
Abstract
Densities in the homogeneous liquid phase of (mono-)ethanolamine (MEA) and diethanolamine (DEA) were investigated using a commercially available high-pressure vibrating-tube densimeter (VTD). Due to the melting point of the experimental materials, the setup of the VTD had to be modified by an insulated housing of the entire piping including the pressure pump. The insulated housing could be heated up by a temperature-controlled heating fan. The liquid samples with a purity of (0.9994 or 0.9950) mole fraction, respectively, were decanted within an inert protective argon atmosphere and further degassed by several freeze–pump–thaw cycles. Density measurements were carried out at temperatures between (293, respectively, 313 and 423) K and at pressures between (5 and 90) MPa. The resulting 140, respectively, 120 (p, ρ, T) data points, explicitly extend the published database for MEA and DEA, with regards to pressure. A comparison with the currently used equations of state for MEA and DEA revealed a maximum relative deviation of – 0.18 % for MEA and – 0.41 % for DEA, each at the highest investigated temperature and pressure. Considering the measurement uncertainties in temperature, pressure, and oscillation period, as well as uncertainties resulting from the calibration and from the impurities of the sample, the combined expanded relative uncertainty (k = 2) in density varied from (0.1027 to 0.1038) % and from (0.1104 to 0.1130) %, respectively. The VTD was previously calibrated by comprehensive measurements of water and helium and had been further validated by measurements with pure propane.
Similar content being viewed by others

1 Introduction
In the context of the aimed at global energy transition, various measures to reduce greenhouse gas emissions are discussed by politics and science. One opportunity is carbon capture and storage processes (CCS), which have a scientifically proven potential for reducing carbon dioxide emissions and which are recognized by the reference scenarios “European Taxonomy for Sustainable Finance” [1] and “Clean Planet for all” [2]. In such CCS applications, amine-based solvents such as (mono-)ethanolamine [3,4,4] (MEA) or diethanolamine [5, 6] (DEA) are used in amine gas-treating processes for the chemical absorption of CO2 from the exhaust gases of power plants or other industrial processes. As a so called end-of-pipe technology, such processes could be attached to the flue gas cleaning of, e.g., any combustion power plant [7]. Furthermore, the two amines MEA and DEA are used to separate CO2 from natural gas or from hydrogen produced by steam reforming [8]. Due to the explained applications, MEA and DEA must be considered potential minor impurities of the mentioned flows of natural gas, hydrogen or typical CO2-rich mixtures descending from CCS processes.
The design of such processes requires data on properties that are obtained by appropriate equations of state. Fundamental equations of state for both MEA and DEA were developed by Herrig [9] and Kortmann [10], respectively, and are implemented in TREND 5.0 [11]. However, these approaches are typical empirical models, and thus, their accuracy in predicting thermodynamic properties depends on the quality of the underlying experimental data. Reviews of the available experimental homogenous density data in the liquid phase, including the number of points as well as the temperature and pressure ranges are summarized in Table 1 for MEA and in Table 2 for DEA. Although numerous references for homogeneous densities of MEA and DEA in the liquid phase can be found in the literature, almost the complete database is limited to single measurement points at ambient pressure. The density measurements by Han et al. [12, 13] are the only existing datasets at pressures higher than ambient pressure. Therefore, the goal of this work was to screen the (p, ρ, T) behavior of liquid MEA and DEA within a comprehensive and consistent dataset extending the ranges of temperature and especially pressure. The distribution of the data from literature is presented in (p, T) diagrams in Fig. 1 for MEA (a) and DEA (b), together with the state points investigated in the scope of this work.

Selection of experimental (p, ρ, T) data for MEA (a) and DEA (b) from literature and (p, T) state points investigated in the present work shown in a pressure versus temperature phase diagram. ○, this work; □, Han et al. [12, 13]; ◊, further data from literature (see Tables 1 and 2); Tm,n, normal melting temperature; ―, phase boundary calculated with the equation of state by Herrig [9] and Kortmann [10], respectively
2 Experimental Section
2.1 Apparatus Description
The density measurements were carried out using a modified commercially available Vibrating Tube Densimeter (VTD). The experimental setup was described in detail within our previous works [100, 101, 107]; however, a brief summary and further small adjustments are presented here.
The essential component of the VTD (Anton Paar, Austria, type: DMA HPM) is the insulated measuring cell that is heated up or cooled down with a circulating bath thermostat (Huber, Germany, type: ministat 125-cc-NR). The thermostat is filled with ethylene glycol to realize measurements in a temperature range between (283 and 423) K. A 100 Ω platinum resistance thermometer (Merz Messfühlertechnik, Germany, type: P100/2528) was calibrated to the ITS-90 and is connected to a digital multimeter (Tektronix, USA, type: DMM 4050), leading to an estimated expanded uncertainty (k = 1.73) in temperature measurements of 60 mK over the investigated temperature range. The pressure measurement is implemented by a calibrated pressure transducer (WIKA, Germany, type: P-30) that enables measurements up to 100 MPa while maintaining an expanded uncertainty (k = 2) of 0.01 MPa (5.0 MPa ≤ p < 35.0 MPa) or 0.03 MPa (35.0 MPa ≤ p ≤ 100.0 MPa), respectively. The pressure is controlled by a hand pump (HiP, USA, type: 50-6-15) and a rotary-vane vacuum pump (Pfeiffer, Germany, type: DUO 016 B) is used for the evacuation of the system. To connect the measuring cell with the pressure transducer, the hand pump, the sample cylinder, and the ventilation outlets, a sample manifold is installed as displayed in Fig. 2. In contrast to our previous measurements [101], the entire piping including the hand pump was now enclosed by an insulated box that can be thermostated to a desired constant temperature by a controlled heating fan. This modification was necessary due to the high normal melting temperatures of MEA (283.45 K) and DEA (301.15 K).

Schematic diagram of the modified vibrating-tube densimeter measuring system
During a measurement, the oscillation period τ of the vibrating tube is measured by a frequency counter that is included in the Anton Paar mPDS 5 system and is displayed with a resolution of 0.001 µs. In accordance with the fundamental measuring principle, the density ρ of the fluid inside the tube is determined by the relation shown in the following equation:
where p and T are the pressure and the temperature of the fluid, respectively. The apparatus parameters A(p, T) and B(p, T), which depend on pressure and temperature, are determined by calibration (see Sect. 3.1).
2.2 Experimental Materials
The experimental materials used in the present work are specified in Table 3. The calibration included measurements with water and helium, while propane was measured to validate the calibrated setup. Helium and propane were provided as gaseous samples of research grade and were used as received with impurities in the ppm-range as displayed in Table 3. In contrast, high-purity water was decanted into a stainless steel sample cylinder that could be attached to the sample manifold of the VTD, as illustrated in Fig. 2 and was further degassed by several freeze–pump–thaw cycles, as described in previous works [100,101,102,103, 107]. Liquid nitrogen was used to freeze the sample, while several impurities remain gaseous, and were evacuated before the sample was thawed again and the cycle could be repeated.
Liquid samples of MEA and DEA were treated analogically to the sample of water, however, the process of decanting was carried out within the dry and inert argon atmosphere of a customized glove-box, which offered two decisive advantages. On the one hand, contact with the humidity of the ambient air was avoided, while a heating fan realized a temperature above 313 K on the other hand, which significantly simplified the treatment of the two rather viscous samples. Subsequently, both samples were degassed as described before. The purities of MEA and DEA were stated by the supplier to be at least as good as (0.9997 and 0.9990) area fraction according to gas chromatography analysis. However, the GC analysis was conducted without being calibrated to any standards of well-known composition. Therefore, no specification of the present impurities and initially no statement on the molar composition could be made. Thus, we investigated the effect of typical impurities of the two samples such as water, ethylene oxide, ammonia and MEA or DEA and assumed that the remaining fraction is entirely dominated by the respective impurity. Subsequently, we assumed an ideal mixing rule for each binary system at the respective state point. This way we found that the purity of MEA was 0.9994 mol fraction with DEA being the dominant impact. In the same way, the purity of DEA was determined to be 0.9950 mol fraction with water being the impurity that affects the result of the measurements the most.
2.3 Experimental Procedures
To fill the VTD, the sample cylinder with the liquid MEA or DEA, respectively, was connected to the evacuated measuring system. The measuring cell, as well as the sample manifold, was thermostated at approximately 313 K, while the sample cylinder was insulated and warmed by use of a heating jacket up to about 323 K. Thus, the vapor pressure of the sample was raised and filling with the opened valves V1, V2 and V6 (see Fig. 2) could be mainly realized by taking advantage of the temperature and pressure gradient as well as by support of the hand pump. The pressure was increased with the hand pump to the highest pressure of 90 MPa, before valve V2 was closed to separate the measuring cell and the pressure transducer from the significantly larger volume of the sample manifold and the hand pump. The measurements were conducted along isotherms with decreasing pressure, while at least 20 min of equilibration time were allowed at each state point. A single measurement extends over a period of 10 min and was started after a temperature fluctuation of less than 2 mK and a pressure stability of 0.01 MPa was reached. The measured values of temperature, pressure and oscillation period were recorded each 10 s and an averaged (p, T, τ) data point was computed for further data analysis.
3 Results and Discussion
3.1 Calibration
The density is determined from the measured oscillation period according to Eq. (1) using the apparatus parameters A(p, T) and B(p, T). Both apparatus parameters are known from calibration measurements with the reference fluids helium and water, using a polynomial based model by Outcalt and McLinden [104] (see Eqs. 2–4) as discussed in our previous works [100, 101, 107]. However, various further models are provided by May et al. [105] or the manufacturer of the VTD [106], a systematical review by Yang et al. [107] confirmed that the best regression is provided by the model by Outcalt and McLinden [104], as long as comprehensive calibration data are available over the entire temperature and pressure range. According to Outcalt and McLinden [104], the apparatus parameters A(p, T) and B(p, T) are regressed by the following relations:
and
where T is the temperature, p is the pressure and τ is the oscillation period.
In a first step, the parameters ci (see Table 4) were determined from measurements with evacuated measuring cell (see supplementary material, Table S1), which are referred to as vacuum measurements. Vacuum measurements were carried out at 15 temperatures between (283 and 423) K and the experimental (T, τ) data were correlated to the second-order polynomial, Eq. (4).
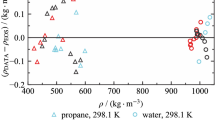
In a second step, the calibration includes measurements with the reference fluids helium and water, to determine the parameters Ai and Bi (see Table 4) according to Eqs. (2) and (3). Densities of both reference fluids were measured over the full temperature range from (283 to 423) K at nine different temperatures. While measurements with the high-purity water also covered the whole pressure range from (5 to 90) MPa, measurements with helium of research grade were limited to a maximum pressure of 60 MPa, thus leading to a set of 90 (p, T, τ) data points for water (see supplementary material, Table S3) and 63 (p, T, τ) data points for helium (see supplementary material, Table S2). The parameters of the model (Eqs. 2 and 3) were fitted with a least-square method, minimizing the difference between densities obtained by the calibration measurements using the working Eq. (1), and reference values calculated from the equation of state by Ortiz-Vega et al. [108] or Wagner and Pruss [109], respectively. For temperatures between (200 and 500) K the uncertainty of densities calculated from the equation of state for helium is given as 0.03 % for pressures below 40 MPa and as 0.1 % for pressures up to 100 MPa. For the equation of state of water, an uncertainty of 0.001 % for pressures up to 10 MPa is stated, while the uncertainty increases up to 0.1 % for higher pressures, with temperatures up to 423 K in both cases. Relative deviations of the experimental densities from densities calculated with the respective equation of state are plotted against pressure and are depicted in Fig. 3 for helium (a) and water (b).

Results of the calibration measurements with helium (a) and water (b) as well as results of the validation measurements with propane (c). Relative deviations of the experimental densities ρexp from values ρEOS calculated with the equation of state by Ortiz-Vega et al. [108] (a), by Wagner and Pruss [109] (b) and by Lemmon et al. [110] (c), respectively, (zero line), are plotted versus pressure. + , T = 283 K; ▷, T = 293 K; ○, T = 303 K; ✱, T = 313 K; □, T = 323 K; ▽, T = 343 K; ◊, T = 363 K; ☆, T = 383 K; △, T = 403 K; ×, T = 423 K
To validate the calibrated setup of the VTD, measurements with propane were conducted over the full temperature and pressure range, as listed in Table S4, which are referred to as validation measurements. The propane of research grade was measured at ten temperatures from (283 to 423) K and pressures between (5 and 90) MPa resulting in a validation data set including 100 (p, ρ, T) measuring points. The relative deviations of the experimental densities of propane from values calculated with the equation of state by Lemmon et al. [110] are plotted versus pressure in Fig. 3(c). All experimental data points with p ≥ 60 MPa are within a maximum relative deviation of ± 0.05 % from the equation of state, just like data points with p ≥ 10 MPa and T ≤ 343 K. The uncertainty of the equation of state is stated as 0.01 % in density in the liquid phase below 350 K and at greater temperatures for pressures above 10 MPa, while uncertainties in density increase in the extended critical region, especially near the critical point [110]. However, values specified above are still within the estimated experimental uncertainty which could reach 0.2%. For lower pressures and higher temperatures, the relative deviations increase significantly. With the critical point of propane at about 370 K and 4.25 MPa, some of the investigated state points are close to the critical point and three isotherms are in the supercritical region. Especially measurements at (383, 403 and 423) K with pressures at 10 MPa and lower belong to the supercritical section with gas-like behavior (see the densities reported in the supplementary material, Table S4). In this range, the sensitivity of measured densities with respect to uncertainties in pressure and temperature measurements increases drastically. In addition, the uncertainty of density determination with a VTD increases at low density as well, see the comparison between the representation of the calibration data for helium and water, Fig. 3(a) and (b). This leads to larger relative deviation of the propane data, which is negligible in terms of the validation for MEA and DEA measurements. Liquid states with densities in the range (900–1000) kg·m−3 were expected for the measurements with MEA and DEA (see Tables 5 and 6), while measurements with propane only reach a maximum density of 609 kg·m−3 at 283 K and 90 MPa.
3.2 Density Measurements of MEA and DEA
Density measurements of MEA and DEA samples as described in Sect. 2.2 were conducted utilizing the Vibrating Tube Densimeter (VTD) depicted in Sect. 2.1 and the apparatus coefficients determined from calibration according to Sect. 3.1. Measurements in the liquid phase of MEA were carried out along 14 isotherms at temperatures between (293.15 and 423.15) K, while the measurements with DEA were restricted to 12 isotherms between (313.15 and 423.15) K due to the higher melting temperature of DEA. For both series of measurements, ten different pressures between (5 and 90) MPa were set on each isotherm. This way, datasets containing 140 and 120 (p, ρ, T) data points were obtained.
In Fig. 4, the absolute experimental densities of MEA (a) and DEA (b) are plotted versus pressure, thus, revealing the course of the related isotherms. For both systems, values of density are increasing with increasing pressure and decreasing temperature following an almost linear trend. The results are reported in Table 5 for MEA and in Table 6 for DEA, stating the specific temperature and pressure of each measuring point, as well as the experimental oscillation period and the resulting experimental density.

Results of the density measurements with MEA (a) and DEA (b). Absolute experimental densities ρexp are plotted versus pressure. ▷, T = 293 K; ○, T = 303 K; ✱, T = 313 K; □ , T = 323 K; ⊥, T = 333 K; ▽, T = 343 K; + , T = 353 K; ◊, T = 363 K; ◁, T = 373 K; ☆, T = 383 K; ⊤, T = 393 K; △, T = 403 K; ∃, T = 413 K; ×, T = 423 K
The densities measured for MEA and DEA were compared with the currently used equations of state for MEA by Herrig [9] and DEA by Kortmann [10] as implemented in the software tool TREND 5.0 [11]. The relative deviations of the experimental densities from the densities calculated with the respective equation of state are plotted versus pressure in Fig. 5 for MEA (a) and DEA (b). For MEA, relative deviations are within about ± 0.05 % up to 40 MPa over the whole investigated temperature range. At pressures above 40 MPa, larger negative relative deviations develop up to a maximum of − 0.18 % at 423 K and 90 MPa. With relative deviations between (− 0.036 and 0.005) %, which are well covered by the uncertainty of the measurements (see Table 5), the isotherm at 293 K is in good agreement with the equation of state over the whole investigated pressure range. The measurements conducted with DEA show larger relative deviations of the experimental densities, reaching a maximum of − 0.41 % at the highest temperature (423 K) and the highest pressure (90 MPa).

Results of the density measurements with MEA (a) and DEA (b). Relative deviations of the experimental densities ρexp from values ρEOS calculated with the equation of state by Herrig [9] and Kortmann [10] (zero line), respectively, are plotted versus pressure. ▷, T = 293 K; ○, T = 303 K; ✱, T = 313 K; □, T = 323 K; ⊥, T = 333 K; ▽, T = 343 K; +, T = 353 K; ◊, T = 363 K; ◁, T = 373 K; ☆, T = 383 K; ⊤, T = 393 K; △, T = 403 K; ∃, T = 413 K; ×, T = 423 K
In literature, a single dataset by Han et al. [12, 13] could be found for both MEA (see Table 1) and DEA (see Table 2), which provide density measurements for the two pure amines at pressures above ambient pressure (see Fig. 1). In Fig. 6, relative deviations of the experimental densities from values calculated with the equations of state by Herrig [9] (a) and Kortmann [10] (b) are plotted versus temperature. For MEA, a similar trend could be observed for both data series (measured at 0.7 MPa by Han et al. [12, 13] and at 5 MPa from this work) with an increasing relative deviation from values calculated with the equation of state when reaching higher temperatures. For DEA, relative deviations decrease while the temperature is increasing. However, both datasets show a similar trend over the temperature again.

Comparison of our datasets for MEA (a) and DEA (b) with the only experimental densities available from literature at pressures above ambient pressure. Relative deviations of the experimental densities ρexp from values ρEOS calculated with the equation of state by Herrig [9] and Kortmann [10] (zero line), respectively, are plotted versus temperature. □, p = 0.7 MPa (Han et al. [12, 13]); ×, p = 5 MPa (this work)
3.3 Uncertainty in Measurements
The combined expanded uncertainty in density Uc(ρ) was estimated according to the “Guide to the Expression of Uncertainty in Measurement” [111], referred to as GUM. Uncertainties attributed to the measurement of the temperature u(T), the pressure u(p) and the oscillation period u(τ) were individually estimated and then summarized to determine umeas(ρ) according to the following equation, considering the sensitivity of density to uncertainties in the independent variables by the appropriate derrivatives:
Further contributions from the uncertainty of the equations of state of water and helium uEOS(ρ) and the applied calibration model umodel(ρ) used in the scope of the calibration as well as the uncertainty assigned to the purity of the respective sample material ux(ρ) were considered according to the following equation with the expansion factor k = 2:
The uncertainty of the calibration model was estimated to be the maximum difference between the density value calculated from the respective equation of state of water or helium and the experimental density value calculated from the applied calibration model among all the measuring points. Uncertainty contributed from the purity of the sample was conservatively estimated by investigating the impurity that affects the measurement result the most and considering this impurity to be the remaining mole fraction of the sample materials composition.
The uncertainty budget for the density measurements in MEA and DEA is exemplified in Table 7 for the case of the median temperature and pressure conditions of each series of measurements. For the measurements with MEA, the combined expanded relative uncertainty becomes 0.1031 % for a density of ρ = 990.43 kg·m−3 at the selected state point with T = 353.19 K and p = 50.00 MPa. Even though the expanded uncertainty differs from (0.944 to 1.076) kg·m−3 over the investigated temperature and pressure region, the combined expanded relative uncertainty is almost constant at about 0.103%. For the measurements with DEA the selected median state point at T = 363.36 K and p = 50.05 MPa has a density of ρ = 1070.87 kg·m−3 which contains a combined expanded uncertainty of 0.1107 %. Again, the combined expanded relative uncertainty varies only slightly between (0.110 and 0.113) % among all investigated state points, while the combined expanded uncertainty differs from 1.137 kg·m−3 at T = 423.29 K and p = 5.05 MPa to 1.232 kg·m−3 at T = 313.12 K and p = 90.13 MPa.
4 Conclusion
The densities of MEA and DEA were measured in the homogenous liquid phase at temperatures from (293, respectively, 313 to 423) K and at pressures between (5 and 90) MPa, significantly extending the existing database with regards to pressure. Measurements were conducted using a commercially available high-pressure vibrating-tube densimeter. The setup of the VTD was modified by implementation of an insulated and heated housing of the entire piping including the pressure pump to enable measurements for fluids with high melting point. A calibration according to Outcalt and Mclinden [104] based on measurements with the evacuated measuring cell as well as with the reference fluids helium and water was conducted over the whole temperature range of the apparatus from (283 to 423) K and at pressures between (5 and 60 or 90) MPa. Experimental densities of MEA and DEA were compared to the respective equations of state by Herrig [9] and Kortmann [10]. Both datasets are in a good agreement with the respective equation of state at low pressures, but were found to reach a maximum relative deviation of − 0.18 % for MEA and − 0.41 % for DEA at 423 K and 90 MPa, thus offering potential for improvement by the development of new equations of state. In addition, our experimental data at the lowest pressure and temperatures between (373 and 423) K is in quite good agreement with the only dataset available from literature with a pressure above ambient pressure by Han et al. [12, 13] and shows a similar behavior when being compared to the respective equation of state. Considering the measurement uncertainties in temperature, pressure and oscillation period, as well as uncertainties resulting from the calibration and the impurities of the sample, the combined expanded uncertainty (k = 2) in density varies from (0.944 to 1.076) kg·m−3 for MEA or from (1.137 to 1.232) kg·m−3 for DEA, respectively.
References
EU Technical Expert Group on Sustainable Finance, Taxonomy Report (2020). https://ec.europa.eu/info/sites/info/files/business_economy_euro/banking_and_finance/documents/200309-sustainable-finance-teg-final-report-taxonomy-annexes_en.pdf. Accessed 17 September 2020
European Comission, A Clean Planet for all (2019). https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:52018DC0773&from=EN. Accessed 17 September 2020
M.R.M. Abu-Zahra, L.H.J. Schneiders, J.P.M. Niederer, P.H.M. Feron, G.F. Versteeg, Int. J. Greenhouse Gas Control (2007). https://doi.org/10.1016/S1750-5836(06)00007-7
S. Baj, A. Siewniak, A. Chrobok, T. Krawczyk, A. Sobolewski, J. Chem. Technol. Biotechnol (2013). https://doi.org/10.1002/jctb.3958
E.B. Rinker, S.S. Ashour, O.C. Sandall, Ind. Eng. Chem. Res. (2000). https://doi.org/10.1021/ie990850r
H. Kierzkowska-Pawlak, A. Chacuk, J. Air Waste Manage. Assoc. (2010). https://doi.org/10.3155/1047-3289.60.8.925
A. Rieder, S. Unterberger, Energy Proc. (2013). https://doi.org/10.1016/j.egypro.2013.06.576
G.T. Rochelle, Science (New York) (2009). https://doi.org/10.1126/science.1176731
S. Herrig, Dissertation, Ruhr-Universität Bochum. https://hss-opus.ub.ruhr-uni-bochum.de/opus4/frontdoor/deliver/index/docId/6284/file/diss.pdf (2018). Accessed 21 September 2020
M. Kortmann, Master thesis, Ruhr-Universität Bochum, 2016
R. Span, R. Beckmüller, S. Hielscher, A. Jäger, E. Mickoleit, T. Neumann, S. Pohl, B. Semrau, M. Thol, TREND. Thermodynamic Reference and Engineering Data 5.0 (Lehrstuhl für Thermodynamik: Ruhr-Universität Bochum, Bochum, 2020)
J. Han, J. Jin, D.A. Eimer, M.C. Melaaen, J. Chem. Eng. Data (2012). https://doi.org/10.1021/je2010038
J. Han, J. Jin, D.A. Eimer, M.C. Melaaen, J. Chem. Eng. Data (2012). https://doi.org/10.1021/je300345m
A.I. Ageev, A.I. Ezrielev, E.S. Roskin, J. Appl. Chem. USSR 43, 2392 (1970)
J. Águila-Hernández, A. Trejo, B.E. García-Flores, R. Molnar, Fluid Phase Equilib. (2008). https://doi.org/10.1016/j.fluid.2008.02.023
E. Álvarez, F. Cerdeira, D. Gómez-Diaz, J.M. Navaza, J. Chem. Eng. Data (2010). https://doi.org/10.1021/je900437b
T.G. Amundsen, L.E. Øi, D.A. Eimer, J. Chem. Eng. Data (2009). https://doi.org/10.1021/je900188m
A. Arce, A. Marchiaro, A. Soto, Fluid Phase Equilib. (2004). https://doi.org/10.1016/j.fluid.2004.09.030
A. Blanco, A. García-Abuín, D. Gómez-Díaz, J.M. Navaza, J. Chem. Eng. Data (2012). https://doi.org/10.1021/je300742t
A. Blanco, A. García-Abuín, D. Gómez-Díaz, J.M. Navaza, Ó.L. Villaverde, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je301123j
S. Bogacheva, K.B. Zemdikhanov, G.K. Mukhamedzyanov, A.K. Sadykov, A.G. Usmanov, Russ. J. Phys. Chem. 54, 838 (1980)
I.S. Bogacheva, K.B. Zemdikhanov, A.G. Usmanov, Chim. Chim. Tech. 25, 182 (1982)
D. Brasoveanu, M. Mihai, M. Belcu, Rev. Chim. 51, 687 (2000)
S.E. Burke, B. Hawrylak, R. Palepu, Thermochim. Acta (2000). https://doi.org/10.1016/S0040-6031(99)00401-3
R. Dean, J. Moulins, A. MacInnis, R.M. Palepu, Phys. Chem. Liq. (2009). https://doi.org/10.1080/00319100701824306
R.M. DiGuilio, R.J. Lee, S.T. Schaeffer, L.L. Brasher, A.S. Teja, J. Chem. Eng. Data (1992). https://doi.org/10.1021/je00006a028
A. García-Abuín, D. Gómez-Díaz, M.D. La Rubia, J.M. Navaza, J. Chem. Eng. Data (2011). https://doi.org/10.1021/je100967k
A.B. López, A. García-Abuín, D. Gómez-Díaz, M.D. La Rubia, J.M. Navaza, J. Chem. Thermodyn. (2013). https://doi.org/10.1016/j.jct.2013.01.020
Y. Geng, S. Chen, T. Wang, D. Yu, C. Peng, H. Liu, Y. Hu, J. Mol. Liq. (2008). https://doi.org/10.1016/j.molliq.2008.06.014
H. Guo, L. Hui, S. Shen, J. Chem. Thermodyn. (2019). https://doi.org/10.1016/j.jct.2018.12.028
H. Herba, G. Czechowski, B. Zywucki, M. Stockhausen, J. Jadiyn, J. Chem. Eng. Data 40, 214 (1995)
M.K.M.Z. Hyder, S. Akhtar, S.H. Mir, A. Khosla, Microsyst. Technol. (2018). https://doi.org/10.1007/s00542-018-3813-6
M.N. Islam, M.M. Islam, M.N. Yeasmin, J. Chem. Thermodyn. (2004). https://doi.org/10.1016/j.jct.2004.06.004
J.H. Jones, H.R. Froning, E.E. Claytor, J. Chem. Eng. Data (1959). https://doi.org/10.1021/je60001a012
H. Ju, W. ElMoudir, A. Aboudheir, N. Mahinpey, J. Chem. Eng. Data (2018). https://doi.org/10.1021/acs.jced.7b01101
U.R. Kapadi, D.G. Hundiwale, N.B. Patil, M.K. Lande, Fluid Phase Equilib. (2002). https://doi.org/10.1016/S0378-3812(02)00095-X
V. Kartsev, M. Rodnikova, V. Tsepulin, V. Markova, J. Phys. Chem. 62, 1152 (1988)
V.N. Kartsev, M.N. Rodnikova, V.V. Tsepulin, K.T. Dudnikova, V.G. Markova, J. Struct. Chem. (1987). https://doi.org/10.1007/BF00754026
V.G. Kozin, A.A. Mukhamadiev, Russ. J. Appl. Chem. (2002). https://doi.org/10.1023/A:1020791608247
S.S. Kurtz, D.L. Camin, A.R. Thompson, J. Chem. Eng. Data (1965). https://doi.org/10.1021/je60027a010
I. Lafontaine, Bull. Soc. Chim. Belges (1958).https://doi.org/10.1002/bscb.19580670308
M.-J. Lee, T.-K. Lin, J. Chem. Eng. Data (1995). https://doi.org/10.1021/je00017a074
M.-J. Lee, T.-K. Lin, Y.-H. Pai, K.-S. Lin, J. Chem. Eng. Data (1997). https://doi.org/10.1021/je9700636
A.G. Leibush, E.D. Shorina, Zh. Prikl. Khim 20, 69 (1947)
M.H. Li, K.P. Shen, J. Chem. Eng. Data (1992). https://doi.org/10.1021/je00007a002
X.-X. Li, G.-C. Fan, Z.-L. Zhang, Y.-W. Wang, Y.-Q. Lu, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je4000372
D. Ma, C. Zhu, T. Fu, X. Yuan, Y. Ma, J. Chem. Thermodyn. (2019). https://doi.org/10.1016/j.jct.2019.06.032
Y. Maham, T.T. Teng, L.G. Hepler, A.E. Mather, Thermochim. Acta (2002). https://doi.org/10.1016/S0040-6031(01)00812-7
Y. Maham, T.T. Teng, L.G. Hepler, A.E. Mather, J. Solution. Chem. (1994). https://doi.org/10.1007/BF00973546
J.B. Matthew, J.F. Summer, E.A. Moelwyn-Hughes, Trans. Faraday Soc. 46, 797 (1950)
M. Moosavi, C.J. Sisco, A.A. Rostami, F.M. Vargas, Fluid Phase Equilib. (2017). https://doi.org/10.1016/j.fluid.2017.06.018
F. Murrieta-Guevara, A. TrejoRodriguez, J. Chem. Eng. Data (1984). https://doi.org/10.1021/je00036a032
F. Murrieta-Guevara, E. Rebolledo-Libreros, A. Trejo, Fluid Phase Equilib. (1993). https://doi.org/10.1016/0378-3812(93)87178-4
A. Nath, E. Bender, J. Chem. Eng. Data (1983). https://doi.org/10.1021/je00034a007
F. Ouaar, A. Negadi, I. Bahadur, R. Phadagi, B. Feddal-Benabed, L. Negadi, J. Chem. Thermodyn. (2018). https://doi.org/10.1016/j.jct.2018.02.018
M. Pagé, J.-Y. Huot, C. Jolicoeur, Can. J. Chem. (1993). https://doi.org/10.1139/v93-142
K.J. Patil, Indian J. Pure Appl. Phys. 16, 608 (1978)
A. Pourmohammadbagher, J.M. Shaw, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je400184t
F. Pouryousefi, R.O. Idem, Ind. Eng. Chem. Res. (2008). https://doi.org/10.1021/ie0709786
R. Ramezani, S. Mazinani, R. Di Felice, Chem. Eng. Sci. (2019). https://doi.org/10.1016/j.ces.2019.05.039
K.R. Reddy, D.B.K. Kumar, G.S. Rao, P.V.S. Sairam, P. Anila, C. Rambabu, J. Chem. Eng. Data (2012). https://doi.org/10.1021/je3002078
R.E. Reitmeier, V. Sivertz, H.V. Tartar, J. Am. Chem. Soc. (1940). https://doi.org/10.1021/ja01865a009
M.S. Shaikh, A.M. Shariff, M.A. Bustam, G. Murshid, J. Chem. Eng. Data (2014). https://doi.org/10.1021/je400830w
S. Shen, Y.-N. Yang, Y. Wang, S. Ren, J. Han, A. Chen, Fluid Phase Equilib. (2015). https://doi.org/10.1016/j.fluid.2015.04.021
J.-H. Song, S.-B. Park, J.-H. Yoon, H. Lee, K.-H. Lee, J. Chem. Eng. Data (1996). https://doi.org/10.1021/je9601366
H.-J. Song, M.-G. Lee, H. Kim, A. Gaur, J.-W. Park, J. Chem. Eng. Data (2011). https://doi.org/10.1021/je101144k
M.S. Sunder, D.H.L. Prasad, J. Chem. Eng. Data (2007). https://doi.org/10.1021/je7001336
M.M. Taib, T. Murugesan, J. Chem. Eng. Data (2010). https://doi.org/10.1021/je1008125
M.M. Taib, T. Murugesan, J. Chem. Eng. Data (2012). https://doi.org/10.1021/je2007204
M.M. Taib, M.M. Akbar, T. Murugesan, J. Mol. Liq. (2013). https://doi.org/10.1016/j.molliq.2013.02.022
H. Tanaka, D. Kodama, R. Yaginuma, M. Kato, Netsu Bussei 15, 182 (2001)
S. Tian, S. Ren, Y. Hou, W. Wu, W. Peng, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je3009073
J. Timmermans, M. Hennaut-Roland, J. Chim. Phys. Phys.-Chim. Biol. 56, 984 (1959)
H. Touhara, S. Okazaki, F. Okino, H. Tanaka, K. Ikari, K. Nakanishi, J. Chem. Thermodyn. (1982). https://doi.org/10.1016/0021-9614(82)90026-X
Y.-M. Tseng, A.R. Thompson, J. Chem. Eng. Data (1964). https://doi.org/10.1021/je60021a043
N.G. Tsierkezos, I.E. Molinou, J. Chem. Eng. Data (1999). https://doi.org/10.1021/je990059p
A. Valtz, C. Coquelet, D. Richon, Thermochim. Acta (2005). https://doi.org/10.1016/j.tca.2004.11.015
Y.W. Wang, S. Xu, F.D. Otto, A.E. Mather, Chem. Eng. J. (1992). https://doi.org/10.1016/0300-9467(92)85004-S
X. Wang, F. Yang, Y. Gao, Z. Liu, J. Chem. Thermodyn. (2013). https://doi.org/10.1016/j.jct.2012.08.021
F. Yang, X. Wang, W. Wang, Z. Liu, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je3013205
M. Yasmin, M. Gupta, J. Solution. Chem. (2011). https://doi.org/10.1007/s10953-011-9731-1
M.I. Zaretskii, V.B. Kogan, N.F. Kononov, V.G. Podolyak, F.K. Tolchinskaya, J. Appl. Chem. USSR 43, 1304 (1970)
J. Aguila-Hernández, A. Trejo, J. Gracia-Fadrique, Fluid Phase Equilib. (2001). https://doi.org/10.1016/S0378-3812(01)00467-8
E. Álvarez, D. Gómez-Díaz, M.D. La Rubia, J.M. Navaza, J. Chem. Eng. Data (2006). https://doi.org/10.1021/je050463q
E. Álvarez, F. Cerdeira, D. Gómez-Diaz, J.M. Navaza, J. Chem. Eng. Data (2010). https://doi.org/10.1021/je900739x
N.M. Cherkasova, I.K. Zhuravleva, E.F. Zhuravlev, V.B. Muhkametshina, Viniti 1181, 1 (1974)
F.I. Chowdhury, S. Akhtar, M.A. Saleh, Phys. Chem. Liq. (2009). https://doi.org/10.1080/00319100802620538
K. Fischer, A. Valtz, D. Richon, ELDATA. Int. Electron. J. Phys. Chem. Data 5, 97 (1999)
A. Haghtalab, A. Shojaeian, J. Chem. Thermodyn. (2014). https://doi.org/10.1016/j.jct.2013.09.001
P.K. Khabibullaev, K. Parpiev, S.S. Aliev, U. Gadaibaev, Russ. J. Phys. Chem. 45, 1220 (1971)
E. Kuss, Z. Angew, Z. Angew. Phys. 7, 372 (1955)
F. Murrieta-Guevara, E. Rebolledo-Libreros, A. Trejo, Fluid Phase Equilib. (1994). https://doi.org/10.1016/0378-3812(94)80068-5
O. Noll, A. Valtz, D. Richon, T. Getachew-Sawaya, I. Mokbel, J. Jose, ELDATA Int. Electron. J. Phys. Chem. Data 4, 105 (1998)
M.E. Rebolledo-Libreros, A. Trejo, J. Chem. Eng. Data (2006). https://doi.org/10.1021/je050462y
S.S. Ashour, E.B. Rinker, O.C. Sandall, Chem. Eng. Commun. (1997). https://doi.org/10.1080/00986449708936617
V.D. Spasojević, S.P. Šerbanović, B.D. Djordjević, M.L. Kijevčanin, J. Chem. Eng. Data (2013). https://doi.org/10.1021/je300938w
F. Yang, X. Wang, Z. Liu, Thermochim. Acta (2012). https://doi.org/10.1016/j.tca.2012.01.007
I.K. Zhuravleva, Zh. Obshch. Khim. 54, 982 (1984)
I.K. Zhuravleva, E.F. Zhuravlev, V.B. Mukhametshina, Z.Z. Khisametdinova, Russ. J. Gen. Chem. 47, 1545 (1977)
C.C. Sampson, X. Yang, J. Xu, M. Richter, J. Chem. Thermodyn. (2019). https://doi.org/10.1016/j.jct.2018.10.016
C.W. Scholz, O. Frotscher, S. Pohl, R. Span, M. Richter, Ind. Eng. Chem. Res. (2021). https://doi.org/10.1021/acs.iecr.0c06248
R. Wegge, M. Richter, R. Span, J. Chem. Eng. Data (2015). https://doi.org/10.1021/je501065g
R. Wegge, M. Richter, R. Span, Fluid Phase Equilib. (2016). https://doi.org/10.1016/j.fluid.2015.10.034
S.L. Outcalt, M.O. McLinden, Ind. Eng. Chem. Res. (2007). https://doi.org/10.1021/ie070791e
E.F. May, W.J. Tay, M. Nania, A. Aleji, S. Al-Ghafri, J.P.M. Trusler, Rev. Sci. Instrum. (2014). https://doi.org/10.1063/1.4894469
Anton Paar GmbH, Instruction Manual, DMA HPM, Density Measuring Cell for High Pressures and High Temperatures (Original Instruction), www.anton-paar.com
X. Yang, C.C. Sampson, O. Frotscher, M. Richter, J. Chem. Thermodyn. (2020). https://doi.org/10.1016/j.jct.2020.106054
Ortiz-Vega, D.O., Hall, K.R., Holste, J.C., Arp, V.D. and Lemmon, E.W., J. Phys. Chem. Ref. Data, to be published (2021)
W. Wagner, A. Pruß, J. Phys. Chem. Ref. Data (2002). https://doi.org/10.1063/1.1461829
E.W. Lemmon, M.O. McLinden, W. Wagner, J. Chem. Eng. Data (2009). https://doi.org/10.1021/je900217v
International Organisation for Standardization, ISO/IEC Guide 98–3:2008 (Switzerland, Geneva, 2008)
Acknowledgements
We thank Prof. Dr. Markus Richter of TU Chemnitz for helpful discussions regarding the present measurements and for useful hints and ideas in the context of planning and conducting the two presented measurement series.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
CWS: experimental work, analysis of results, validation, visualization, and writing—original draft. RS: conceptualization, supervision, resources, writing—review and editing.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scholz, C.W., Span, R. Measurement of the (p, ρ, T) Behavior of Liquid MEA and DEA at Temperatures from (293.15 to 423.15) K and Pressures up to 90 MPa. Int J Thermophys 42, 70 (2021). https://doi.org/10.1007/s10765-021-02808-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-021-02808-x