
Abstract
• Key message
Understanding the adaptive mechanisms of forest species is vital to ensure their survival in a climate change scenario. This study aimed at uncovering the relationship between genetic variability and environmental variables in natural Castanea sativa populations, unveiling how different climate scenarios drove local adaption processes using a landscape genomics approach. Our findings provide useful data for future management of this species.
• Context
Temperate forest species, such as chestnut (Castanea sativa Mill.), are currently threatened by increasing temperature together with disruption and reduction of precipitation due to climate change. In this context, understanding the adaptation processes of species will help to manage and ensure the conservation of forests.
• Aims
We studied the relationship between genetic variability and climate variables in natural populations of C. sativa using a landscape genomics approach aimed to identify local adaption processes.
• Methods
Using five genomic SSRs and eight functional EST-SSRs markers, 268 individuals belonging to ten different natural European chestnut populations distributed in contrasting climatic sites were genotyped. In addition, associations between allelic variation and climatic variables (environmental association analyses approach) were performed using Samβada and LFMM.
• Results
Results highlighted a strong inter-relationship between climate variables and evolutionary processes resulting in adaptive variation. STRUCTURE analysis based on functional markers split the populations in three separate gene pools (K = 3), mostly in agreement with the different climatic conditions existing in the studied areas. Divergent spatial patterns of genetic variation between rainy and arid areas were found. We detected a total of 202 associations with climate among 22 different alleles, 9% of which related with the outlier locus FIR059, known to be implicated in regulatory mechanisms during water stress adaptation processes.
• Conclusion
Landscape genomics analyses revealed a pattern of adaptive variation, where specific climatic variables influenced the frequencies distribution and fixation of several alleles, resulting in local adaptation processes of the populations in the investigated areas. Our findings underline the close inter-relationship existing between climate and genetic variability and indicate how this approach could provide valuable information for the management of forest species in a rapidly changing environment.
Similar content being viewed by others

1 Introduction
Past and current climate changes have affected the pattern of genetic diversity and genetic structure of extant tree species. However, there is evidence that the intensity of the current climate change is higher than the ability of trees to adapt to changing conditions (Davis et al. 2005; Aitken et al. 2008). The survival of forests is highly dependent on the genetic variation within and among populations (Barrett and Schluter 2008), and for this reason, it is essential to evaluate the genetic variability of trees. Adaptive genetic variation is defined as the variation found between the genomes of individuals resulting from natural selection. Local environmental conditions can induce spatially varying selective pressure, which directly affects adaptive genetic variation by favoring different alleles in different spatial localities (Hedrick et al. 1976; Schoville et al. 2012; Richardson et al. 2014). In view of current climate change, the potential ability of trees to adapt to different environmental conditions should also be studied.
The Fst outlier test is a widely used approach to study local adaptation by detecting loci putatively under divergent selection. Loci exhibiting a non-neutral pattern of variation, with a higher or lower genetic differentiation than expected under neutrality, are to be considered under selection (Narum and Hess 2011). However, the key drawback of this type of approach is the risk of detecting false positives. In fact, loci with Fst values that deviate significantly from neutrality may be due to locus-specific effects (i.e., adaptive selection, mutation, assortative mating, and recombination), or due to genome-wide effects (i.e., genetic drift, bottlenecks, and gene flow). To avoid this uncertainty, Fst outlier analysis can be combined and supported by a landscape genomics approach. Landscape genomics focuses on understanding the interactions between environmental heterogeneity and adaptive genetic variation in natural populations. This is possible through environmental association analysis (EAA), which identifies genetic variants associated with particular environmental factors and has the potential to uncover adaptive patterns that are not discovered by traditional tests for the detection of outlier loci based on population genetic differentiation (Eckert et al. 2009a, 2010b; Schoville et al. 2012). In other words, EAA relates genomic information with environmental variables in order to reveal signatures of adaptive genetic variation and evolutionary processes (Joost et al. 2007; Coop et al. 2010; Stucki et al. 2017). Several statistical models to perform EAA have been developed, and at least two models should be combined in order to obtain reliable results (Rellstab et al. 2015). Recent landscape genomics studies on forest species combined outlier locus detection with environmental association analyses (Bradbury et al. 2013; Cuervo-Alcaron et al. 2018; Martins et al. 2018). Additional models considering the interaction between adaptive genetic variation and multiple environmental gradients have also been developed. One such tool is Gradient Forests (Ellis et al. 2012) which models and maps turnover in allele frequencies along environmental gradients.
The sweet chestnut is the only species in the genus Castanea in Europe and is widespread throughout the Mediterranean basin, ranging from the Black Sea region to the Atlantic coast of the Iberian Peninsula (Fineschi et al. 2000; Maurer and Fernández-López 2001). Its current distribution is the consequence of natural colonization together with a long history of human intervention, with the first documented domestication attempts dating back to the Roman Empire and Ancient Greece (Conedera et al. 2004; Mattioni et al. 2013). Sweet chestnut has a high economic value, mainly due to the consumption of its fruits and the production of timber. Because of the existing climatic variability along the distribution of chestnut in Europe and because the susceptibility of chestnut to climate change events (Camisón et al. 2020), it is an ideal species for studying neutral and adaptive genetic variability.
Microsatellite markers (simple sequence repeat (SSR)) are powerful tools for genetic diversity and evolutionary process studies in forest species (Tuskan et al. 2004; Yin et al. 2004; Varshney et al. 2005). A vast number of expressed sequence tags (ESTs) are available for many plant species, some of them including expressed sequence tag (EST)-SSR markers linked to transcribed regions of the genome, with known or suggested functions (Scott et al. 2000; Krutovskii and Neale 2001; Kalia et al. 2011). Although EST-SSR are less polymorphic compared to genomic SSRs, they are more suited to reveal functional diversity in relation to adaptive variation (Varshney et al. 2005; Yatabe et al. 2007) and are thus used to understand local adaptation processes. EST-SSRs associated to drought stress have been reported in Quercus spp. (Sullivan et al. 2013; Lind and Gailing 2013), chestnut (Martín et al. 2010; Alcaide et al. 2019), and walnut (Torokeldiev et al. 2019). To study the adaptive variability and the processes involved in local adaptation of sweet chestnut, genomic and EST-SSR markers, and natural populations selected in areas with considerable differences in climatic conditions were used.
The main objectives of this study were to (1) perform simulations of chestnut distribution in view of climate change; (2) identifying signatures of adaptive variation of the populations in relation with the local climatic variables; (3) identify markers with signatures of selection performing outlier tests analysis; (4) associate these markers with climate variables of the population through a landscape genomics approach.
2 Materials and methods
2.1 Tree populations
Sweet chestnut has a very fragmented distribution within Europe, ranging from the regions of the Caucasus to the Atlantic coast of Portugal. In this study, 268 individuals from 10 different natural populations located in four European countries (Turkey, Greece, Italy, and Spain) were analyzed (Table 1; Fig. 1). The chestnut populations were chosen based on their geographic location (Castellana 2020) and considering the different climatic conditions for each location in terms of rainfall events and temperatures (Appendix Fig. 5). The populations from Northern Italy, Northern Spain, and East Turkey (Villar Pellice IT08, Costa Atlántica SP03, Hopa TR03) are located in humid environments, characterized by heavy rainfall and low-to-moderate temperatures throughout the year. The southern Italian (Madonie IT01), the Spanish (Castanyet SP02, Hervás SP06, Sierra Norte SP14), the two Greek (Holomontas GR01; Hortiatis GR02), and western Turkish (Bursa TR11) populations are characterized by low rainfall and moderate-to-high temperatures throughout the year. All the populations studied are part of the germplasm collection of European chestnut populations conserved at the Institute of Research on Terrestrial Ecosystems; the samples have been collected in the field during several years and subsequently stored at − 80 °C.

Geographical distribution of the 10 Castanea sativa populations studied
2.2 Climatic scenarios of populations
To compare present and future scenarios of tree populations, in view of climate change, twenty-one climatic variables for each population were used (Table 2). Data were obtained from WORLDCLIM “bioclimatic variables” digital dataset (Fick and Hijmans 2017). For present data, version 2.1 has been used; the future projection data used in this study are CMIP6 downscaled from Global Climate Model (GCM) BCC-CSM2-MR and Shared Socio-economic Pathways (SSP) 245. The spatial resolution used was 2.5 min. The use of a large number of environmental factors may increase the number of statistical tests during association analysis, which needs to be considered in order that autocorrelation could be reduced. Furthermore, we looked for correlation between variables through Pearson’s correlation coefficient (|r| > 0.8) using the “ggpubr” package implemented in R (R Development Core Team 2019). Graphical output was achieved with the package “corrplot” (Appendix Fig. 6). Subsequently to highlight correlations, a principal components analysis (PCA) was performed with the “FactoMineR” package (Lê et al. 2008).
Using the climatic data in the DIVA-GIS software (Hijmans et al. 2004), it was possible to represent present and future climatic scenarios for Europe (Appendix Fig. 5). Present and future climatic conditions referred to years 2020 and 2050, respectively. For each of the ten sampling locations, average minimum monthly temperatures, average maximum monthly temperatures, average minimum annual temperature, average maximum annual temperature, average monthly rainfall, and average annual rainfall were collected. Through the “Ecocrop” function implemented in DIVA-GIS, it was possible to determine the areas in which chestnut performs best in present and future climatic conditions (Fig. 2). EcoCrop module uses FAO’s EcoCrop database (FAO 2019) of the environmental requirements of a long list of plant species, including sweet chestnut.

Representation of the areas with best suitability for chestnut development in reference to 2020 (a) and 2050 (b)
2.3 DNA isolation, SSR amplification, and genotyping
DNA extraction was performed with the DNAeasy 96 Plant Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s protocol. The amount and quality of extracted DNA was visualized through electrophoresis in agarose gel at 1% with 5 × TAE as running buffer. All individuals were genotyped with 13 SSR markers (Castellana 2020). Five were gSSR markers developed in C. sativa (Appendix Table 7) (Marinoni et al. 2002; Buck et al. 2003) and eight were EST-SSR markers. The EST-SSR markers were developed from gene expression during tree exposure to water stress in Quercus robur (Durand et al. 2010) and selected based on information about their polymorphism linkage group and potential transferability to C. sativa (Alcaide et al. 2019) (Appendix Table 8). Three different PCR amplification multiplexes were assembled based on the size of products, using fluorescent dye-labelled primers (6-FAM, VIC, NED, PET; Applied Biosystems, Foster City, California, USA). Mixtures were arranged as follows: mix (A) including CsCAT1, CsCAT3, CsCAT6, (B) CsCAT16, EMCs38 (C) FIR080, GOT004, GOT021, VIT057, and (D) FIR059, FIR094, GOT045, and VIT033 primers. Amplifications were performed with the Type-It® Microsatellite PCR Kit (Qiagen, Valencia, CA, USA). PCR mix consisted of 4 µL of genomic DNA, 6.25 µL of 2× Type-it Multiplex PCR Master Mix, 1.25 µL of 10× Primer Mix (2 µM of each primer) and 1 µL of RNase-free water for a total volume of 12.5 µL. Amplification conditions were as follows: an initial heat activation step at 95 °C for 5 min, followed by 27 cycles consisting of a denaturation step at 95 °C for 30 s, an annealing step at 57° for 1.5 min, and an extension step at 72 °C for 30 s. A final extension step at 60 °C for 30 min was executed. PCR fragments have been run on an ABI PRISM® 3130 XL Genetic Analyzer for separation and sizing. GeneScan 250 LIZ was used as an internal size standard. Genotyping was performed using GeneMapper v4.0 software (Applied Biosystems, Foster City, USA).
2.4 Genetic diversity and structure of populations, and Fst outlier test
The probability of null alleles (Fnull) for each of the 5 gSSR and 8 EST-SSR loci analyzed was tested using the software FreeNA (Chapuis and Estoup 2007).
For each population and separately for neutral markers (gSSR) and EST-SSR, the number of alleles (Na), observed and expected heterozygosity (Ho, He), fixation index (Fis), and pairwise Fst were calculated using GeneAlex 6.503 (Peakall and Smouse 2012). Allelic richness (Ar) was evaluated through the use of HP-Rare (Kalinowski 2005). Significance test for the Fis values and the molecular variance (AMOVA) were calculated with Arlequin 3.1.1 software (Excoffier et al. 2005). Population structure was inferred using a Bayesian approach as implemented in the STRUCTURE 2.3.4 software (Pritchard et al. 2000), separately for the genomic gSSR and EST-SSR. Both analyses used the admixture model with correlated allele frequencies. Parameters were set for a burn-in period of 100,000 and a MCMC (Markov chain Monte Carlo) with 200,000 iterations. The range of K tested was equal to the number of the populations analyzed plus one, i.e., 11. Potential clusters (K) were tested using 20 iterations. To determine the most likely number of K, the ΔK method by Evanno et al. (2005) was applied using STRUCTURE HARVESTER (Dent and Von Holdt 2012) A graphical representation of the STRUCTURE results was performed using CLUMPAK (Kopelman et al. 2015).
Detection of Fst outliers was performed using BayeScan 2.1 (Foll et al. 2008). The eight EST-SSR and five gSSR loci were tested for the evidence of the effect of natural selection among all populations. The underlying geographic genetic structure was assessed during the detection of the Fst outlier; hence, analyses were ran first based on all populations together, then by comparing eastern vs western populations and finally by comparing the different genetic pools highlighted by the structure analyses. The program has been executed with twenty pilot runs with a length of 5,000 and a burn-in length of 50,000. The thinning interval was set to 50. Significant loci with positive alpha values were considered candidates for diversifying selection, according to Jeffrey’s scale of evidence (Jeffrey 1961).
2.5 Environmental association analysis of populations
To perform environmental association analysis (EAA), two different models were used: a logistic regression implemented in the Samβada software (Stucki et al. 2017), and a Bayesian mixed hierarchical model implemented in the software Latent Factor Mixed Model (LFMM) (Frichot and Francois 2013, 2015). The associations between climate variables and genetic variability were tested for all gSSR and EST-SSR markers. Both analyses were performed considering the underlying neutral genetic structure; this step is of fundamental importance; not taking into account the underlying neutral genetic structure can lead to false positives discoveries. If not corrected for the neutral genetic structure, the identification of associations could be the result of spatial arrangement and the demographic history of the populations, and not a sign of local adaptation; the underlying neutral genetic structure can mimic patterns expected under non-neutral processes. With the results obtained from STRUCTURE 2.3.4 software for gSSR markers, a multivariate analysis using the coefficients of membership (Q) for each individual was run, and the G scores to assess significance were calculated. As a second approach, a latent factor mixed model (LFMM) implemented in the R package LEA was performed. In this model, neutral population structure is introduced as a “latent factor.” The number of detected clusters (K) calculated by STRUCTURE were applied. Ten LFMM repetitions with 100,000 iterations and 10,000 burn-in were performed for each climate variable. Z-scores of multiple runs were combined using the median value, and p-values were adjusted for expected FDR at 0.05.
Although Samβada and LFMM are able to reveal associations between genetic variation and climate variables, none of these two approaches quantifies the contribution of each variable in the overall genetic structure of chestnut populations. Through the use of the “gradientForest” package (Ellis et al. 2012) implemented in R, it was possible to quantify the contribution of each climate variable on the allelic frequency variation in chestnut. GradientForest is capable of partitioning the allele frequency data at split values along climate gradients, allowing the exploration of nonlinear associations of climatic and allelic variables.
3 Results
3.1 Present and future climate conditions
A comparison between present and future climate scenarios within chestnut populations (Appendix Fig. 5) revealed an average increase of mean temperatures of 2.21 °C. Areas with the highest increase in temperatures were coincident with Bursa (TR11) and Sierra Norte (SP14) populations and showed an increase of 2.45 °C. All populations except Madonie (IT01), with an increase of 27 mm of rainfall, showed a decrease in rainfall of 85 mm in average. Costa Atlántica (SP03) showed the highest reduction of rainfall, i.e. 227 mm. The EcoCrop module showed a considerable reduction of areas with the best conditions for the development of chestnut in Europe (Fig. 2). Generally, areas with the best conditions for chestnut development were displaced to higher latitudes and altitudes.
3.2 Genetic diversity, population structure, and outlier loci
For both gSSR and EST-SSR loci, no high frequency of null alleles was detected; Fst including null alleles (INA) was 0.1063 and 0.2272 for gSSR and Est-SSR, respectively, while the Fst excluding null alleles (ENA) was 0.1015 and 0.2174 for gSSR and EST-SSR, respectively. Overall, neutral markers (gSSR) showed higher values for the genetic diversity indices per population compared to EST-SSR. Expected heterozygosity (He) for gSSR ranged from 0.639 (Hortiatis GR02) to 0.824 (Hervás SP06) with a mean value of 0.764 (Table 3). EST-SSR expected heterozygosity (He) ranged from 0.284 (Holomontas GR01) to 0.501 (Costa Atlántica SP03) with a mean value of 0.389 (Table 4). The fixation index (Fis) was significantly different from zero (p < 0.01, p < 0.001) in several populations, both for gSSR (Bursa TR11, Hopa TR03, Villar Pellice IT08, Holomontas GR01) and EST-SSR (Hervás SP06, Bursa TR11, Hopa TR03, Villar Pellice IT08). Overall, allelic richness (Ar) showed higher mean values for neutral markers (Ar = 7.95) compared to functional markers (Ar = 3.01). The highest values of Ar were observed in the Hopa (TR03) population, for both neutral and functional markers. The results provided by STRUCTURE, considering both the delta K method by Evanno et al. (2005) and the distribution of the posterior log-likelihood (Appendix Figs. 7 and 8 ) have been checked. Based on the higher deltaK and on the lowest variance of the L(K) distribution, the most likely number of clusters (K) were K = 2 for gSSRs and K = 3 for EST-SSRs.
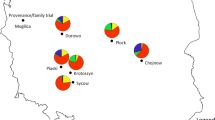
Results for the population structure based on the gSSR highlighted two core genetic population groups, separating east (Greek and Turkish populations) from west populations (Italian and Spanish) (Fig. 3a). On the other hand, the population genetic structure revealed by the EST-SSR was congruent with the different climatic conditions of the study areas (Fig. 3b, Appendix Fig. 5). Group I (blue colored in Fig. 3b) comprised Bursa (TR11), Holomontas (GR01), Hortiatis (GR02), Hervás (SP06), and Sierra Norte (SP14) populations, located in areas characterized by low precipitation and high temperature throughout the year (Appendix Fig. 5). The Madonie (IT01) population fell within group II (purple colored in Fig. 3b); this population is located at 1.100 m MSL, and the site is characterized by a low temperature and low precipitations and a xerothermic index (Xi) of 110 (Mattioni et al. 2008). The Hopa (TR03) population, located in a highly rainy area, belonged to the group III (orange color). The Castanyet (SP02) population, found in an area characterized by low mean annual temperature and low precipitations, showed some admixture with groups I and II, while Villar Pellice (IT08) and Costa Atlántica (SP03), located in areas with low mean annual temperature and high precipitations, showed some admixture with groups II and III. All the less significant structures and the sub-structures are reported in Appendix Fig. 9. For all of the approaches tested for the detection of outlier loci, BayeScan 2.1 analysis highlighted only the locus FIR059 as putatively under positive selection (Table 5). On the basis of Jeffrey’s scale of evidence, locus FIR059 was identified as a ‘decisive’ outlier candidate for diversifying selection. No sign of selection was highlighted for any of the other EST-SSR and gSSR markers (Appendix Fig. 10).

Population genetic structure of ten sweet chestnut populations grouped into clusters I (blue color) and II (orange) according to gSSRs markers (K = 2) (a) and grouped into clusters I (blue), II (purple) and III (orange) according to EST-SSR markers (K = 3) (b)
3.3 Environmental associations
To reduce the risk of false positive loci discoveries, (i) EAA analyses was performed with two different models, (ii) associations between genetic variation and climate variables were corrected for neutral genetic structure, (iii) only associations for the outlier locus FIR059 were considered, and (iv) for FIR059, only alleles showing signs of associations with a climate variable in both Samβada and LFMM software applications were considered. The variables highlighted by Pearson’s test as being highly correlated (i.e., with |r| > 0.8; Appendix Fig. 6) were found to be the same as those of the PCA (Appendix Fig. 11). Pearson’s analysis revealed highly correlated climatic variables (|r| > 0.8), and for each set of correlated variables only one variable was selected. The resulting set of associated climatic variables and the ones selected for the analysis are shown in the Appendix Table 9. Variables AnnPrec and MeanAnnT were removed from subsequent analyses as they were highly correlated with all variables related to rainfall and temperatures. Samβada highlighted 14 alleles out of 54 that were associated with at least one climate variable, across seven EST-SSR. No associations were found for the GOT004 marker. Most alleles showed an association with more than one climate variable. LFMM analyses identified 20 out of the 54 alleles associated with at least one climate variable across eight EST-SSR. Again, no significant associations were found for the GOT004 marker. A total of 98 significant associations were highlighted by the two models, 27 of which were shared between Samβada and LFMM (Appendix Table 10). Seven associations for three different alleles were observed for FIR059 according to Samβada (Table 6). For the same locus, LFMM revealed 19 associations across 9 alleles, showing a higher potential for discovery than Samβada (Table 6).
3.4 Contribution of climate variables in structuring genetic variation
GradientForest revealed climatic variables related to precipitation as key predictors for genetic variation at the locus FIR059. Of these, precipitation during the wettest quarter (PrecWeQ) was the most relevant (i.e., highest R2 values), followed by precipitation during the driest quarter (PrecDQ) (Fig. 4). The variables showing a greater contribution to the variation in allele frequencies, overlapped with those that showed significant signs of association by Samβada and LFMM. Allelic changes along the environmental gradients showed an important variation along the variable “PrecWeQ,” with a spike in the variability between 350 and 550 mm of rainfall (Fig. 4b).

(a) Importance in terms of R2 weight of climate variables related to alleles of locus FIR059. (b) Cumulative importance of allelic change for the locus FIR059 along most important environmental gradients
4 Discussion
We evaluated the adaptive genetic variability of sweet chestnut populations and identified genomic regions that might be involved in local adaptation processes in response to climate conditions. In order to prevent spurious correlations not directly related to adaptation (Novembre and Di Rienzo 2009), both gSSR and EST-SSR markers were used to assess neutral and adaptive components of genetic variability. Previous studies on chestnut have identified outlier loci related to bud burst (Martín et al. 2010), tolerance to drought (Alcaide et al. 2019), and resistance to Phytophthora cinnamomi (Alcaide et al. 2020).
The growing interest in the impact of climate change on forest ecosystems has produced a vast amount of documentation over the last twenty years. Forest ecosystems are at great risk, including trees with an economic value in terms of production, such as the sweet chestnut. The estimated loss of areas with suitable conditions for the growth of chestnut, due to climate change (Fig. 2b) provides additional information in forestry and underlines the importance of studying adaptation in tree species. From an agricultural perspective, many nut trees require a certain amount of winter chill hours for proper fruit development (Byrne and Bacon 1992). The effect of prolonged water stress, prolonged waterlogging and chill hour reduction due to climate change on nut trees has already been documented (Luedeling et al. 2011; Camisón et al. 2020). Combined with this, a rainfall deficit and extreme summer heat in Europe can lead to a severe reduction in nut productivity (Ciais et al. 2005).
4.1 Genetic diversity and population structure of chestnut in Europe
Higher genetic diversity was observed for neutral markers in comparison to adaptive ones. As EST-SSR markers are linked to coding regions, they tend to be more conserved and, consequently, less polymorphic than neutral markers (Varsheney et al. 2005; Ellis and Burke 2007). Several other studies assessing the genetic diversity of plants species confirmed the higher polymorphism of genomic markers in comparison to functional markers (Şelale et al. 2013; Torokeldiev et al. 2019). The genetic diversity indices indicate slightly higher values for the western populations compared to those of the east; this result is in contrast to previous studies carried out on vascular plants in Europe, which see a tendential decrease in genetic diversity moving from east to west (Fady and Conord 2010; Conord et al. 2012). Here, the studied chestnut populations are a subsample of others genotyped in a previous work by Mattioni et al. (2017), in which higher values of He and Ar were observed in the central area of the species distribution. Hence, higher values of genetic diversity for the western populations can be a consequence of several colonization routes from different refugees, as observed in other species (Petit et al. 2006) as well as of human intervention.
The positive values of Fis observed in some populations for both types of molecular markers are probable due to factors such as the fragmentation and isolation of populations. It is notable that in some populations, considered as refugia during the last glaciation, the allelic richness and private allelic richness at neutral loci is high. This result is in agreement with those obtained in a previous paper on C. sativa (Mattioni et al. 2017) and Fagus selvatica (Comps et al. 2001), and it is supported by the hypotheses of Widmer and Lexer (2001) on high values of allelic richness on rifugia areas. On the other hand, the high value of private allelic richness at EST-SSr loci observed especially for Hopa population could reflect a selection of specific alleles due to adaptation.
STRUCTURE analyses produced different results for neutral and functional markers. When considering gSSR, STRUCTURE highlighted a congruence between the genetic diversity and geographical location of the populations: clear separation between eastern and western European populations. This demonstrates that the Carpathian and Balkanian mountain chain acted as a barrier, blocking the gene flow between two macro-regions. In Mattioni et al. (2017), where a large number of natural sweet chestnut populations have been genotyped with neutral markers, similar results were reported. Three different genetic groups were highlighted by the EST-SSR markers. The resulting structure clustered together populations collected in areas characterized by similar climatic conditions. Trees from Hopa (TR03), located in a region with heavy rainfall throughout the year (annual average of 1,227 mm) and moderate-to-low temperatures, were included in a well-defined genetic cluster (III). Similar climate conditions, referring to precipitations, can be found in Galicia (SP03) and Piemonte (IT08) regions; this could explain the membership of several individuals from these populations to the group III. Moreover, the two Spanish (SP02, SP03) and the Italian (IT08) populations are located in areas with low mean annual temperature, as well as the population IT01, that is located at 1100 m of altitude. We can suppose that a climatic variable as the low temperature could explain the genetic similarity of some individuals of these populations.
Results suggest that the selected EST-SSR markers were informative and able to differentiate populations based on climatic conditions. Indeed, we have to underline that our results have been achieved with a limited numbers of functional markers; further research, using a more consistent set of markers ideally found within transcribed regions of the genome are needed to better understand adaptive processes of chestnuts populations to different environmental conditions.
4.2 Environmental association analysis of chestnut in Europe
Compared to the Fst outlier tests, EAA analyses are more sensitive to subtle changes in allele frequencies and generally tend to be more robust (De Mita et al. 2013; Ahrens et al. 2018). Combining Fst with EAA was the best approach to maximize the probability of finding significant associations and to minimize the risk of detecting false positives (Rellstab et al. 2015). None of the five neutral markers showed association with climatic variables. This lack of association reinforces the hypothesis of the neutrality of the selected markers. As they are linked to non-coding regions of the genome, they do not show signs of intervention in adaptive processes. However, EAA highlighted 98 significant associations between EST-SSR markers and climatic variables. LFMM showed a slightly higher potential for discovering environmental associations than Samβada (59 vs. 39). Among the nine alleles, for which associations with climate variables were found for the locus FIR059, three of these were shared with the LFMM and Samβada, specifically for alleles 152, 181 and 185. The associations found for allele 152 were mainly related to rainfall. Interestingly, allele 152 showed an almost total exclusivity for individuals from the Hopa (TR03) population (Appendix Fig. 12), which belongs to the area with the highest precipitation. It could be possible that the fixation of this allele for individuals of the Hopa (TR03) population was due to the climatic conditions of the region. The associations found for allele 181 were again related to precipitation. In this case, rain associated events were related to the driest period of the year (PrecDQ), and the fixation of this allele may respond to environmental scenarios related to warm and dry areas. In terms of allelic presence (Appendix Fig. 12), none of the trees from the Hopa (TR03) region showed the allele 181, in contrast to Hervás (SP06), Holomontas (GR01), and Madonie (IT01) trees, located in dry regions, which showed the highest frequencies. The associations found for allele 185 were linked to temperature variables, namely MeanTCQ, TSeas, and ATR. In this case, this allele showed frequencies distributed among seven different populations, with the highest frequencies in chestnuts from Castanyet (SP02) and Costa Atlántica (SP03). Galicia and Catalonia regions have a very mild climate, with average minimum and maximum temperatures of 11.0 vs. 11.7 and 18.2 vs. 18.6 °C, respectively. The fixation of allele 185 could therefore be linked to this particular temperature range.
4.3 The relevance of the FIR059 marker
The Fst test used here to identify outliers implemented in BayeScan 2.1 is more conservative than other methods (Narum and Hess 2011). Out of 13 markers tested for evidence of selection, FIR059 was detected as an outlier locus with “decisive” signs of selection. The FIR059 marker is putatively linked to the RH7 gene, belonging to the DEAD-box-RNA helicase family. This family of helicases is involved in the metabolic processes of RNA, such as transcription, splicing and translation of mRNA, and degradation of the DNA (Huang et al. 2015; Liu et al. 2016). RNA helicases have been associated with different functions linked to the correct development of the plant and biotic and abiotic stress responses (Kim et al. 2008). Specifically, the RH7 helicase plays an important role during the embryonic phase development of plants and in their tolerance to heat, frost and drought stress (Vashisht and Tuteja 2006; Macovei et al. 2012). It is possible that the fixation of the alleles for the locus FIR059 found to be associated with climatic variables may be the result of selective processes due to the different climatic conditions in the various regions.
Our results are in agreement with a study by Alcaide et al. (2019) on sweet chestnut, which identified the locus FIR059 as an outlier locus also putatively under selection. The locus was found to be involved in the tolerance and the susceptibility of chestnut to drought stress. Specifically, FIR059 showed three private alleles for drought-susceptible individuals and two private alleles for drought-tolerant individuals. Of these five alleles, three (152, 160, and 176) were found here to be associated with climatic variables, of which allele 152 related to precipitation. In the study by Alcaide et al. (2019) allele 152 was found to be private in drought-tolerant plants, reinforcing the hypothesis of a link between this allele and the plant’s exploitation of water resources. Associations for alleles 160 and 176 were not explored as they were highlighted only by LFMM. Allele 176, highlighted as private in drought-tolerant chestnuts (Alcaide et al. 2019), showed here an association with the mean temperature of the wettest quarter. Allele 160, also highlighted as a private allele in drought-susceptible chestnuts (Alcaide et al. 2019), showed associations with mean annual temperature and annual minimum temperature, and precipitation seasonality. This provides evidence about the putative involvement of alleles 152, 160, and 176 in mechanisms responding to abiotic stresses. Results strongly suggest that FIR059 is a marker of considerable interest for the identification of genotypes adapted to different climatic conditions.
5 Conclusions
Climate predictions will induce a change in suitable areas for sweet chestnut distribution. Here, we have reported a restricted case, represented by a limited number of chestnut population, we have been able of highlighting regions of the genome that have been putatively affected by climate pressures. The identification of alleles related to climatic variables could be relevant for understanding adaptation of this tree species in the future. It has been proven here that landscape genomics and association analyses were capable of identifying in chestnut loci involved in mechanisms of tolerance and adaptation to different environmental challenges. This work aimed to emphasize the close inter-relationship existing between trees and environment, and how climate variables were able to shape their genetic diversity and adaptive variation.
Data availability
The datasets generated and/or analyzed during the current study are available in the Zenodo repository, https://doi.org/10.5281/zenodo.4384029
References
Aitken SN, Yeaman S, Holliday JA, Wang T, Curtis-McLane S (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol Appl 1:95–111. https://doi.org/10.1111/j.1752-4571.2007.00013.x
Alcaide F, Solla A, Mattioni C, Castellana S, Martín MA (2019) Adaptive diversity and drought tolerance in Castanea sativa assessed through EST-SSR genic markers. Forestry 92:287–296. https://doi.org/10.1093/forestry/cpz007
Alcaide F, Solla A, Cherubini M, Mattioni C, Cuenca B, Camisón Á, Martín MÁ (2020) Adaptive evolution of chestnut forests to the impact of ink disease in Spain. J Syst Evol 58, https://doi.org/10.1111/jse.12551
Ahrens C, Rymer PD, Stow A, Bragg J, Dillon S, Umbers KDL, Dudaniec RY (2018) The search for loci under selection: trends, biases and progress. Mol Ecol 27:1342–1356. https://doi.org/10.1111/mec.14549
Barrett RD, Schluter D (2008) Adaptation from standing genetic variation. Trends Ecol Evol 23:38–44. https://doi.org/10.1016/j.tree.2007.09.008
Bradbury D, Smithson A, Krauss SL (2013) Signatures of diversifying selection at EST-SSR loci and association with climate in natural Eucalyptus populations. Mol Ecol 22:5112–5129. https://doi.org/10.1111/mec.12463
Buck EJ, Hadonou M, James CJ, Blakesley D, Russell K (2003) Isolation and characterization of polymorphic microsatellites in European chestnut (Castanea sativa Mill.). Mol Ecol Notes 3:239–241. https://doi.org/10.1046/j.1471-8286.2003.00410.x
Byrne DH, Bacon TA (1992) Chilling estimation: its importance and estimation. The Texas Horticulturist 18:8–9
Camisón A, Martín MÁ, Dorado FJ, Moreno G, Solla A (2020) Changes in carbohydrates induced by drought and waterlogging in Castanea sativa. Trees 34:579–591. https://doi.org/10.1007/s00468-019-01939-x
Castellana S (2020) Signatures of local adaptation to climate in natural populations of sweet chestnut (Castanea sativa Mill.) from southern Europe. [dataset]. Zenodo. V1. https://doi.org/10.5281/zenodo.4384029
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621 631. https://doi.org/10.1093/molbev/msl191
Cho SK, Ryu MY, Song C, Kwak JM, Kim WT (2008) Arabidopsis PUB22 and PUB23 are homologous U-Box E3 ubiquitin ligases that play combinatory roles in response to drought stress. Plant Cell 20:1899–1914. https://doi.org/10.1105/tpc.108.060699
Ciais P, Reichstein M, Viovy N, Granier A, Ogee J, Allard V et al (2005) Europe-wide reduction in primary productivity caused by the heat and drought in 2003. Nature 437:529–533. https://doi.org/10.1038/nature03972
Cohen D, Bogeat-Triboulot MB, Tisserant E (2010) Comparative transcriptomics of drought responses in Populus: a meta-analysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genomics 11:630. https://doi.org/10.1186/1471-2164-11-630
Comps B, Gomory D, Letouzey J, Thiebaut B, Petit RJ (2001) Diverging trends between heterozygosity and allelic richness during postglacial colonization in european beech. Genetics 157:389–397, PMC1461495
Conedera M, Manetti MC, Giudici F, Amorini E (2004) Distribution and economic potential of the sweet chestnut (Castanea sativa Mill.) in Europe. Ecologia Mediterranea 30:179–193
Conord C, Gurevitch J, Fady B (2012) Large-scale longitudinal gradients of genetic diversity: a meta-analysis across six phyla in the Mediterranean basin. Ecol Evol 2(10):2600–14. https://doi.org/10.1002/ece3.350
Coop G, Witonsky D, Di Rienzo A, Pritchard J (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics 185:1411–1423. https://doi.org/10.1534/genetics.110.114819
Cuervo-Alarcon LC, Arend M, Müller M, Sperisen C, Finkeldey R, Krutovsky KV (2018) Genetic variation and signatures of natural selection in populations of European beech (Fagus sylvatica L.) along precipitation gradients. Tree Genet Genomes 14:84. https://doi.org/10.1007/s11295-018-1297-2
Davis MB, Shaw RG, Etterson JR (2005) Evolutionary responses to changing climate. Ecology 86:1704–1714
De Mita S, Thuillet AC, Gay L (2013) Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol Ecol 22:1383–1399. https://doi.org/10.1111/mec.12182
Dent E, Von Holdt M (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Desikan R, Horák J, Chaban C, Mira-Rodado V, Witthoft J (2008) The histidine kinase AHK5 integrates endogenous and environmental signals in Arabidopsis guard cells. PLoS One 3:e249. https://doi.org/10.1371/journal.pone.0002491
Durand J, Bodénès C, Chancerel E, Frigerio JM, Vendramin GG, Sebastiani F et al (2010) A fast and cost-effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genomics 11:570. https://doi.org/10.1186/1471-2164-11-570
Eckert AJ, Bower AD, González-Martínez SC, Wegrzyn JL, Coop G, Neale DB (2010) Back to nature: ecological genomics of loblolly pine (Pinus taeda, Pinaceae). Mol Ecol 19:3789–3805. https://doi.org/10.1111/j.1365-294X.2010.04698.x
Eckert AJ, Bower AD, Wegrzyn JL (2009) Asssociation genetics of Coastal Douglas Fir (Pseudotsuga menziesii var. menziesii, Pinaceae) I Cold-fardiness related traits. Genetics 182:1289–1302. https://doi.org/10.1534/genetics.109.102350
Ellis JR, Burke JM (2007) EST-SSRs as a resource for population genetic analyses. Heredity 99:125–132. https://doi.org/10.1038/sj.hdy.6801001
Ellis N, Smith SJ, Pitcher CR (2012) Gradient forests: Calculating importance gradients on physical predictors. Ecology 93:156–168. https://doi.org/10.1890/11-0252.1
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2005) Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fady B, Conord C (2010) Macroecological patterns of species and genetic diversity in vascular plants of the Mediterranean Basin. Divers. Distrib 16:53–64. https://doi.org/10.1111/j.1472-4642.2009.00621.x
Fang Y, Xiong L (2015) General mechanisms of drought response and their application in drought resistance improvement in plants. Cell Mol Life Sci 72:673–689. https://doi.org/10.1007/s00018-014-1767-0
FAO (2019) Ecocrop database. FAO, Rome, Italy. http://ecocrop.fao.org/ecocrop
Fineschi S, Laurchini D, Villani F, Vendramin GG (2000) Chloroplast DNA polymorphism reveals little geographical structure in Castanea sativa Mill. (Fagaceae) throughout southern European Countries. Mol Ecol 9:1495–1503. https://doi.org/10.1046/j.1365-294x.2000.01029.x
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1km spatial resolution climate surfaces for global land areas. Int J Climatol 37. https://doi.org/10.1002/joc.5086
Foll M, Gaggiotti OE (2008) A genome scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Frichot E, Schoville SD, Bouchard G, François O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30:1687–1699. https://doi.org/10.1093/molbev/mst063
Frichot E, Francois O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929. https://doi.org/10.1111/2041-210X.12382
Hedrick PW (1976) Genetic variation in a heterogeneous environment. II Temporal heterogeneity and directional selection. Genetics 84:145–150
Hijmans RJ, Guarino L, Bussink C, Mathur P, Cruz M, Barrentes I, Rojas E (2004) DIVA‐GIS. Vsn. 5.0. A geographic information system for the analysis of species distribution data. http://www.divagis.org.
Huang C, Shen Y, Huang L, Wu S, Yeh CH, Lu C (2015) The DEAD-box RNA helicase AtRH7/PRH75 participates in pre-rRNA processing, plant development and cold tolerance in Arabidopsis. Plant Cell Physiol 57:174–191. https://doi.org/10.1093/pcp/pcv188
Jeffreys H (1961) Theory of Probability, 3rd ed. Oxford University Press
Joost S, Bonin MW, Bruford L, Després C, Conord G, Taberlet P (2007) A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol 16:3955–3969. https://doi.org/10.1111/j.1365-294X.2007.03442.x
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK (2011) Microsatellite markers: An overview of the recent progress in plants. Euphytica 177:309–334
Kalinowski ST (2005) HP-Rare: a computer program for performing rarefaction on measures of allelic diversity. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kim JS, Kim KA, Oh TR, Park CM, Kang H (2008) Functional characterization of dead-box RNA helicases in Arabidopsis thaliana under abiotic stress conditions. Plant Cell Physiol 49:1563–1571. https://doi.org/10.1093/pcp/pcn125
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Krutovskii KV, Neale DB (2001) Forest genomics for conserving adaptive genetic diversity. Forest Genetics Resources 29:7–9
Lê S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw 25:1–18. https://doi.org/10.18637/jss.v025.i01
Lind JF, Gailing O (2013) Genetic structure of Quercus rubra L. and Quercus ellipsoidalis E.J. Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree Genet Genomes 9:707–722. https://doi.org/10.1007/s11295-012-0586-4
Liu Q, Umeda M, Uchimiya H (1994) Isolation and expression analysis of two rice genes encoding the major intrinsic protein. Plant Mol Biol 26:2003–2007. https://doi.org/10.1007/BF00019511
Liu Y, Tabata D, Imai R (2016) A cold-inducible DEAD-box RNA helicase from Arabidopsis thaliana regulates plant growth and development under low temperature. PLoS One 11:e0154040. https://doi.org/10.1371/journal.pone.0154040
Luedeling E, Girvetz EH, Semenov MA, Brown PH (2011) Climate change affects winter chill for temperate fruit and nut trees. PLoS One 6:e20155. https://doi.org/10.1371/journal.pone.0020155
Macovei A, Vaid N, Tula S, Tuteja N (2012) A new DEAD-box helicase ATP binding protein (OsABP) from rice is responsive to abiotic stress. Plant Signal Behav 7:1–6. https://doi.org/10.4161/psb.21343
Mattioni C, Cherubini M, Micheli E, Villani F, Bucci G (2008) Role of domestication in shaping Castanea sativa genetic variation in Europe. Tree Genet Genomes 4:563–574. https://doi.org/10.1007/s11295-008-0132-6
Marinoni D, Akkak A, Bounous G, Edwards KJ, Botta R (2003) Development and characterization of microsatellite markers in Castanea sativa (Mill.). Mol Breeding 11:127–136. https://doi.org/10.1023/A:1022456013692
Martin MA, Mattioni C, Cherubini M, Taurchini D, Villani F (2010) Genetic diversity in European chestnut populations by means of genomic and genic microsatellite markers. Tree Genet Genomes 6:735–744. https://doi.org/10.1007/s11295-010-0287-9
Martins K, Gugger PF, Llanderal-Mendoza J, Gonzalez-Rodriguez A, Fitz-Gibbon ST, Zhao JL et al (2018) Landscape genomics provides evidence of climate-associated genetic variation in Mexican populations of Quercus rugosa. Evol Appl 11:1842–1858. https://doi.org/10.1111/eva.12684
Mattioni C, Martin MA, Pollegioni P, Cherubini M, Villani F (2013) Microsatellite markers reveal a strong geographical structure in European populations of Castanea sativa (Fagaceae): evidence for multiple glacial refugia. Am J Bot 100:951–961. https://doi.org/10.3732/ajb.1200194
Mattioni C, Martin MA, Chiocchini F, Cherubini M, Gaudet M, Pollegioni P et al (2017) Landscape genetics structure of European sweet chestnut (Castanea sativa Mill): indications for conservation priorities. Tree Genet Genomes 13:39. https://doi.org/10.1007/s11295-017-1123-2
Maurer WD, Fernández-López J (2001) Establishing an international sweet chestnut (Castanea sativa Mill.) provenance test: preliminary steps. Journal of Forest Snow and Landscape Research 76:482–486
Narum SR, Hess JE (2011) Comparison of FST outlier tests for SNP loci under selection. Mol Ecol Resour 11:184–194. https://doi.org/10.1111/j.1755-0998.2011.02987.x
Novembre J, Di Rienzo A (2009) Spatial patterns of variation due to natural selection in humans. Nat Rev Genet 10:745–755. https://doi.org/10.1038/nrg2632
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Petit R, Hampe A (2006) Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37:187–214. https://doi.org/10.1146/annurev.ecolsys.37.091305.110215
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Core Team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24:4348–4370. https://doi.org/10.1111/mec.13322
Richardson JL, Urban MC, Bolnick DI, Skelly DK (2014) Microgeographic adaptation and the spatial scale of evolution. Trends Ecol Evol 29:165–176. https://doi.org/10.1016/j.tree.2014.01.002
Schoville SD, Bonin A, Francois O (2012) Adaptive genetic variation on the landscape: methods and cases. Annu Rev Ecol Evol Syst 43:23–43. https://doi.org/10.1146/annurev-ecolsys-110411-160248
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ (2000) Analysis of SSRs derived from grape ESTs. Theor Appl Genet 100:723–726. https://doi.org/10.1007/s001220051344
Şelale H, Çelik I, Gültekin V, Allmer J, Doğanlar S, Frary A (2013) Development of EST-SSR markers for diversity and breeding studies in opium poppy. Plant Breed 132:344–351. https://doi.org/10.1111/pbr.12059
Silveira RD, Abreu FR, Mamidi S, McClean PE, Vianello RP, Lanna AC (2015) Expression of drought tolerance genes in tropical upland rice cultivars (Oryza sativa). Genet Mol Res 14:8181–8200. https://doi.org/10.4238/2015.July.27.6
Stucki S, Orozco-terWengel P, Forester BR, Duruz S, Colli L, Masembe C, Joost S (2017) High performance computation of landscape genomic models including local indicators of spatial association. Mol Ecol Resour 17:1072–1089. https://doi.org/10.1111/1755-0998.12629
Sullivan A, Lind JF, McCleary T, Romero-Severson J, Gailing O (2013) Development and characterization of genomic and gene-based microsatellite markers in North American red oak species. Plant Mol Biol Report 31:231–239. https://doi.org/10.1007/s11105-012-0495-6
Torokeldiev N, Ziehe M, Gailing O (2019) Genetic diversity and structure of natural Juglans regia L. populations in the southern Kyrgyz Republic revealed by nuclear SSR and EST-SSR markers. Tree Genet Genomes 15:5. https://doi.org/10.1007/s11295-018-1311-8
Tuskan GA, Gunter LE, Yang ZK, Yin T, Sewell MM, DiFazio SP (2004) Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa. Can J For Res 34:85–93. https://doi.org/10.1139/x03-283
Van Hove J, De Jaeger G, De Winne N, Guisez Y, Van Damme EJM (2015) The Arabidopsis lectin EULS3 is involved in stomatal closure. Plant Sci 238:312–322. https://doi.org/10.1016/j.plantsci.2015.07.005
Vashisht AA, Tuteja N (2006) Stress responsive DEAD-box helicases: a new pathway to engineer plant stress tolerance. J Photochem Photobiol, B 84:150–160. https://doi.org/10.1016/j.jphotobiol.2006.02.010
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55. https://doi.org/10.1016/j.tibtech.2004.11.005
Widmer A, Lexer C (2001) Glacial refugia: sanctuaries for allelic richness, but not for gene diversity. Trends Ecol Evol 16:267–269. https://doi.org/10.1016/j.tibtech.2004.11.005
Yatabe Y, Kane NC, Scotti-Saintagne C, Rieseberg LH (2007) Rampant gene exchange across a strong reproductive barrier between the annual sunflowers Helianthus annuus and H petiolaris. Genetics 175:1883–1893. https://doi.org/10.1534/genetics.106.064469
Yin T, DiFazio SP, Gunter LE, Riemenschneider D, Tuskan GA (2004) Large‐scale heterospecific segregation distortion in Populus revealed by a dense genetic map. Theor Appl Genet 109:451–463. https://doi.org/10.1007/s00122-004-1653-5
Acknowledgments
The authors are grateful for the support in the spatial and statistical analyzes provided by Dr. Francesca Chiocchini and Dr. Isacco Beritognolo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Bruno Fady
Contribution of the co-authors Conceptualization: Simone Castellana; Claudia Mattioni; methodology: Simone Castellana; Claudia Mattioni; software: Simone Castellana; David Neale; validation: Simone Castellana; Claudia Mattioni; David Neale; formal analysis: Simone Castellana; David Neale; Claudia Mattioni; investigation: Simone Castellana; data curation: Simone Castellana; writing—original draft: Simone Castellana; writing—review and editing: Claudia Mattioni; David Neale; Alejandro Solla; Maria Angela Martin; visualisation: Simone Castellana.
Appendix
Appendix

Present (a) and future (b) mean annual temperature, and present (c) and future (d) mean annual rainfall across the ten Castanea sativa populations examined in the study

Pearson’s correlation test plot. The degree of correlation between the variables is shown on the ordinate axis (from red − 1 to blue 1); variables with a degree of correlation |< 0.8| were classified as “highly correlated”

∆K Evanno plot for the gSSR

∆K Evanno plot for the EST-SSR

Bar-plots of STRUCTURE analysis results for gSSR and EST-SSR from K = 1 to K = 8

Results of BayeScan Fst outlier analysis for gSSR and EST-SSRs; FIR059 shows “decisive” signs of diversifying selection

Principal component analysis (PCA) of the environmental variables; all of the environmental variables refer to the ID in Table 2 (V1–V21); percentage of explained variance for the two axes are elucidated in the figure

Allele frequencies for the locus FIR059 by population
Rights and permissions
About this article

Cite this article
Castellana, S., Martin, M.Á., Solla, A. et al. Signatures of local adaptation to climate in natural populations of sweet chestnut (Castanea sativa Mill.) from southern Europe. Annals of Forest Science 78, 27 (2021). https://doi.org/10.1007/s13595-021-01027-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13595-021-01027-6