ho, and He of the SCG, calculated with SNPs in our study, were higher than those of the NCG, corresponding to the higher genetic diversity of the SCG. This finding showed that gene exchange in the SCG was more frequent than in the other cultivar groups.
It was originally speculated from historical records that the Chinese plum originated in the Yangtze River basin in China (Hartmann and Neumulle 2009). A palynological study showed that the P. salicina in South China is more primitive than that in North China (Guo 2006). Chinese plums cultivated in southwest China were tested with high genetic diversity using SSR markers (Wei et al. 2019): the high Shannon index of diversity, the high value of average sufficient allele, and the high expected heterozygosity, combined with the fact that there were wild plum populations discovered in Yunnan, Sichuan, and Guizhou Provinces, suggested high genetic diversity (Wei et al. 2020). In the present study, the genetic diversity of the SCG was higher than that of the NCG, while some cultivars from the southwest region were distributed in the other three groups. We therefore suggest that southwestern China was a primitive domestication center of the Chinese plum; the genetic diversity of the SCG was lower than that of the FG and MG, which could be explained by the presence of artificially hybridized cultivars in the FG and MG.
The clustering results in Fig. 2a showed that, for the five newly bred cultivars from the NGRPA in the MG, one of their parents was from the USA (“Blackamber,” “Friar”) or Japan (“Akihime”) and the other parent was native to China, and also that gene flow occurred between the SCG and FG (Fig. 4). Thus, we assumed that the MG cultivars were genetically related. As mentioned in other studies, including a dendrogram analysis (Liu et al. 2006; Liu et al. 2007; Wei et al. 2019), most of the Chinese plum cultivars from Japan and the improved Chinese plum hybrids from the USA were distributed across the Chinese indigenous plum group. The P. salicina-predominant genetic components of improved Chinese plum hybrids (Faust and Suranyi 1999) the view that plum cultivars, originating in China, were initially introduced to Japan, and then exported from Japan to the USA in 1870 by Luther Burbank (Hartmann and Neumulle 2009).
Genetic differentiation between populations is considered moderate when the FST value is higher than 0.05, and highly differentiated when the FST value is higher than 0.15 (Xu 2009). In our study, the NCG-FG pair possessed the highest FST value, followed by the SCG-FG pair. In contrast, the FST value of the MG-FG was significantly lower, as there were five newly selected plum cultivars by the NGRPA in the MG with foreign lineage from the FG. Human intervention, artificially selecting favorable phenotypic traits to enhance production and improve desirable agronomic traits, can reduce the levels of genetic variability and skew allele frequencies (Tajima 1989).
The present study is the first in which we constructed high-quality SNPs from 67 accessions in the Chinese plum by whole-genome resequencing, with the peach genome as the reference. Our findings show that the domestication center of origin of the Chinese plum was in the southwest region of China. This study, in providing genetic variation features of plum cultivars and an analysis of their genetic diversity and population structure, lays a foundation for breeders to use diverse germplasm and allelic variants to improve Chinese plum varieties.
References
Acuña CV, Rivas JG, Brambilla SM, Cerrillo T, Frusso EA, García MN, Villalba PV, Aguirre NC, Sabio y García JV, Martínez MC, Hopp EH, Marcucci Poltri SN (2019) Characterization of genetic diversity in accessions of Prunus salicina Lindl: keeping fruit flesh color ideotype while adapting to water stressed environments. Agronomy 9:487
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Arús P, Verde I, Sosinski B, Zhebentyayeva T, Abbott AG (2012) The peach genome. Tree Genet Genomes 8:531–547
Ben Tamarzizt H, Ben Mustapha S, Baraket G, Abdallah D, Salhi-Hannachi A (2015) Assessment of genetic diversity and relationships among wild and cultivated Tunisian plums (Prunus spp.) using random amplified microsatellite polymorphism markers. Genet Mol Res 14:1942–1956
Cao K, Zheng Z, Wang L, Liu X, Zhu G, Fang W, Cheng S, Zeng P, Chen C, Wang X, Xie M, Zhong X, Wang X, Zhao P, Bian C, Zhu Y, Zhang J, Ma G, Chen C, Li Y, Hao F, Li Y, Huang G, Li Y, Li H, Guo J, Xu X, Wang J (2014) Comparative population genomics reveals the domestication history of the peach, Prunus persica, and human influences on perennial fruit crops. Genome Biol 15:1–15
Cao K, Zhou Z, Wang Q, Guo J, Zhao Pei, Zhu Gengrui, Fang Weichao, Chen Changwen, Wang Xinwei, Wang Xiaoli, Tian Zhixi, Wang Lirong (2016) Genome-wide association study of 12 agronomic traits in peach. Nat Commun 7. https://doi.org/10.1038/ncomms13246
Carrasco B, González M, Gebauer M, García-González R, Maldonado J, Silva H (2018) Construction of a highly saturated linkage map in Japanese plum (Prunus salicina L.) using GBS for SNP marker calling. PLoS One 13:1–14
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, Vean GM, Durbin R (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Ken K, Buckler ES, Mitchell SE, Orban L (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:1–10
Faust M, Suranyi D (1999) Origin and dissemination of plum. Hortic Rev 23:179–231
Food and Agriculture Organization of the United Nations (2018) World production of plums and sloes. http://www.fao.org/faostat/zh/#data/QC. Accessed 23 April 2020
Guo Z (2006) Collection and utilization of Prunus salicina germplasm resource in South China. [Doctor’s thesis]. [Nanjing]: Nanjing Agricultural University
Hartmann W, Neumulle M (2009) Plum breeding. In: Jain SM, Priyadarshan PM (eds) Breeding Plantation Tree Crops: Temperate Species. Springer Science Business Press, Media, pp 161–231
Hazzouri KM, Flowers JM, Visser HJ, Khierallah HSM, Rosas U, Pham GM, Meyer RS, Johansen CK, Fresquez ZA, Masmoudi K, Haider N, El Kadri N, Idaghdour Y, Malek JA, Thirkhill D, Markhand GS, Krueger RR, Zaid A, Purugganan MD (2015) Whole genome resequencing of date palms yields insights into diversification of a fruit tree crop. Nat Commun 6:8824
Kawuki RS, Ferguson M, Labuschagne M, Herselman L, Kim D (2009) Identification, characterisation and application of single nucleotide polymorphisms for diversity assessment in cassava (Manihot esculenta Crantz). Mol Breed 23:669–684
Khush G (2002) Molecular genetics-plant breeder’s perspective. In: Jain SM, Brar DS, Ahloowalia BS (eds) Molecular techniques in crop improvement. Kluwer Academic Press, Dordrecht, pp 1–8
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079
Li W, Liu L, Wang Y, Zhang Q, Fan G, Zhang S, Wang Y, Liao K (2020) Genetic diversity, population structure, and relationships of apricot (Prunus) based on restriction site-associated DNA sequencing. Hortic Res 7:69
Li Y, Cao K, Zhu G, Fang W, Chen C, Wang X, Zhao P, Guo J, Ding T, Guan L, Zhang Q, Guo W, Fei Z, Wang L (2019) Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history. Genome Biol 20:36
Liu C, Feng C, Peng W, Hao J, Wang J, Pan J, He Y (2020) Chromosome-level draft genome of a diploid plum (Prunus salicina). GigaScience 9(12):130
Liu W (2005) Studies on the genetic diversity among plum germplasm resources and the phylogenetic relationships of main plum species. [Doctor’s thesis]. [Beijing]: China Agricultural University
Liu W, Liu D, Feng C, Zhang A, Li SH (2006) Genetic diversity and phylogenetic relationships in plum germplasm resources revealed by RAPD markers. J Hortic Sci Biotechnol 81:242–250
Liu W, Liu D, Zhang A, Feng C, Yang J, Yoon J, Li SH (2007) Genetic diversity and phylogenetic relationships among plum germplasm resources in China assessed with inter-simple sequence repeat markers. J Am Soc Hortic Sci 132(5):619–628
Marti AFI, Saski CA, Manganaris GA, Gasic K, Crisosto CH (2018) Genomic sequencing of Japanese plum (Prunus salicina Lindl.) mutants provides a new model for rosaceae fruit ripening studies. Front. Plant Sci 9:21
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8(11):e1002967
Pinosio S, Marroni F, Zuccolo A, Vitulo N, Mariette S, Sonnante G, Aravanopoulos FA, Ganopoulos I, Palasciano M, Vidotto M, Magris G, Iezzoni A, Vendramin GG, Morgante M (2020) A draft genome of sweet cherry (Prunus avium L.) reveals genome-wide and local effects of domestication. Plant J 103(4):1420–1432
Pop R, Hârţa M, Szabo K, Zanescu M, Sisea CR, Catana C, Pamfil D (2018) Genetic diversity and population structure of plum accessions from a Romanian germplasm collection assessed by simple sequence repeat (SSR) markers. Not Bot Hort Agrobot 46(1):90–96
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Salazar JA, Pacheco I, Silva C, Zapata P, Shinya P, Ruiz D, Martínez-Gómez P, Infante R (2019) Development and applicability of GBS approach for genomic studies in Japanese plum (Prunus salicina Lindl.). J. Hortic. Sci. Biotechnol. 94(3):284–294
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Topp BL, Russell DM, Neumuller M, Dalbo M, Liu WS (2012) Plum. In: Badenes ML, Byrne DH (eds) Fruit Breeding. Springer Science Business, Media, pp 571–621
Urrestarazu J, Errea P, Miranda C, Santesteban LG, Pina A, Pang XM (2018) Genetic diversity of Spanish Prunus domestica L. germplasm reveals a complex genetic structure underlying. PLoS One 13:1–21
Verde I, Abbott AG, Scalabrin S, Jung S, Shu SQ, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F, Zuccolo A, Rossini L, Jenkins J, Vendramin E, Meisel LA, Decroocq V, Sosinski B, Prochnik S, Mitros T, Policriti A, Cipriani G, Dondini L, Ficklin S, Goodstein DM, Xuan P, Fabbro CD, Aramini V, Copetti D, Gonzalez S, Horner DS, Falchi R, Lucas S, Mica E, Maldonado J, Lazzari B, Bielenberg D, Pirona R, Miculan M, Barakat A, Testolin R, Stella A, Tartarini S, Tonutti P, Arús P, Orellana A, Wells C, Main D, Vizzotto G, Silva H, Salamini F, Schmutz J, Morgante M, Rokhsar DS (2013) The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45(5):487–494
Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E (2009) EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19:327–335
Wang K, Li M, Hakonarson H (2010) ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164
Wei X, Zhang Q, Liu N, Zhang Y, Liu S, Zhang Y, Ma X, Liu W (2019) Genetic diversity of the Prunus salicina L. from different Sources and their related species. Sci Agric Sin 52(3):568–578. https://doi.org/10.3864/j.issn.0578-1752
Wei X, Zhang Q, Liu W (2020) Research progress on plum germplasm resources in China. Acta Hortic Sin 47(6):1203–1212
Wu W, Chen F, Yeh K, Chen J (2019) ISSR analysis of genetic diversity and structure of plum varieties cultivated in Southern China. Biology (Basel) 8:1–13
Xu G (2009) Plant population genetics. Science Press, Beijing
Yu X, Zhang Q, Liu W, Sun M, Liu N, Zhang Y, Xu M (2011) Genetic diversity analysis of morphological and agronomic characters of Chinese plum (Prunus salicina Lindl.) germplasm. J. Plant. Genet. Resour. 12(3):402–440
Zhang J (1990) Report on the investigation for national plum and apricot resources. China Fruits 4:29–34
Zhang J, Zhou E (1998) China fruit-plant monographs, plum flora. China Forestry Press, Beijing
Zhang Q, Wei X, Liu W, Liu N, Zhang Y, Xu M, Liu S, Zhang Y, Ma X, Dong W (2018) The genetic relationship and structure of some natural interspecific hybrids in Prunus subgenus Prunophora, based on nuclear and chloroplast simple sequence repeats. Genet Resour Crop Evol 65:625–636
Zhang Q, Wei X, Liu N, Zhang Y, Xu M, Zhang Y, Ma X, Liu W (2020) Construction of an SNP-based high-density genetic map for Japanese plum in a Chinese population using specific length fragment sequencing. Tree Genet Genomes 16:18
Zhebentyayeva T, Shankar V, Scorza R, Callahan A, Ravelonandro M, Castro S, DeJong T, Saski CA, Dardick C (2019) Genetic characterization of worldwide Prunus domestica (plum) germplasm using sequence-based genotyping. Hortic Res 6:12
Data archiving statement
All of the raw reads of the plum accessions generated in this study have been deposited in the public database of National Center of Biotechnology Information under PRJNA659814.
Funding
This work was supported by National Natural Science Foundation of China (31401826), the Program of Conservation and Utilization of Crop Germplasm Resources (2018NWB003) and National Crop Germplasm Resources Platform of China (NICGR2018-056).
Author information
Authors and Affiliations
Contributions
ZQP and LWS conceived and designed the experiments. WX and SF performed the experiments. WX, SF and ZQP analyzed the data and wrote the paper. LN, ZYP, XM, LS, MXX, and ZYJ collected samples and managed the materials. LWS revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by W.-W. Guo
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, X., Shen, F., Zhang, Q. et al. Genetic diversity analysis of Chinese plum (Prunus salicina L.) based on whole-genome resequencing. Tree Genetics & Genomes 17, 26 (2021). https://doi.org/10.1007/s11295-021-01506-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-021-01506-x